- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

In vitro Assay for Bacterial Membrane Protein Integration into Proteoliposomes
Published: Vol 10, Iss 10, May 20, 2020 DOI: 10.21769/BioProtoc.3626 Views: 5932
Reviewed by: David A. CisnerosAgnieszka ZienkiewiczSandeep Dave
Original research article
The authors used this protocol in:

Dec 2019
Advertisement




Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
It is important to experimentally determine how membrane proteins are integrated into biomembranes to unveil the roles of the integration factors, and to understand the functions and structures of membrane proteins. We have developed a reconstitution system for membrane protein integration in E. coli using purified factors, in which the integration reaction in vivo is highly reproducible. This system enabled not only analysis of membrane-embedded factors including glycolipid MPIase, but also elucidation of the detailed mechanisms underlying membrane protein integration. Using the system, the integration of membrane proteins can be evaluated in vitro through a protease-protection assay. We report here how to prepare (proteo)liposomes and to determine the activities of membrane protein integration.
Keywords: Membrane protein integrationBackground
Membrane proteins and presecretory proteins, synthesized in the cytosol, are integrated into and translocated across biomembranes, respectively, to be localized at their destinations and to express their functions. Cells possess systems to integrate membrane proteins into and translocate presecretory proteins across biomembranes, these proteins being commonly conserved from bacteria to higher eukaryotes.
In E. coli, a model organism, some integration factors were identified through genetic approaches, these factors including translocon SecYEG (Newitt and Bernstein, 1998), and signal recognition particle (SRP) and its receptor SR (Ulbrandt et al., 1997), and membrane protein insertase/chaperone YidC (Samuelson et al., 2000). These genetic studies were complemented by biochemical approaches. The molecular mechanisms underlying membrane protein integration have been studied extensively using an in vitro system, in which the protein integration reactions were reproduced in test tubes.
Inverted membrane vesicles (INV) can be prepared by disrupting E. coli cells with a French press, followed by sucrose gradients (Alami et al., 2002). The outside surface of INV corresponds to the cytoplasmic face of the inner membrane. Therefore, presecretory proteins are translocated into the lumen of INV. Membrane proteins, synthesized outside of INV using the cell-free translation system, are integrated into INV. The roles of the above-mentioned factors have been analyzed using INV prepared from the respective mutant cells (Koch et al., 1999; Koch and Muller, 2000). However, since most of the factors are essential for cell growth, depletion of each of the factors causes unexpected pleiotropic effects. To exclude such secondary effects, a reconstitution system has been developed using purified factors. Proteoliposomes can be prepared by mixing membrane proteins, solubilized and purified in a detergent solution, and phospholipids, followed by removal of the detergent. The reconstituted proteoliposomes are essentially the same as INV and thus they can be directly used in the integration assay in vitro. On the other hand, disordered spontaneous integration of membrane proteins into liposomes formed of phospholipids is a serious problem, since such spontaneous integration is an in vitro artefact that does not reflect the reaction in vivo. We have solved this problem by introducing diacylglycerol (DAG) into the liposomes (Nishiyama et al., 2006; Kawashima et al., 2008). As a consequence, DAG blocked spontaneous integration at the physiological content, which enabled reconstitution of membrane protein integration. By using such a reconstitution system we identified a glycolipid named MPIase (Membrane Protein Integrase) as an essential factor for membrane protein integration (Nishiyama et al., 2006; Nishiyama et al., 2010). Based on the observation that MPIase catalyzes membrane protein integration (Nishiyama et al., 2010), we proposed that MPIase is a “glycolipozyme” (Nishiyama et al., 2012). We also determined the MPIase structure, in which a glycan chain composed of 9~11 repeats of a unit of three N-acetylated amino sugars, GlcNAc, ManNAcA and Fuc4NAc, is connected to DAG through a pyrophosphate linker (Nishiyama et al., 2012). Thus, membrane protein integration can be reproduced by introducing lipid components such as DAG and MPIase into reconstituted proteoliposomes.
Recently, we found another problem when reconstituting proteoliposomes. DAG is not only solubilized by a series of detergents such as dodecyl maltoside (DDM) and dodecyl phosphocholine (DPC), but also forms wax-like complexes with them (Sasaki et al., 2019). Since these wax-like complexes induce the spontaneous integration, the detergents must be removed completely from the reconstituted proteoliposomes. On the other hand, DDM and DPC are frequently used to purify the SecYEG complex (Collinson et al., 2001) and YidC (Stiegler et al., 2011; Welte et al., 2012). Therefore, the membrane components should be properly reconstituted without the formation of the unexpected complexes that induce spontaneous integration.
