- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

NMR waterLOGSY as An Assay in Drug Development Programmes for Detecting Protein-Ligand Interactions–NMR waterLOGSY
Published: Vol 10, Iss 13, Jul 5, 2020 DOI: 10.21769/BioProtoc.3666 Views: 6948
Reviewed by: Chiara AmbrogioIvanhoe LeungMostafa Rahnama
Original research article
The authors used this protocol in:

Jul 2018
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
In drug development programmes, multiple assays are needed for the determination of protein-compound interactions and evaluation of potential use in assays with protein-protein interactions. In this protocol we describe the waterLOGSY NMR method for confirming protein-ligand binding events.
Keywords: DrugsBackground
As more altered protein forms are found in disease cells, there has been an increase in drug discovery programmes that rely on primary screening of target proteins with small molecule libraries followed by further medicinal chemistry campaigns to increase the potency of the chemicals emanating from the screens. After primary screens, there is a raft of orthogonal assays that contribute to verification of hit chemical matter and allow stratification of compounds for selecting the best ones to take into hit-to-lead stages and onwards to lead optimization. Among these assays is the NMR-based water Ligand Observed via Gradient SpectroscopY (waterLOGSY) method (Dalvit et al., 2000 and 2001). waterLOGSY is especially useful for detecting the binding of ligands that interact relatively weakly with the target proteins (i.e., dissociation constants in the µM to low mM range), such as would be expected to be associated with initial hits from a large chemical or fragment library screen (Lepre, 2011) prior to medicinal chemistry to improve potency and drug-like properties.
How WaterLOGSY works
The waterLOGSY method makes use of 1H NMR observation of small molecules (ligands) for the detection of ligand-macromolecule binding (Huang and Leung, 2019). It relies on the transfer of proton (1H) magnetization from excited water molecules to ligands either via 1) their direct interaction or 2) indirectly through initial transfer to protons at a protein surface and then relayed onto a protein-bound ligand. The direct interaction of the ubiquitous water molecules with free (unbound) ligands (route 1) leads to an increase in the observed ligand signal intensity due to a direct (through-space) magnetic interaction between water and ligand protons known as a positive nuclear Overhauser effect (nOe). In contrast, magnetization transfer from water to protein then relayed onto a receptor bound ligand (route 2) yields a net decrease in the ligand signal intensity (here due to a negative nOe). This difference in transfer behavior originates in the different tumbling rates of molecules in solution (i.e., their rotational correlation rates) which are “fast” for small, unbound ligands but “slow” for macromolecular receptors and for their bound ligands. Thus, a comparison of ligand signal intensities in the absence and presence of a protein receptor may indicate whether ligand binding has occurred, with a difference in signal intensity being suggestive of binding. Note that a third magnetization transfer mechanism may also occur for exchangeable (acidic) ligand protons due to their dynamic interchange with water protons, which yields the same signal response as the negative nOe regardless of the presence of protein. Thus, responses from exchangeable protons should be ignored for ligand screening purposes.
The waterLOGSY experiment detects only the signals of the ligand(s) when free in solution so relies on the dissociation of the ligand-bound complex and the release of ligand which then carries the negative nOe with it for detection. This process requires that the ligand dissociation rate is sufficiently high for transfer of the ligands into solution for nOe detection prior to it being lost through natural relaxation processes that are always operative. It similarly requires that ligand residence times on the receptor are sufficiently long for the magnetization transfer itself to take place prior to ligand release. As such, waterLOGSY is best suited to the detection of moderate to weak affinity binders, with dissociation constants in the µM to low mM range. The method has proven especially popular in the screening of libraries of small molecule fragments since these typically have weak binding affinities. Strong binding ligands (KD < μM) have residence times on the protein that are too long and their binding is less likely to be detected, leading to the possibility of false negatives in such cases.
Applications of the waterLOGSY method
In this article we present waterLOGSY protocols for evaluation of protein-ligand binding and show how protein-protein interactions can be employed to inhibit protein-ligand binding, thereby confirming the ligand location on the target protein.
