- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Single Cell Volume Measurement Utilizing the Fluorescence Exclusion Method (FXm)

(*contributed equally to this work) Published: Vol 10, Iss 12, Jun 20, 2020 DOI: 10.21769/BioProtoc.3652 Views: 4857
Reviewed by: Samantha E. R. DundonFarah HaqueAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Oct 2018
Advertisement




Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
The measurement of single cell size remains an obstacle towards a deeper understanding of cell growth control, tissue homeostasis, organogenesis, and a wide range of pathologies. Recent advances have placed a spotlight on the importance of cell volume in the regulation of fundamental cell signaling pathways including those known to orchestrate progression through the cell cycle. Here we provide our protocol for the Fluorescence Exclusion Method (FXm); references to the development of FXm; and a brief outlook on future advances in image analysis which may expand the range of problems studied utilizing FXm as well as lower the barrier to entry for groups interested in adding cell volume measurements into their experimental repertoire.
Keywords: Single cellBackground
The first patent for an impedance-based flow cytometry device was issued in 1953 to Wallace H. Coulter (Coulter, 1953), and “Coulter counters” are still used today in the measurement of cell number/concentration as well as to approximate cell volume. They function on the principle that the electrical resistance of a small volume of electrolyte solution changes when a particle is present, displacing its volume of the electrolyte solution. These devices are typically very high throughput and easy to use; however, the measurement of absolute cell volume (in contrast to relative volume across conditions or cell types) requires both a calibration step with an object of known volume (e.g., glass bead) as well as a good approximation of the electrical properties of the cell. If these properties differ greatly from that of the reference object, the absolute volume may be inaccurate. We found these effects to limit the precision of the technique when comparing biological repeats across multiple days (Perez Gonzalez, 2019).
The next thirty years saw the introduction of optical flow cytometers resembling those in common use today and methods for approximating cell size from observed light scattering. While convenient (forward/side scatter is routinely obtained during fluorescent cell sorting) we have found these measurements to be more variable than those obtained through use of a Coulter counter (Perez Gonzalez, 2019). In 1983, Gray et al. (1983) developed a method based on the exclusion of a fluorescent dye within the cell suspension fluid of a flow cytometer which laid the foundation for the Fluorescence Exclusion Method (FXm) discussed here.
The method described by Gray et al. (1983) contains all the basic components of FXm: the cell is surrounded by a membrane-impermeable fluorescent dye, and having excluded some volume of fluid, the total light intensity of an image containing the cell will be darker than an image of an empty frame. The amount of dye excluded, and thus the intensity drop, is proportional to the volume of fluid excluded–the volume of the cell. The biggest difference between this method and FXm, is that in utilizing a flow apparatus, this method requires the cell to be in suspension. Suspending the cell dramatically changes cell physiology and cell shape, likely leading to a change in volume (Tao, 2015; Perez Gonzalez, 2018). Recently, increased interest in single cell volume has motivated a variety of novel methods to replace or supplement flow cytometry in volume measurements. In 2011, Bottier et al. (2011) introduced FXm in the form discussed here, utilizing a microfluidic device and epifluorescence microscope in place of a flow cytometer and eliminating the need for cell suspension. Recent publications by ourselves and others (Zlotek-Zlotkiewicz, 2015; Cadart et al., 2017 and 2018) have extensively adopted FXm and we hope both this protocol and future improvements of this method will encourage more groups to conduct cell volume measurements.
Materials and Reagents
A. Microfluidic Supplies
A thin microfluidic channel is required. This channel is usually constructed out of a sandwich between a polydimethylsiloxane (PDMS) top and a glass coverslip bottom; however, two glass coverslips separated by adhesive tape have also been used (Bottier, 2011). We will cover the use of PDMS in this protocol. The stiffness of the bottom may also be modified by coating with a thin layer of PDMS. Following this protocol, to include the manufacture of custom PDMS chips (which may alternatively be purchased (Bottier, 2011), the following materials will be required:
- Printed mask, (2D) negative of the mold (FineLineImaging, self-designed, may be designed with AutoCAD)
- PDMS for the chip (Sylgard 184)
- 12.6 kPa PDMS for coating (Dow Corning Toray, catalog numbers: CY52-276A and CY52-276B for 12.6 kPa) (Bergert et al., 2016)
- 3 kPa PDMS for coating (Dow Corning Toray, catalog numbers: CY52-276A and CY52-276B) (Style et al., 2014)
- 0.4kPa PDMS for coating (Quantum Silicones, catalog numbers: QGel 920A and QGel 920B) (Gutierrez et al., 2011)
- Glass substrate (50-mm glass-bottom Petri dishes, FlouroDish Cell Culture Dish; World Precision Instruments)
- Blunt-tipped 21-gauge needle and syringe (McMaster Carr, catalog number: 75165A681)
- Isopropyl alcohol (IPA)
B. Cell Culture Supplies
Culture media and supplies are largely standard with the major exception being the fluorescent dye. The dye must be conjugated to something which is sufficiently membrane-impermeable and water soluble so to be uniformly distributed throughout the culture media and excluded from the cell. We tried a variety of conjugates and settled on a large dextran group.