In this protocol, we report a method for reconstituting proteoliposomes containing SecYEG and YidC, but free of the complex of DAG and DDM/DPC. To achieve this, SecYEG and/or YidC were first reconstituted into DAG-free proteoliposomes, followed by membrane fusion with liposomes containing DAG and MPIase by freezing-thawing-sonication, which yielded unilamellar proteoliposomes (Sasaki et al., 2019). Such proteoliposomes are ready for the membrane integration assay. This assay system includes a Pure System, a reconstituted translation system, to in vitro synthesize substrate membrane proteins (Nishiyama et al., 2010; Shimizu et al., 2001). The integration activity was determined by analyzing the membrane protected fragments (MPF) (Koch et al., 1999). The MPFs generated upon protease digestion after the integration reaction reflect the membrane integration.
Materials and Reagents
- Plastic tubes (1.5 ml) (Greiner, catalog number: 616201 )
- Ultracentrifuge tubes (1.5 ml) (Beckman Coulter, catalog number: 357448 )
- Glass vials (2 ml) (AS ONE, catalog number: 9-852-01 )
- Dialysis tubes (Tokyo Garasu Kikai Co., Ltd, catalog number: 0 3272337 )
- E. coli polar lipid extract (phospholipids) (Avanti Polar Lipids, Inc., catalog number: 100600C )
- 1,2-Dioleoyl-sn-glycerol (DAG) (Merck, catalog number: D0138 )
- 2-[4-(2-Hydroxyethyl)-1-piperazinyl]ethanesulfonic acid (HEPES) (Dojindo Laboratories, catalog number: 342-01375 )
- Dithiothreitol (Wako, catalog number: 048-29224 )
- Chloroform, Guaranteed Reagent (Wako, catalog number: 038-02606 )
- Acetone, Guaranteed Reagent (Wako, catalog number: 012-00343 )
- n-Octyl-β-D-glucopyranoside (OG) (Dojindo Laboratory, catalog number: O001 )
- [35S]-EXPRESS Protein Labeling Mix (PerkinElmer Inc, catalog number: NEG072 )
- Pure System (Gene Frontier, catalog number: PF001-0.25 )
- Proteinase K (Roche, catalog number: 03 115 879 001 )
- Trichloroacetic acid, Guaranteed Reagent (Wako, catalog number: 204-02405 )
- Acrylamide, Electrophoresis grade (Wako, catalog number: 011-08015 )
- N,N'-Methylenebis(acrylamide), Electrophoresis grade (Wako, catalog number: 130-06031 )
- Sodium dodecyl sulfate (SDS), for Molecular Biology (Wako, catalog number: 194-13985 )
- 2-Amino-2-hydroxymethyl-1,3-propanediol (Tris), Guaranteed Reagent (Wako, catalog number: 207-06275 )
- Na2HPO4, Guaranteed Reagent (Wako, catalog number: 197-02865 )
- NaH2PO4, Guaranteed Reagent (Wako, catalog number: 192-02815 )
- Glycine, Guaranteed Reagent (Wako, catalog number: 077-00735 )
- Bromophenol Blue (Wako, catalog number: 021-02911 )
- β-Mercaptoethanol (Wako, catalog number: 131-14572 )
- Glycerol, Guaranteed Reagent (Wako, catalog number: 075-00611 )
- Ffh, purified as described (Eisner et al., 2003)
- FtsY, purified as described (Koch et al., 1999)
- YidC, purified as described (Nishikawa et al., 2017)
- SecYEG, purified as described (Moser et al., 2013)
- MPIase, purified as described (Nishiyama et al., 2010 and 2012)
- TALON Metal Affinity Resin (Clontech, catalog number: 635503 )
- Imaging plates (GE, BAS IP MS 2025 E, catalog number: 28-9564-75 )
- Buffer A (see Recipes)
- OG stock (see Recipes)
- Sodium phosphate buffer (pH 7.2) (see Recipes)
Equipment
- Bath-type sonicator (Branson, model: Bransonic 12 )
- Freeze-dryer (EYELA, model: FD-80 )
- Vacuum pump (ULVAC, model: GCD-051X )
- Ultracentrifuge (Beckman Coulter, model: Optima TL )
- Rotor (Beckman Coulter, model: TLA-55 )
- Incubator (Eppendorf, model: Thermomixer Comfort )
- Phosphorimager (GE, model: Storm 820 )
- -80 °C freezer
Software
- ImageQuant software (GE Healthcare)
Procedure
- Preparation of proteoliposomes containing proteinaceous factors such as SecYEG and YidC
- Place the phospholipid solution (25 mg/ml in chloroform) in a glass vial. For one reconstitution sample, use 200 μg of phospholipids. Dry phospholipids up under a N2 gas stream until the bulk of chloroform is evaporated, and then place in a freeze-dryer under vacuum for 1 h.