WaterLOGSY is a versatile method that allows to asses qualitatively the binding of small ligands to proteins. This can be done with only one ligand present (see basic protocol) but it can also be carried out with multiple ligands present (see screening protocol), therefore allowing it to be used as a medium throughput assay. In this case, it is important to use DMSO solutions of ligand instead of the DMSO-d6 solution used in the basic protocol. Too much DMSO-d6 may prevent the NMR instrument from locking onto the D2O. DMSO could also lead to interference with the protein structure itself. Generally, a maximum of 10% v/v of DMSO-d6 or DMSO should be used.
The first step in any waterLOGSY experiment is to determine that the ligand does not aggregate (see aggregation protocol). Aggregation of small molecules causes a false positive response in the waterLOGSY experiment because the aggregate adopts the tumbling behavior of a macromolecular species and thus gives waterLOGSY responses as if the ligand were bound. This means that every ligand should be tested without protein to eliminate the presence of confounding aggregation. Conversely, this behavior leads to waterLOGSY being a very useful method to assess aggregation of small molecules.
Materials and Reagents
- Norell® Select SeriesTM 3 mm NMR tubes (Sigma-Aldrich, catalog number: NORS36008 )
- Eppendorf Tube® volume (0.5 ml), PCR clean (Sigma-Aldrich, catalog number: EP0030124537-500EA )
- Soda lime Pasteur pipettes unplugged, capacity 2 ml, 230 mm length (VWR, catalog number: 612-1702 )
- Pipette teats, MBL® natural rubber (SciLabware, catalog number: BIBBBPP004 )
- Pipette tips:
10 µl graduated tips (Starlab, catalog number: S1111-3800 )
200 µl yellow tips (Starlab, catalog number: S1111-0806)
1,000 µl blue tips (Starlab, catalog number: S1111-6801) - Phosphate buffered saline tablet (Sigma-Aldrich, catalog number: P4417 )
- Distilled water (in-house)
- D2O (deuterium oxide 99.9 atom % D) (Sigma-Aldrich, catalog number: 151882 )
- DMSO-d6 (dimethyl sulfoxide-d6 "100%", 99.96 atom % D) (Sigma-Aldrich, catalog number: 156914 )
Equipment
- Hand centrifuge without rotor 2 x 15 ml Swing Out Conical Tube Rotor 90° for Hand Centrifuge (Satorius, model: C1011 )
- Scientific Industries SITM Vortex-GenieTM 2 (Scientific Industries, model: SI-0266 , catalog number: 15557335 )
- -80 °C freezer
- pH meter (Jenway, catalog number: 351001 )
- Gilson pipettes, single channel:
Pipetman L, 2-20 µl (Gilson, catalog number: FA10003M )
Pipetman L, 20-200 µl (Gilson, catalog number: FA10005M )
Pipetman L, 100-1,000 µl (Gilson, catalog number: FA10006M )
Procedure
Note: It is assumed local knowledge of NMR spectrometer operation exists for 1H NMR spectroscopy or can be sourced appropriately. Commands listed here relate to the operation of Bruker NMR spectrometers–follow equivalent protocols for other vendors.
- Aggregation protocol
- Dissolve the ligand (compound in DMSO-d6 to obtain a 10 mM stock solution).
Weigh out the compound of interest on an accurate balance and add the DMSO-d6 using a micropipette to dissolve the compound and obtain a stock solution. If the compound was not fully soluble at first, gentle heating (around 40-50 °C) with a hair dryer can be applied for less than 1 min at a time followed by 30 s of vortexing. This can be repeated until the solid is fully dissolved. The resulting solution should be clear with no floating particles of material and not cloudy. - Prepare a fresh buffer solution of 10 mM PBS buffer corrected to pH 7.4.
Dissolve a phosphate buffered saline tablet in 100 ml of water and correct the resulting solution to pH 7.4 by mean of a pH meter. - Add 10 µl of the stock 10 mM ligand solution to a 0.5 ml Eppendorf tube [the following steps (A3-A5) are intended to make a compound dilution to a final concentration of 500 µM].
Use a micropipette and tip to add the solution of ligand to the clean new Eppendorf. Discard the tip. Put the Eppendorf on an Eppendorf stand. - Add 170 µl of the buffer to the Eppendorf and vortex the solution for 30 s.