- DMEM (Corning)
- PBS (phosphate-buffered saline solution)
- Penicillin/streptomycin (Gibco)
- FBS (Sigma-Aldrich)
- Matrix Protein (50 μg/ml of type I rat-tail collagen) (Corning, catalog number: 354236)
- Alexa Fluor 488 Dextran molecular weight 2,000 kD (Sigma-Aldrich, catalog number: FD2000S-1G)
Equipment
- Silicon wafers (Wafer World, catalog number: 2886)
- Photoresist (SU8-3000 and developer, Kayaku Advanced Materials)
- Oven (VWR, Gr Con 2.3CF)
- Plasma cleaner (Harrick Plasma, pdc-32g)
- Microscopy/Cell Incubation
The method below utilizes three imaging modalities: confocal, epifluorescence, and differential interference contrast (DIC) microscopy. Only an epifluorescence microscope is essential for this technique; however, we will emphasize the value of a confocal scope for the measurement of microfluidic channel height during device calibration. Other transmitted light images (e.g., phase contrast) may be substituted for the DIC images in this protocol, though we expect some steps to be easier to implement with DIC. Standard cell incubation equipment is required, dependent on the cell type. - Microfabrication
We manufactured our PDMS chips using standard photolithographic equipment; however, due to the relatively large feature size, devices may be assembled to a satisfactory quality outside of a clean room (e.g., a biosafety level 1 space is appropriate). Additionally, this device is a single layer and thus its construction does not require a mask aligner. A simple UV lightsource, spin-coater, and hot plate should be sufficient for mold construction. Bonding of the substrate and PDMS chip is achieved by exposing one or both components to oxygen plasma. A variety of plasma sources may be used. An oven is useful for quickly curing PDMS and ensuring bonding of the PDMS chip to the substrate; however PDMS will eventually cure/bond at room temperature. A sonicator is useful for cleaning the PDMS chips before they are bonded to the substrate but this step may be skipped if necessary.
Software
- MATLAB (MathWorks)
- ZEN 3.0 (Zeiss)
Procedure
A. Construct mold (Pattern silicon wafer)
- Design mask: Our mask, (2D) negative of the mold shown in Figure 1A), is a simple array of straight channels, approximately 17 mm long and 1.5 mm wide with slightly larger circles at each end where the holes used for media/cell injection are punched.
- Center silicon wafer on spin coater and pour photoresist over center of wafer until approximately 2/3rds of the wafer is coated. The wafer may be exposed to oxygen plasma first to aid adhesion of the photoresist if desired. Spin two cycles, first at 500 rpm for 7 s with an acceleration of 100 rpm/s, and again at 2,000 rpm for 30 s with an acceleration of 300 rpm/s. This yields a post-exposure height of approximately 15 μm when using SU8-3000 photoresist and manufacturers’ recommendations.
- Bake (soft bake) on hotplate for approximately 40 min at 80 °C.
- Place mask on top of wafer and expose wafer to UV light (duration will depend on intensity of the light source).
- Wash wafer in developer, rinse (in IPA for SU8-3000), and dry.
Note: With use, the features of the mold degrade. We store our molds in a cool, dry place but do not restrict exposure to air. Additional steps may be taken to slow feature degradation; however, regardless of the steps taken to protect the features of the mold, it is very likely the height will change over time. For this reason we suggest that beyond measuring the height of the mold initially after fabrication (via profilometer etc.), the height of the final PDMS channel itself is measured using confocal microscopy as described below. Additionally, it should be noted that the mold is likely to stick to PDMS during the first few uses and extra care should be taken with a new mold to maintain pattern integrity.