- Suspend the phospholipids (200 µg/one reconstitution sample) in buffer A (50 mM HEPES-KOH, pH7.5, 1 mM dithiothreitol) at 10 mg/ml, followed by bath-sonication (about 10 s) to form liposomes until a slightly opaque suspension is obtained.
- Solubilize liposomes thus prepared in 1.5% [w/v] OG in buffer A by adding 15% [w/v] stock.
- Mix the solubilized phospholipids (200 µg) with SecYEG (~10 μg) and/or YidC (~10 μg) solubilized in 1.5% [w/v] OG. Adjust the volume to ~100 µl, keeping the OG concentration at 1.5% [w/v]. Incubate the mixture on ice for 20 min.
- Change the detergents used to solubilize SecYEG and YidC to OG, if they were solubilized in DDM or DPC. Apply the sample to a TALON column, and then wash with buffer containing 1.5% [w/v] OG, followed by elution with buffer containing 100 mM imidazole. In the case of SecYEG, add 40% [w/v] glycerol to the buffer. Keep the DDM/DPC concentration as low as possible.
- Dialyze the mixture against 500 ml of buffer A at 4 °C for at least 3 h to form proteoliposomes by removing OG. The proteoliposome suspension becomes a bit turbid.
- Add 0.9 ml of buffer A to the dialyzed sample, followed by recovery of proteoliposomes by centrifugation (170,000 x g, 1 h, 4 °C). Resuspend the pellets in 50 µl of buffer A.
- Make proteoliposomes unilamellar by freezing (in liquid nitrogen)-thawing (at room temperature)-sonication (~10 s) once. Make sure that the suspension is not heated when it is sonicated.
- Store frozen proteoliposomes at -80 °C, if necessary, before thawing and sonication.
- Preparation of liposomes containing DAG
- Mix phospholipids (1 mg) and DAG (0.05 mg), and dry the lipid mixture up as in Step A1.
- Suspend the dried residue in buffer A at 10 mg phospholipids/ml, followed by bath-sonication (~10 s).
- Preparation of liposomes containing DAG and MPIase
- Mix phospholipids and DAG in chloroform, and dry the mixture up, as described in Step B1. Typically, add DAG at 25% [w/w] to phospholipids.
- Dissolve the mixture in solvent C (chloroform/ethanol/water = 3/7/4), and then mix with MPIase dissolved in solvent C, typically at 25% [w/w] as to phospholipids.
- Evaporate solvent C under a N2 stream and then under vacuum.
- Suspend the dried residue in buffer A at 10 mg phospholipids/ml, followed by bath-sonication (~10 s).
- Fusion of proteoliposomes with MPIase/DAG-liposomes to incorporate MPIase and DAG
- Mix the proteoliposome suspension obtained in Procedure A with MPIase/DAG-liposomes (Procedure C), typically in the ratio of 4:1, which yields proteoliposomes containing 5% [w/w] DAG and 5% [w/w] MPIase.
- Repeat the cycle of freezing (in liquid nitrogen)-thawing (at room temperature)-sonication (~10 s) three times to allow liposome fusion. With this way, proteoliposomes containing SecYEG, YidC, and MPIase/DAG can be prepared.