Use a micropipette and tip to add the buffer solution to the Eppendorf containing the ligand solution. Discard the tip. Close and vortex the Eppendorf at setting 8 for 30 s. Re-open the Eppendorf and put it back on its stand. - Add 20 µl of D2O to the Eppendorf and vortex the solution for 30 s.
Use a micropipette and tip to add D2O to the Eppendorf containing the ligand solution and buffer. Discard the tip. Close the Eppendorf and vortex it at setting 8 for 30 s. Re-open the Eppendorf and put it back on its stand. - Transfer the resulting solution into a 3 mm NMR tube.
Carefully draw the solution in a long Pasteur pipette with the aid of a rubber teat. Then transfer the solution carefully to the tube, putting the pipette completely in and taking it out as the teat is pressed gently to release the solution. Flick the NMR tube a couple of time (towards the floor) to make sure that the liquid moves completely to the bottom of the tube. - Cap and centrifuge the NMR tube.
Cap the NMR tube, place it in a manual hand centrifuge and centrifuge it for a couple of minutes to ensure no bubbles at the surface (as this can cause poor field optimisation (shimming) in the NMR experiment). - Place the tube in the NMR spectrometer for waterLOGSY 1-D NMR detection.
Place the NMR tube in the 3 mm spinner (turbine) and use the appropriate NMR depth gauge to position the tube correctly in the spinner. Transfer the sample/spinner into the NMR spectrometer magnet either via manual placement or via use of a sample transfer robot attached to the magnet cryostat.
- Dissolve the ligand (compound in DMSO-d6 to obtain a 10 mM stock solution).
- waterLOGSY basic protocol
The basic WaterLOGSY is exemplified here with using an initial stock protein solution of C = 50 µM. The concentration of the protein determines the amount of buffer that is needed in the experiment.
Other protein concentrations can be used with buffer amount changed accordingly.- Dissolve the ligand (compound in DMSO-d6 to obtain a 10 mM stock solution).
Weigh out the compound of interest on an accurate balance and add the DMSO-d6 using a micropipette to dissolve the compound and obtain a stock solution. If the compound was not fully soluble at first, gentle heating with a hair dryer can be applied for less than 1 min at a time followed by 30 s of vortexing. This can be repeated until the solid is fully dissolved. - Prepare a fresh buffer solution of 10 mM PBS, 5 mM MgCl2 buffer corrected to pH 7.4.
This buffer is the one required for the protein used in this protocol; the appropriate buffer for the protein under study should be prepared here. Buffers with low proton content, such as PBS, or that are deuterated (e.g., Tris-d11) are preferred to minimise background signals from the buffer. Similarly, concentrations of protonated buffers should be minimised. - Thaw the protein just before the preparation of the sample.
Take the protein out of the -80 °C freezer and place it in a box of dry ice to be carried to the NMR instrument. Take the Eppendorf containing the protein out of the ice and leave it to stand at room temperature to thaw completely. Do not put the protein back in ice until the end of all experiments. In some cases the protein can be frozen back, but only if it is stable enough to do so. For this protocol, we prepared careful aliquots to prevent any protein remaining after carrying out the experiments. - Add 10 µl of the ligand solution to a 0.5 ml Eppendorf tube.
Use a micropipette and tip to add the solution of ligand to the clean new Eppendorf. Discard the tip. Put the Eppendorf on an Eppendorf stand. - Add 130 µl of the buffer to the Eppendorf and vortex the solution for 30 s.
Use a micropipette and tip to add the buffer solution to the Eppendorf containing the ligand solution. Discard the tip. Close and vortex the Eppendorf at setting 8 for 30 s. Re-open the Eppendorf and put it back on its stand. - Add 20 µl of D2O to the Eppendorf and vortex the solution for 30 s.
Use a micropipette and tip to add D2O to the Eppendorf containing the ligand solution and buffer. Discard the tip. Close the Eppendorf and vortex it at setting 8 for 30 s. Re-open the Eppendorf and put it back on its stand. - Add 40 µl of the protein (concentration 50 µM) to the Eppendorf (giving an end volume of 200 µl). The final protein concentration in the assay is 10 µM, the final ligand concentration is 500 µM.