B. Construct microfluidic chip
- Thoroughly mix (stir manually for approximately 1 minute) a 10:1 ratio of PDMS (Sylgard 184) silicone elastomer:curing agent, vacuum degas, pour onto a mold, and cure in an oven at 80 °C for 45 min.
- Peel PDMS from the mold and cut devices into the desired dimensions. We typically arranged the channels so 2-4 independent channels could be bonded to a single substrate and imaged simultaneously.
- Create inlet and outlet ports for injecting cells/media.
Note: This may be accomplished by simply punching holes using a blunt-tipped 21-gauge needle, McMaster Carr. - Wash devices. We sonicate in 100% IPA for 15 min, rinse with water, and dry using a compressed air gun. We found the IPA wash to be particularly useful in clearing the channel of debris when compared to washing with water alone (Figure 1A).
C. Prepare substrate
- Rinse glass substrate with water and dry. We use 50-mm glass-bottom Petri dishes (FlouroDish Cell Culture Dish; World Precision Instruments) which work well for coating with PDMS. If a PDMS coating is desired, to modify the stiffness of the substrate, place the glass dish on a spin-coater.
- For a 12.6 kPa coating, mix a 0.9:1 weight ratio of CY52-276A and CY52-276B.
- For a 3 kPa coating mix a 1:1 weight ratio of CY52-276A and CY52-276B.
- For a 0.4 kPa coating, mix a 1:1 weight ratio of QGel 920A and QGel 920B.
- Cover approximately 2/3rds of the glass with PDMS and spin. Duration and velocity of the spin coater will vary depending on the PDMS and glass substrate used. The goal of spin coating is to obtain an even, flat, and optically transparent layer. The final thickness of the coating needs only to be thin enough that it does not exceed the working distance of the objective used and thick enough that the bulk properties of the underlying glass do not impact the cell (i.e., the cell’s mechanical environment is equivalent to what it would be if placed on a bulk quantity of the coating).
- Expose the PDMS chip to oxygen plasma for approximately 1 min (time will vary by plasma source). This is done to change the surface of the PDMS from hydrophobic to hydrophilic and enables bonding with glass/PDMS. We find plasma treatment wrinkles the PDMS coating on the substrate and do not expose coated substrates to plasma prior to bonding. Uncoated, glass substrates, however, may be exposed to plasma in addition to the PDMS chips to aid bonding.
- Bake bonded devices in an oven at 80 °C for 45 min to ensure bonding (Figure 1B).
D. Seed devices with cells and image
- Expose devices to oxygen plasma for 30 s. This is done to aid collagen/matrix protein deposition; however, as noted above, it can wrinkle PDMS coated substrates.
- Inject matrix protein solution and incubate. We use 50 μg/ml of type I rat-tail collagen (Corning) incubated for 1 h at 37 °C.
- Inject PBS to rinse.
- Prepare cell media. The cells may be seeded in either standard cell culture media (DMEM supplemented with 10% FBS and 1% antibiotic solution, 10,000 units/ml penicillin and 10,000 μg/ml streptomycin [P/S]) or media prepared for imaging which includes the dye, 0.1 μg/ml of Alexa Fluor 488 Dextran molecular weight 2,000 kD. If the dye is not included during the initial seeding, it must be introduced later which risks perturbing and potentially removing cells adhered to the channel. This may be required if long-duration, pre-imaging treatment is conducted after the cells have adhered which would otherwise remain incomplete by the time significant endocytosis occurs. Additionally, it may be noted that while increasing dye concentration increases the signal-to-noise ratio, high dye concentrations tend to photobleach rapidly. Lack of uniformity of this photobleaching may impact the volume calculation.
- Count cells and inject approximately 50 k cells per channel. This estimate requires knowing both the cell concentration in the injected media as well as the approximate volume of the channel.
- Immerse devices in cell culture media to prevent evaporation and incubate (at 37 °C with 5% CO2 and 90% relative humidity) until cells are adhered. Imaging should be conducted as quickly as possible to avoid complications due to endocytosis. We usually completed imaging within 5h of seeding.