- Reactions for (3L-)Pf3 coat integration (Figure 1)
- Translate in vitro the substrate proteins, Pf3 coat and the mutant 3L-Pf3 coat (Kiefer and Kuhn, 1999; Serek et al., 2004), using the Pure System, a reconstituted cell-free system for in vitro translation (Nishiyama et al., 2010; Shimizu et al., 2001). The reaction mixture of the Pure System (20 µl) comprises [35S] methionine (2.5~10 MBq/ml; ~200 nM), plasmid DNA (pT7-3L-Pf3 or pT7-Pf3 [Kawashima et al., 2008; Serek et al., 2004]; ~20 µg/ml), and (proteo)liposomes (0.4 mg phospholipids/ml) (Table 1). When a high level of synthesis is necessary, add cold methionine (0.3 mM) to the mixture. Under these conditions, a ~200-fold increase in the synthesis level is observed. The Pure System is commercially available from Gene Frontier, but adjust the buffer system customly as described (Nishiyama et al., 2010). To avoid aggregation of proteoliposomes, make sure that the Mg concentration is 9 mM or less, and that PEG and polyamines such as putrescine and spermidine are not contained in the reaction mixtures.
- Allow the translation/integration reaction at 37 °C for 30 min.
- Terminate the reaction by chilling on ice for 5 min. Withdraw 3 µl and 15 µl of the mixture (20 µl) into new tubes.
- Add 3 µl of trichloroacetic acid (10% [w/v]) to an aliquot (3 µl) to precipitate the synthesized proteins.
- Mix an equal volume of proteinase K (1 mg/ml) with another aliquot (15 µl), and then incubate at 25 °C for 25 min to generate MPF.