Vortex the Eppendorf containing the protein for 30 s at setting 8. Use a micropipette and tip to add the protein to the Eppendorf containing the ligand solution, buffer and D2O. Discard the tip. Close the Eppendorf and vortex it at setting 8 for 30 s. Re-open the Eppendorf and put it back on its stand. - Transfer the resulting solution into a 3 mm NMR tube.
Carefully draw the solution in a long Pasteur pipette with the aid of a rubber teat. Then transfer the solution carefully to the tube, putting the pipette completely in and taking it out as the teat is pressed gently to release the solution. Flick the NMR tube a couple of time (towards the floor) to make sure that the liquid moves completely to the bottom of the tube. - Cap and centrifuge the NMR tube.
Cap the NMR tube, place it in a manual hand centrifuge and centrifuge it for a couple of minutes to ensure no bubbles at the surface (as this can cause poor field optimisation (shimming) in the NMR experiment). - Place the tube in the NMR spectrometer for waterLOGSY 1-D NMR detection.
Place the NMR tube in the 3 mm spinner (turbine) and use the appropriate NMR depth gauge to position the tube correctly in the spinner. Transfer the sample/spinner into the NMR spectrometer magnet either via manual placement or via use of a sample transfer robot attached to the magnet cryostat.
- Dissolve the ligand (compound in DMSO-d6 to obtain a 10 mM stock solution).
- waterLOGSY protocol for library screening
For the library screening, it is important to choose carefully the compounds that are put together in the experiment. Usually three ligands can be used at once and solutions in DMSO are used–the amount of buffer changed accordingly. The library screening protocol is exemplified here with using an initial stock protein of C = 50 µM.- Dissolve the ligand (compound in DMSO-d6 to obtain a 10 mM stock solution).
Weigh out the compound of interest on an accurate balance and add the DMSO-d6 using a micropipette to dissolve the compound and obtain a stock solution. If the compound was not fully soluble at first, gentle heating with a hair dryer can be applied for less than 1 min at a time followed by 30 s of vortexing. This can be repeated until the solid is fully dissolved.
Repeat this step up to two more times in order to add all the ligands needed to be screened. - Prepare a fresh buffer solution of 10 mM PBS, 5 mM MgCl2 buffer corrected to pH 7.4.
This buffer is the one required for the protein used in this protocol; the appropriate buffer for the protein under study should be prepared here. Buffers with low proton content, such as PBS, or that are deuterated (e.g., Tris-d11) are preferred to minimise background signals from the buffer. Similarly, concentrations of protonated buffers should be minimised. - Add 10 µl of the ligand solution to a 0.5 ml Eppendorf tube.
Use a micropipette and tip to add the solution of ligand to the clean new Eppendorf. Discard the tip. Put the Eppendorf on an Eppendorf stand. Repeat this step for adding additional ligand solutions. - Add 130 µl of the buffer to the Eppendorf and vortex the solution for 30 s (if using 3 ligands).
Use a micropipette and tip to add the buffer solution to the Eppendorf containing the ligand solution. Discard the tip. Close the Eppendorf and vortex it at setting 8 for 30 s. Re-open the Eppendorf and put it back on its stand. - Add 20 µl of D2O to the Eppendorf and vortex the solution for 30 s.
Use a micropipette and tip to add D2O to the Eppendorf containing the ligand solution and buffer. Discard the tip. Close the Eppendorf and vortex it at setting 8 for 30 s. Re-open the Eppendorf and put it back on its stand. - Add 40 µl of the protein (concentration 50 µM) to the Eppendorf (giving an end volume of 200 µl). The final protein concentration in the assay is 10 µM, the final ligand concentration is 500 µM (each).
Vortex the Eppendorf containing the protein for 30 s at setting 8. Use a micropipette and tip to add the protein to the Eppendorf containing the ligand solution, buffer and D2O. Discard the tip. Close the Eppendorf and vortex it at setting 8 for 30 s. Re-open the Eppendorf and put it back on its stand. - Transfer the resulting solution into a 3 mm NMR tube.