- For each region of interest (ROI), two images are taken–one DIC image and one epifluorescence image. Care must be taken in selection of the objective used. Objectives which focus light from a narrow vertical region (distance from the substrate) may not accurately report the total fluorescence emitted from the dye contained in the column of the channel specified by the ROI. Stated another way, the desired total fluorescence signal includes “out of focus” light. This tends to be problematic for higher magnification objectives and when this happens, the below calculation for channel and cell height will be inaccurate. We tested three Zeiss objectives with three different magnifications (63x oil, 0.8 NA; 20x air, 0.45 NA; and 10x air, 0.25 NA) and found that using a 20x air objective, our volume calculations were relatively insensitive to variability in focal plane indicating we are capturing the light emitted from the entire column specified by the ROI. We additionally find this magnification and numerical aperture (NA) to be sufficient for robust shape reconstruction (Figure 1C).
- Set up timelapse experiment if desired. Note that the imaging frequency may be limited by photobleaching or phototoxicity.
- At the completion of the experiment, we recommend capturing a quick-scan (a short pixel-dwell time is sufficient) confocal image at 1 μm interval z-stacks covering the entire depth of the channel at a few extreme ROIs. We typically select 4 ROI’s at the four “corners” of the channel. These stacks may be used to better measure the channel height of the PDMS channel itself which may differ from the original height of mold.
Data analysis
Note: Software utilized: MATLAB (MathWorks) and ZEN 3.0 (Zeiss).
- Measure channel height from confocal stacks. For each ROI at each z-plane, sum the total intensity. Find z value representing the upper and lower local maxima of the second derivative of the intensity as a function of height. We found the distance between these local maxima to be consistent with the channel height. Additionally, we found gross variation among the calculated channel heights for each ROI to indicate problems with channel geometry (bonding, debris, etc.) and typically discarded data from these problematic channels (Figure 1E).
- Identify cell ROIs. Our treatment of the background first requires local bounding rectangles to be identified, each including only one (whole) cell. We do this semi-automatically by cropping each cell into an individual rectangle typically taking under a second of manual work per cell and allowing for a brief visual inspection of the results; however, local minima in the smoothed epifluorescence images could easily be identified to serve as the centroids for automatically generated bounding boxes if desired.
- Smooth epifluorescence image for each ROI. The raw images are very noisy and are not amenable to visualization. We found a 2-D Gaussian image filter (“imgaussfilt” in MATLAB) to be quick and effective provided a reasonably small standard deviation is selected.
- Subtract background from each DIC ROI. As the intensity of the DIC image is not quantified in this protocol (it is in subsequent work, see Future Directions), any method increasing the contrast within the cell and the uniformity outside the cell will work. We typically fit the entire DIC ROI to a 2nd or 3rd order polynomial and subtract that surface from the original image or alternatively smooth the DIC image using a 2-D Gaussian image filter with a large standard deviation and subtract the smoothed image.
- Identify rough cell boundary from the smoothed epifluorescence image. Fit a plane to pixels corresponding to the background of the image. This may be done by fitting a plane to only bright pixels (top 20%, 50%) or conversely excluding dark pixels based on intensity distribution. Create a binary image (mask) representing pixels above/below that plane and identify the largest “blob” in the binary image.
Note: Given only epifluorescence images, this rough cell boundary may be used for volume calculation as the majority of the cell volume will be included; however, thin, protrusive regions of the cell may be lost which will affect shape reconstruction and potentially account for a significant total volume fraction in some cell types. - Identify high contrast regions of the DIC image to expand the cell boundary and include thin, protrusive regions. This can be done by identifying regions of high local standard deviation, or a Euclidean distance transformation (e.g., “bwdist” in MATLAB).
- Combine the DIC and epifluorescence masks. Connect and trim “blobs” as appropriate (e.g., apply “strel”, “imerode” in MATLAB). Remove smaller “blobs” unattached to largest “blob” and fill holes (e.g., “bwareaopen”, “imfill” in MATLAB). The order in which these morphological functions are implemented will affect the final mask. Identify the boundary of the cell from that of the primary “blob” (Figure 1D).
- Cell height calculation. To calculate the volume and later surface area as well as shape reconstruction the height of the cell at each pixel is determined. The height of the cell, hcell at any point is simply.