- After proteinase K digestion, add 30 µl of 10% [w/v] trichloroacetic acid, followed by incubation at 56 °C for 5 min to inactivate proteinase K. Incubate on ice for at least 10 min to precipitate MPF.
- Recover the precipitates (both Steps E4 and E6) by centrifugation (10,000 x g, for 5 min), and then wash them with 100% acetone. After centrifugation (10,000 x g, for 5 min), dry the precipitates up at room temperature for 20~30 min.
- Solubilize the precipitates in 10 µl of SDS sample loading buffer A, followed by boiling for 3 min. The SDS sample loading buffer A is composed of 25 mM sodium phosphate (pH 7.2), 2.5% [w/v] SDS, 25% [w/v] glycerol, 2.5% [w/v] β-mercaptoethanol, and 0.0125% [w/v] bromophenol blue.
- Apply samples onto SDS-gels containing 6 M urea as described in Nishiyama et al. (2006). The gel is composed of 12.5% [w/v] acrylamide-0.27% [w/v] N,N'-methylenebis(acrylamide) in 100 mM sodium phosphate buffer (pH 7.2) as described in Hussain et al. (1980).
- After electrophoresis, soak the gels in 10% [v/v] acetic acid for 10 min, and then dry them.
- Expose the gels to the imaging plates in a cassette overnight.
Table 1. Composition of the Pure System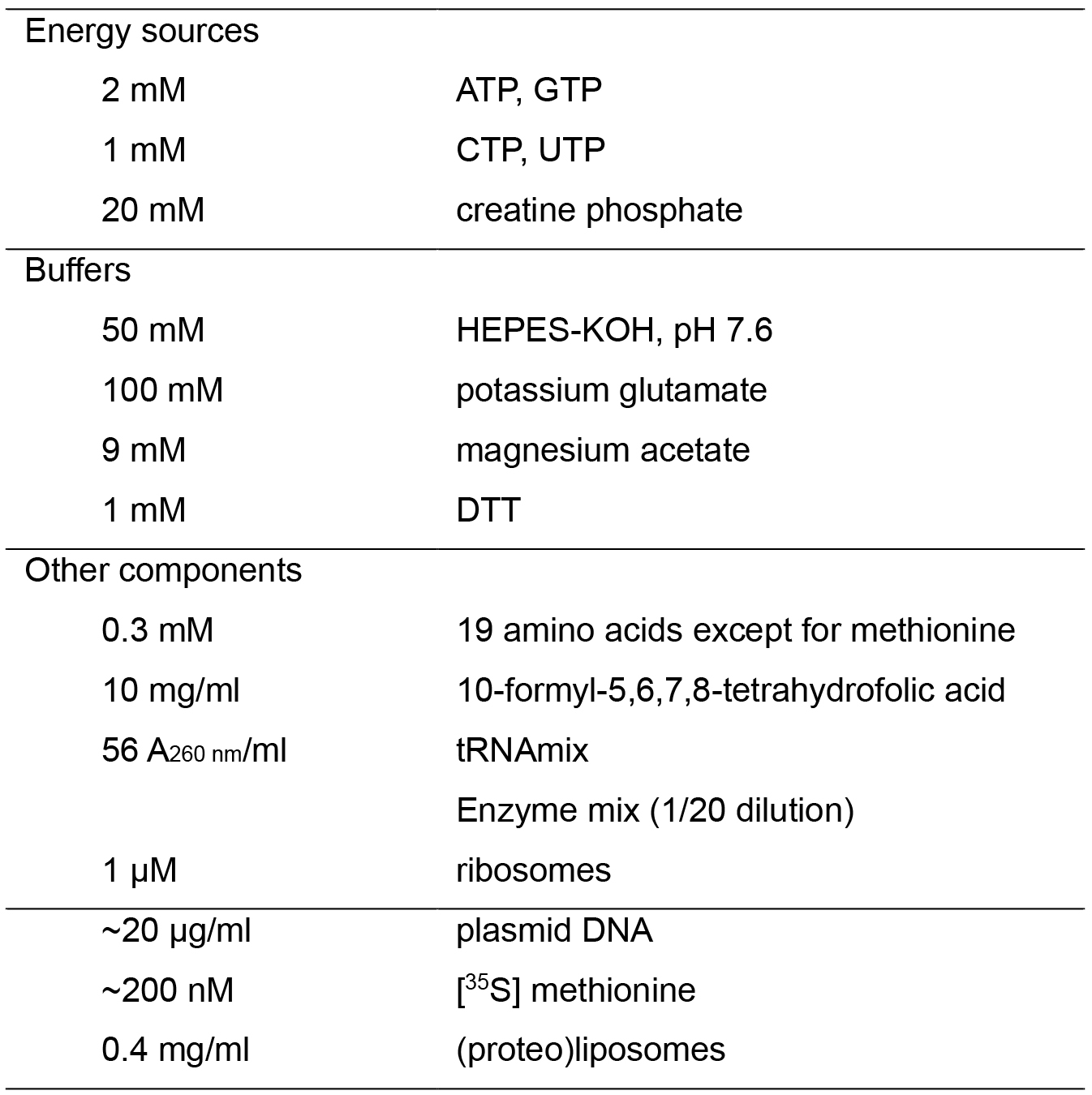
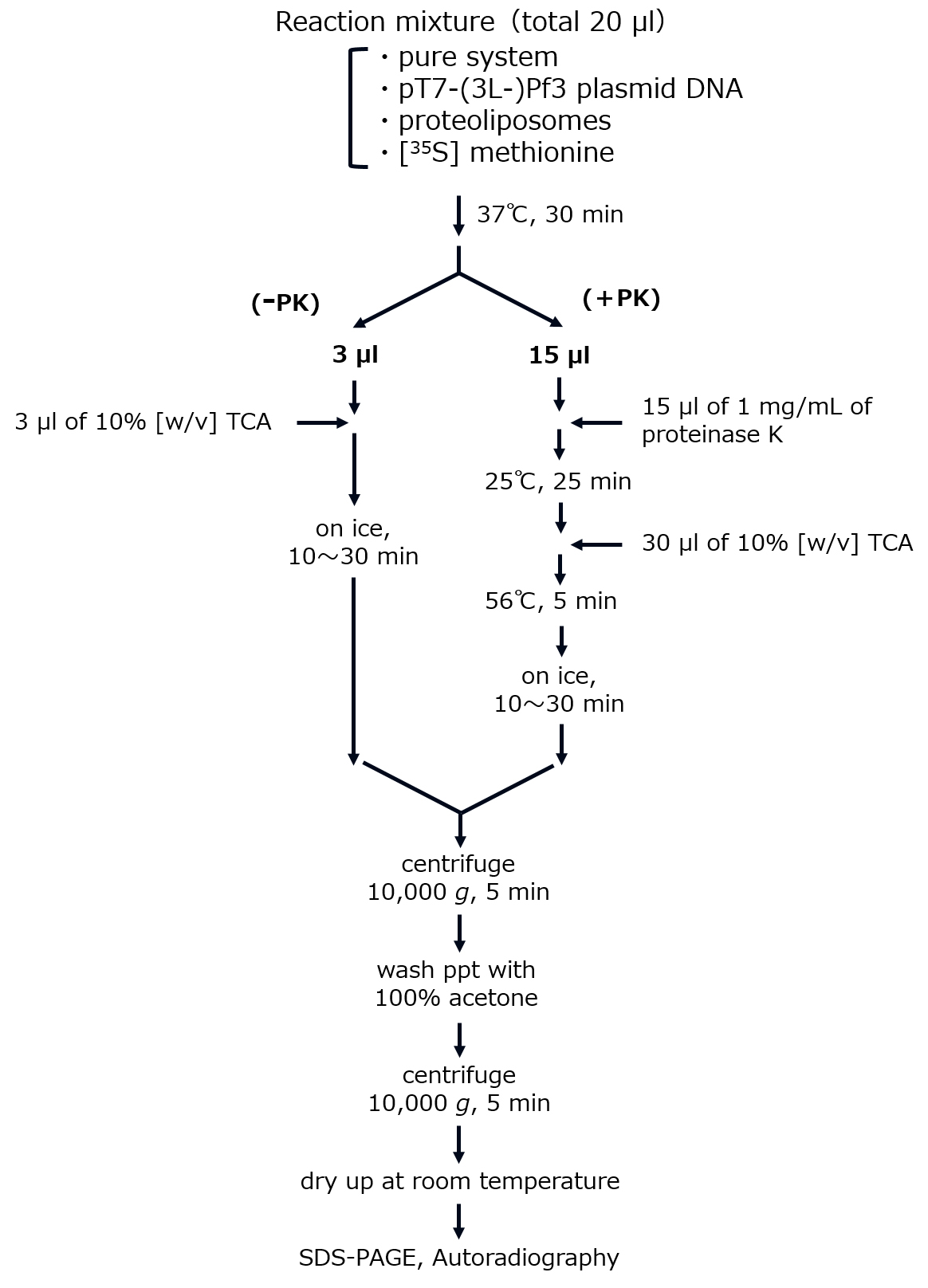
Figure 1. Scheme of the assay for (3L-)Pf3 coat integration
- Reactions for MtlA (mannitol permease) integration