Carefully draw the solution in a long Pasteur pipette with the aid of a rubber teat. Then transfer the solution carefully to the tube, putting the pipette completely in and taking it out as the teat is pressed gently to release the solution. Flick the NMR tube a couple of time (towards the floor) to make sure that the liquid moves completely to the bottom of the tube. - Cap and centrifuge the NMR tube.
Cap the NMR tube, place it in a manual hand centrifuge and centrifuge it for a couple of minutes to ensure no bubbles at the surface (as this can cause a bad shimming in the NMR experiment). - Place the tube in the NMR spectrometer for waterLOGSY 1-D NMR detection.
Place the NMR tube in the 3 mm spinner (turbine) and use the appropriate NMR depth gauge to position the tube correctly in the spinner. It is then transferred into the NMR spectrometer magnet either via manual placement or via use of a sample transfer robot attached to the magnet cryostat.
- Dissolve the ligand (compound in DMSO-d6 to obtain a 10 mM stock solution).
- waterLOGSY protocol for competition
For a competition experiment using Y6-ScFv VH, the preparation was carried out in a similar manner as the basic waterLOGSY protocol; it is exemplified here with using an initial stock protein of C = 312 µM and an initial stock antibody of C = 116.6 µM (protein and antibody are in a 1:1 ratio). The concentration of the proteins determines the amount of buffer that is used in the experiment. Other protein concentrations can be used with buffer amount changed accordingly.- Repeat Steps B1 and B2.
- Thaw the protein and the antibody just before the preparation of the sample.
Take the protein and the antibody out of the -80 °C freezer and place them in a box of dry ice to be carried to the NMR instrument. Take the Eppendorf containing the proteins out of the ice and leave it to stand at room temperature to thaw completely. Do not put the proteins back in ice until the end of all experiments. In some cases the protein can be frozen back, but only if it is stable enough to do so. For this protocol, we prepared careful aliquots to prevent any protein remaining after carrying out the experiments. - Repeat Step A3.
- Add 146.4 µl of the buffer to the Eppendorf and vortex the solution for 30 s.
- Use a micropipette and tip to add the buffer solution to the Eppendorf containing the ligand solution. Discard the tip. Close the Eppendorf and vortex it at setting 8 for 30 s. Re-open the Eppendorf and put it back on its stand.
- Repeat Step A5.
- Add 6.4 µl of the protein (concentration 311.8 µM) to the Eppendorf.
Vortex the Eppendorf containing the protein for 30 s at setting 8. Use a micropipette and tip to add the protein to the Eppendorf containing the ligand solution, buffer and D2O. Discard the tip. Close the Eppendorf and vortex it at setting 8 for 30 s. Re-open the Eppendorf and put it back on its stand. - Add 17.2 µl of the antibody (concentration 116.6 µM) to the Eppendorf (giving an end volume of 200 µl).
Vortex the Eppendorf containing the antibody for 30 s at setting 8. Use a micropipette and tip to add the protein to the Eppendorf containing the ligand solution, protein, buffer and D2O. Discard the tip. Close the Eppendorf and vortex it at setting 8 for 30 s. Re-open the Eppendorf and put it back on its stand. - Repeat Steps A6-A8.
Data analysis
- Instrumentation
Solution-phase NMR spectrometer operating at high field (400+ MHz) equipped with a probe suitable for 1H detection and with z-axis pulsed field gradient capabilities and with active sample temperature regulation (default 298 K). The probe temperature can be altered according to protein requirements and stability.
The use of 3 mm diameter NMR tubes is optimal for 5 mm cryoprobes (or 3 mm microprobes) but can also be used with conventional 5 mm NMR probes, albeit with some sensitivity loss. If sample transport spinners (turbines) for 3 mm tubes are not available, these narrow tubes can be placed directly within conventional 5 mm tubes for transportation with 5 mm spinners. Alternatively, 5 mm tubes alone may be employed which requires all sample volumes in this protocol be scaled three-fold to yield final volumes for analysis of 600 µl.
For library screening, a robotic sample changer is desirable to enable high sample throughput, unattended operation and overnight data collection.