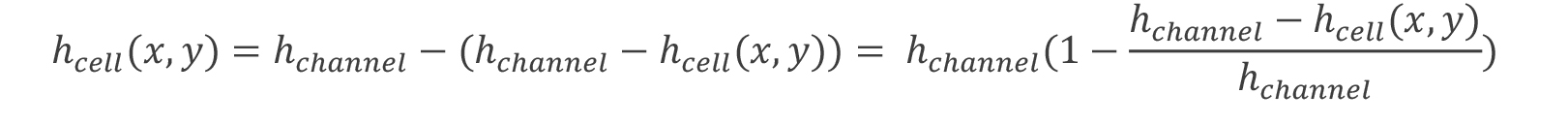
Since the fluorescence intensity at each pixel, I(x,y) is proportional to the amount of dye, we may rewrite this to be: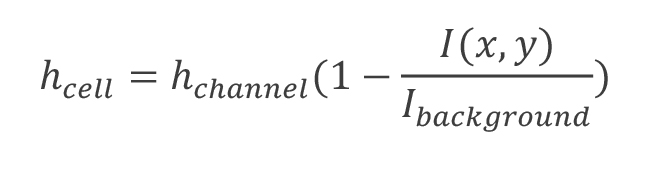
The height of the channel has been obtained from the confocal imaging, and the epifluorescence image has been smoothed. Now what remains is the calculation of the background intensity and any normalization desired across the ROI. - Epifluorescence intensity normalization. We have found it impractical to maintain a uniform background intensity across an entire ROI. Nonuniformity can be caused by variations in the channel height itself as well as any object within the lightpath of the microscope (debris in the field actually displacing dye, smudged glass, impurities in the PDMS, etc.). For this reason, we focused on establishing the optimal neighborhood outside the cell boundary for averaging background intensity. We chose an annulus with an inner boundary specified by dilating the cell boundary by 10 pixels and an outer boundary by 25 pixels and took the mean intensity within this annulus to be Ibackground. Alternatively, a plane may be fit to this annulus and Ibackground may be specified as a function of space; however, we found that in cases where specifying Ibackground this way resulted in a significantly different calculated volume, irregularities in the ROI (large debris etc.) were often observed and these ROI were discarded.
- Volume calculation. Given the cell height at each pixel, the volume of the cell was calculated by simply summing the heights and multiplying by the pixel “area”. All pixels within a boundary dilated 20 pixels from the cell boundary were included in the summation to account for light scattering.
- Surface area calculation. First calculate the surface normal given the 3D coordinates of every point known on the surface–pixel positions for (x,y) and heights for z (e.g., “surfnorm” in MATLAB). Next calculate:
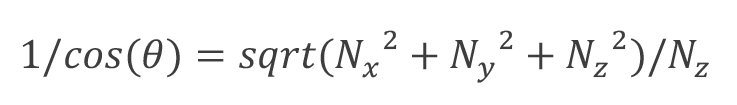
Where theta is the angle between the normal vector (Nx,Ny,Nz) and its z component. The “surface area” for each pixel is the “area” of each pixel multiplied by this value. When the surface is horizontal, and the normal vector points in the z direction, θ = 0,1/cos(θ) = 1. These values may be summed within the cell boundary or a numerical integration scheme may be applied (e.g., “trapz” in MATLAB) to improve accuracy (as we did). Note the normal vectors for the outer perimeter pixels corresponding to the “edge” of the cell should be excluded in this calculation. - Cell shape reconstruction. Given the cell height map and DIC image, 3D illustrations of the cell may be constructed for each frame, as well as GIFs for timelapse experiments. For example, one may use the “surface” function in MATLAB, specifying the height map for z data, “texturemap” for “FaceColor”, “none” for “EdgeColor”, and the DIC image for “Cdata”. Taking care to stabilize the center of the ROI and DIC brightness over time can create compelling visuals of cell migration and shape change using this method (Figure 1F).
- Cell shape statistics. In addition to standard cell shape statistics for the 2D projected area (e.g., ratio of perimeter to area), given the height map, the apical surface of the cell may be similarly analyzed across populations. In particular, we have found for cell types which have sufficiently round 2D projected shapes, reporting the mean height as a distance from the center of volume (equivalent to center of mass) of the cell is useful for comparing phenotypes. Analogous to the difference between cells which are elongated or circular in their 2D projected shapes, cells which have slowly or quickly decaying height functions from the center of mass are flatter (more “pancake-like”) or more hemispherical.
Future Directions
The greatest challenges towards the implementation of FXm today remain the limited duration of timelapse experiments due to endocytosis, the limited timestep of timelapse experiments due to photobleaching/phototoxicity, and the limited range of experimental protocols which may be realized within the microdevice. In an effort to widen the range of problems which may be studied utilizing FXm, current work from our group (Yao et al., 2019) presents a new method which leverages data collected via this protocol to train a neural network capable of predicting cell height maps directly from DIC images (Figure 1G). While the collection of experiment-specific or cell type-specific training data may still be required at this stage, we hope that further development of this method may lower the barrier to entry for groups interested in adding cell volume measurements into their experimental repertoire.