- Synthesize MtlA as in E, using pET-MtlA (Kuruma et al., 2005) and the Pure System. Add Ffh (50 µg/ml), FtsY (17 µg/ml), and cold methionine (30 µM) to the reaction mixture.
- After the translation/integration reaction at 37 °C for 30 min, add 5% [w/v] trichloroacetic acid to an aliquot (9 µl), and digest another aliquot (9 µl) with proteinase K as described in Steps E4-E5. Recover the precipitate as described in Step E5.
- Solubilize the dried precipitates in 10 µl of SDS sample loading buffer B, followed by boiling for 3 min. The SDS sample loading buffer B is composed of 62.5 mM Tris-HCl (pH 6.8), 3% [w/v] SDS, 10% [w/v] glycerol, 5.5% [w/v] β-mercaptoethanol, and 0.01% [w/v] bromophenol blue.
- Load samples onto SDS-gels as described (Laemmli, 1970). The gel is composed of 15% [w/v] acrylamide-0.4% [w/v] N,N'-methylenebis(acrylamide).
- After electrophoresis, soak gels in 10% [v/v] acetic acid for 10 min, and then dry them. Place the gels on the imaging plates.
Data analysis
Determination of the integration activity
- Detect radioactive bands of the synthesized substrates and MPF using a Phosphorimager.
- Determine the radioactivity of each band using the ImageQuant software (Figure 2). Subtract the background level manually with the software.
- Determine the integration activities by dividing the MPF level with that of the synthesized substrate (Figure 2, bottom of each gel image). Consider the numbers of methionine in substrates and MPF.