Commands listed here relate to the operation of Bruker NMR spectrometers- follow equivalent protocols for other vendors. This protocol is suitable for Bruker AVIII spectrometers running TOPSPIN 3.5, or later generations. - Bruker set-up for each sample (+/- protein)
- Record standard 1H NMR spectrum with H2O water suppression
- Load parameter set WATERSUP (1D NOESY preset sequence).
- Perform standard instrument set-up protocol for 1H NMR detection: With sample in probe: tune 1H channel of probe (atma), lock sample (lock H2O/D2O), shim sample (gradient shim routine, e.g., topshim), calibrate 1H pulse (pulsecal).
- Collect 1H reference spectrum and check authenticity of ligand(s) present.
- Record 1H waterLOGSY NMR spectrum
- Load parameter set: SCREEN_WLOGSY.
- Load 1H pulse calibration determined from above set-up protocol.
- Recommended parameters for waterLOGSY screening:
Pulse program: ephogsygpno.2 (ephogsy was an original implementation of this experiment used for protein hydration studies and known as enhanced protein hydration observed through gradient spectroscopy, the name of which is still retained by Bruker for the waterLOGSY sequence)
Recovery delay (d1): 2 s
Water selective 180° pulse for water excitation (p7:sp21): 7.5 ms Gaussian (Gaus1_180i.1000)
Mixing time (d8): 1s
Water suppression 180° selective pulse (p12:sp1): 2 ms sinc (sinc1.1000) or square profile (Squa100.1000)
Spectral width: 16 ppm
Acquisition time (AQ): 2 s
Number of acquired transients (NS): ≥ 128 (dependent on instrument sensitivity)
Do not spin samples.
- Record standard 1H NMR spectrum with H2O water suppression
- Data processing
Process data with 1 Hz line broadening lb = 1 (command ef to process).
Phase spectra such that resonances of bound small molecules are negative (inverted). Protein signals should appear with negative intensity (Figure 1).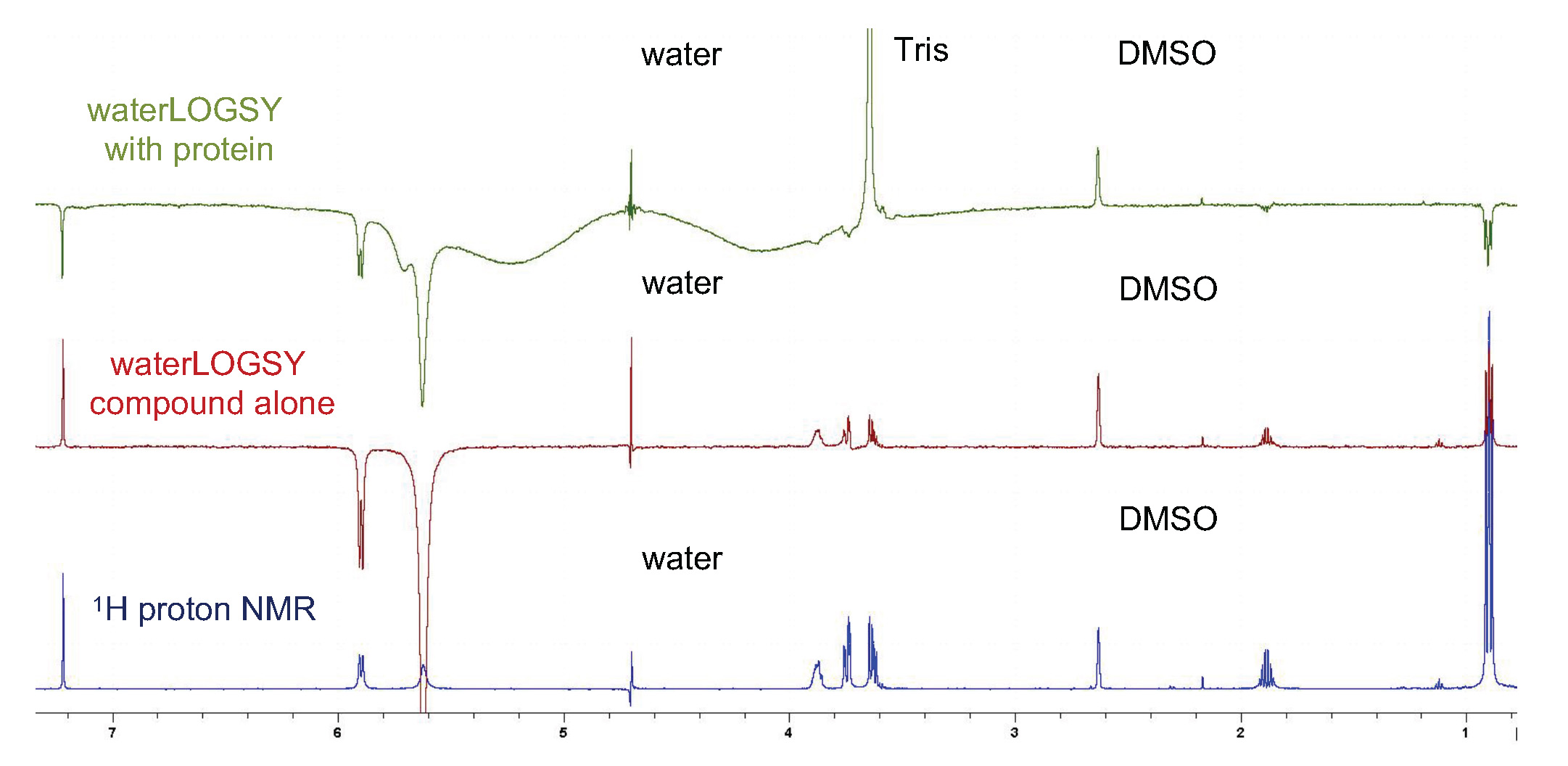