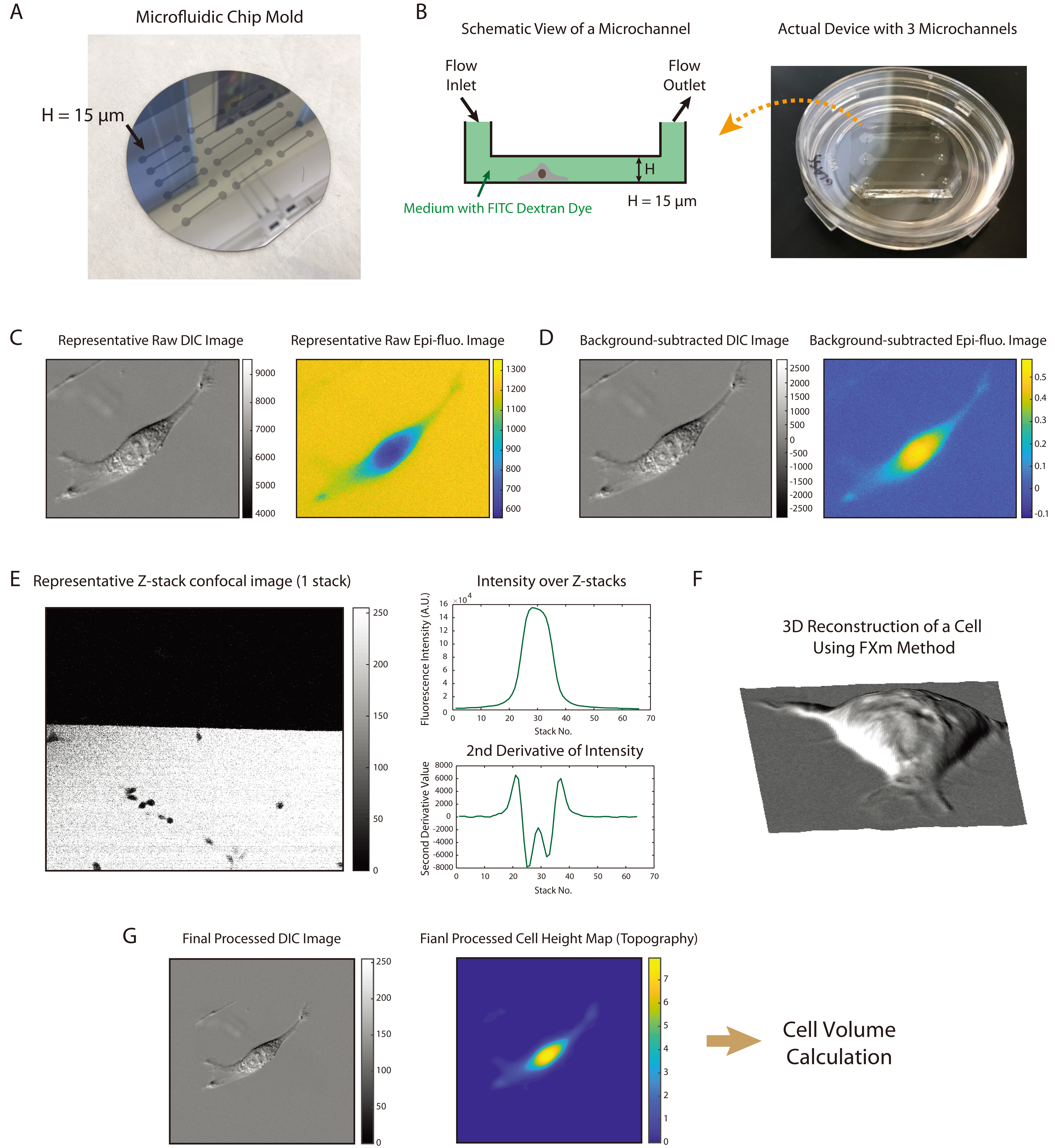
Figure 1. Illustrated Walkthrough of FXm. Beginning with microfabrication and ending with image analysis and future directions.
Acknowledgments
This work has been funded in part by National Institutes of Health grants R01GM114674 and U54CA210172. “Dynamic measurement of the height and volume of migrating cells by a novel fluorescence microscopy technique” was a primary influence for the development of this protocol Bottier et al. (2011).
Competing interests
The authors have no conflicts of interest or competing interests to report.
References
- Bergert, M., Lendenmann, T., Zundel, M., Ehret, A. E., Panozzo, D., Richner, P., Kim, D. K., Kress, S. J., Norris, D. J., Sorkine-Hornung, O., Mazza, E., Poulikakos, D. and Ferrari, A. (2016). Confocal reference free traction force microscopy. Nat Commun 7: 12814.
- Bottier, C., Gabella, C., Vianay, B., Buscemi, L., Sbalzarini, I. F., Meister, J. J. and Verkhovsky, A. B. (2011). Dynamic measurement of the height and volume of migrating cells by a novel fluorescence microscopy technique. Lab Chip 11(22): 3855-3863.
- Cadart, C., Monnier, S., Grilli, J., Sáez, P. J., Srivastava, N., Attia, R., Terriac, E., Baum, B., Cosentino-Lagomarsino, M. and Piel, M. (2018). Size control in mammalian cells involves modulation of both growth rate and cell cycle duration. Nat Commun 9(1): 3275.
- Cadart, C., Zlotek-Zlotkiewicz, E., Venkova, L., Thouvenin, O., Racine, V., Le Berre, M., Monnier, S. and Piel, M. (2017). Fluorescence eXclusion Measurement of volume in live cells. Methods Cell Biol 139: 103-120.
- Coulter, W. H. (1953). Means for counting particles suspended in a fluid. U.S. Patent No. 2,656,508. Washington, DC: U.S. Patent and Trademark Office.
- Gray, M. L., Hoffman, R. A. and Hansen, W. P. (1983). A new method for cell volume measurement based on volume exclusion of a fluorescent dye. Cytometry 3(6): 428-434.
- Gutierrez, E., Tkachenko, E., Besser, A., Sundd, P., Ley, K., Danuser, G., Ginsberg, M. H. and Groisman, A. (2011). High refractive index silicone gels for simultaneous total internal reflection fluorescence and traction force microscopy of adherent cells. PLoS One 6(9): e23807.
- Perez Gonzalez, N. A., Rochman, N. D., Yao, K., Tao, J., Le, M. T., Flanary, S., Sablich, L., Toler, B., Crentsil, E., Takaesu, F., Lambrus, B., Huang, J., Fu, V., Chengappa, P., Jones, T. M., Holland, A. J., An, S., Wirtz, D., Petrie, R. J., Guan, K. L. and Sun, S. X. (2019). YAP and TAZ regulate cell volume. J Cell Biol 218(10): 3472-3488.
- Perez Gonzalez, N., Tao, J., Rochman, N. D., Vig, D., Chiu, E., Wirtz, D. and Sun, S. X. (2018). Cell tension and mechanical regulation of cell volume. Mol Biol Cell 29(21).
- Style, R. W., Boltyanskiy, R., German, G. K., Hyland, C., MacMinn, C. W., Mertz, A. F., Wilen, L. A., Xu, Y. and Dufresne, E. R. (2014). Traction force microscopy in physics and biology. Soft Matter 10(23): 4047-4055.
- Tao, J. and Sun, S. X. (2015). Active biochemical regulation of cell volume and a simple model of cell tension response. Biophys J 109(8): 1541-1550.
- Yao, K., Rochman, N. D. and Sun, S. X. (2020). CTRL - a label-free artificial intelligence method for dynamic measurement of single-cell volume. J Cell Sci 133(7).
- Zlotek-Zlotkiewicz, E., Monnier, S., Cappello, G., Le Berre, M. and Piel, M. (2015). Optical volume and mass measurements show that mammalian cells swell during mitosis. J Cell Biol 211(4): 765-774.
Article Information
Copyright
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Rochman, N. D., Yao, K., Perez Gonzalez, N. A., Wirtz, D. and Sun, S. X. (2020). Single Cell Volume Measurement Utilizing the Fluorescence Exclusion Method (FXm). Bio-protocol 10(12): e3652. DOI: 10.21769/BioProtoc.3652.
Category
Cell Biology > Cell-based analysis > Flow cytometry
Cell Biology > Cell imaging > Fluorescence
Cell Biology > Cell imaging > Microfluidics
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.