Figure 1. Typical result from a compound not aggregating and binding to the protein. In blue is the proton NMR of the compound. In red is the waterLOGSY of the compound alone. All peaks are still up (positive), showing no aggregation, apart from NHs and OHs (5.6 to 6.0 ppm), which exchange with the water and therefore display negative signals. In green is the waterLOGSY of the compound with protein. All the peaks from the compound are negative, indicating binding. The positive peaks seen in the spectra are the DMSO peak at 2.6 ppm and the residual water peak (still observed even after suppression at 4.7 ppm). This particular solution contained residual Tris from the protein preparation, which is observed at 3.7 ppm.
Acknowledgments
The work of CJRB and THR was supported by a grant from Bloodwise (12051) and THR also by grants from the Medical Research Council (MR/J000612/1) and the Wellcome Trust (100842/Z/12/Z).
Competing interests
The authors have no conflicts of interest to declare.
References
- Dalvit, C., Pevarello, P., Tato, M., Veronesi, M., Vulpetti, A. and Sundstrom, M. (2000). Identification of compounds with binding affinity to proteins via magnetization transfer from bulk water. J Biomol NMR 18(1): 65-68.
- Dalvit, C., Fogliatto, G., Stewart, A., Veronesi, M. and Stockman, B. (2001). WaterLOGSY as a method for primary NMR screening: practical aspects and range of applicability. J Biomol NMR 21(4): 349-359.
- Lepre, C. A. (2011). Practical aspects of NMR-based fragment screening. Methods Enzymol 493: 219-239.
- Huang, R. and Leung, I. K. H. (2019). Protein-small molecule interactions by WaterLOGSY. Methods Enzymol 615: 477-500.
Article Information
Copyright
![]() Bataille et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Bataille et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Bataille, C. J. R., Rabbitts, T. H. and Claridge, T. D. W. (2020). NMR waterLOGSY as An Assay in Drug Development Programmes for Detecting Protein-Ligand Interactions–NMR waterLOGSY. Bio-protocol 10(13): e3666. DOI: 10.21769/BioProtoc.3666.
- Bery, N., Cruz-Migoni, A., Bataille, C. J. R., Quevedo, C. E., Tulmin, H., Miller, A., Russell, A., Phillips, S. E. V., Carr, S. B. and Rabbitts, T. H. (2018). BRET-based RAS biosensors that show a novel small molecule is an inhibitor of RAS-effector protein-protein interactions. eLife 7: e37122.
Category
Biophysics > NMR spectroscopy > NMR imgaing
Cancer Biology > General technique > Drug discovery and analysis
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.