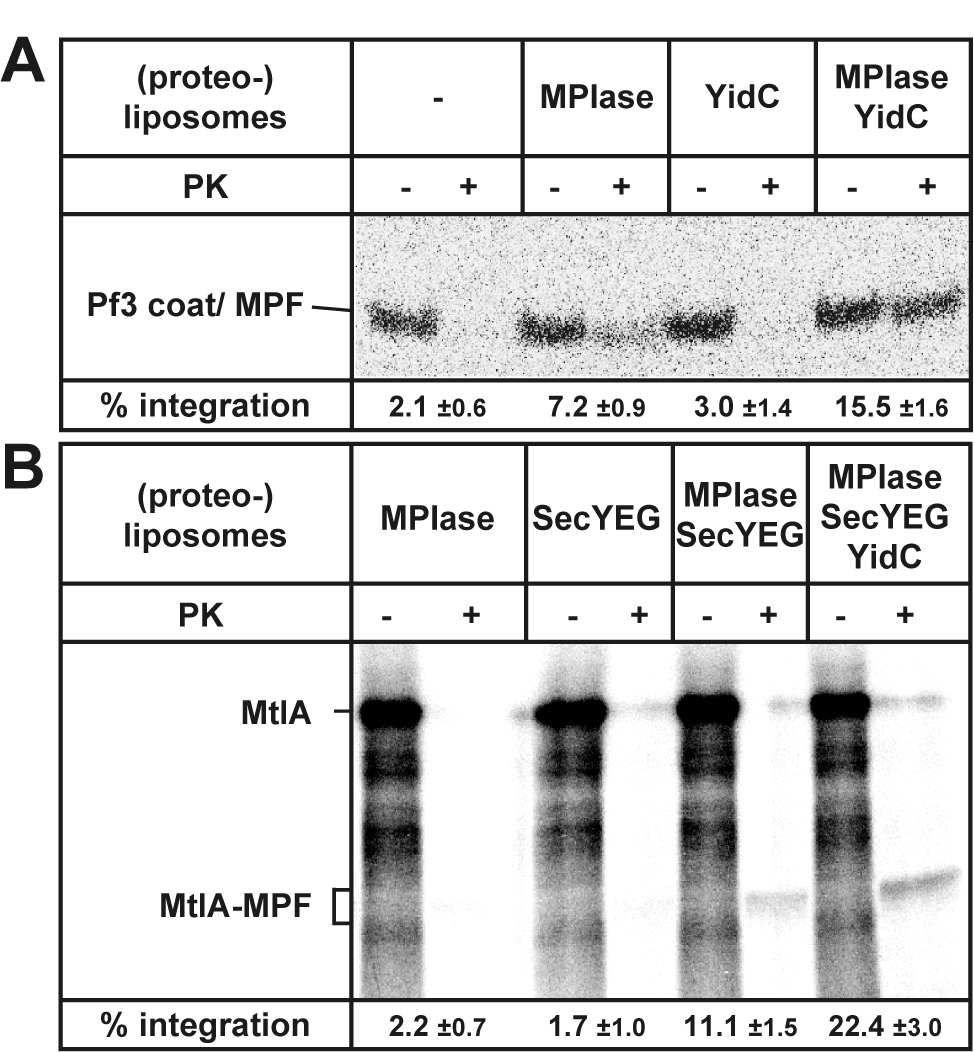
Figure 2. Examples of the results of the integration assay. A. Pf3 coat integration depends on MPIase and is stimulated by YidC. B. Both MPIase and SecYEG are essential for MtlA integration. Moreover, YidC stimulates the MPIase/SecYEG-dependent integration of MtlA. The numbers of methionine (25 for MtlA and 18 for MtlA-MPF) were taken into account for activity determination. Images were taken from Sasaki et al., 2019 (Figures 3C and 3D).
Recipes
- Buffer A
Dilute 1 M HEPES-KOH (pH 7.2) and 1 M dithiothreitol solutions to 50 mM and 1 mM, respectively, with ddH2O - OG stock
Dissolve OG in ddH2O at 15% [w/v], and store at 4 °C - Sodium phosphate buffer (pH 7.2)
- Prepare 1 M NaH2PO4 and 1 M Na2HPO4 solutions
- Mix these, monitoring the pH, to yield 1 M sodium phosphate buffer (pH 7.2)
- Stock this solution at room temperature
Acknowledgments
This work was supported by Japan Society for the Promotion of Science Grants-in-Aid 18J21847 (to H. N.); 15KT0073, 16H01374, 16K15083, 17H02209 and 18KK0197 (to K. N.). This protocol was derived from our report (Sasaki et al., 2019).
Competing interests
We declare that we have no competing interests.
References
- Alami, M., Trescher, D., Wu, L. F. and Muller, M. (2002). Separate analysis of twin-arginine translocation (Tat)-specific membrane binding and translocation in Escherichia coli. J Biol Chem 277(23): 20499-20503.
- Collinson, I., Breyton, C., Duong, F., Tziatzios, C., Schubert, D., Or, E., Rapoport, T. and Kuhlbrandt, W. (2001). Projection structure and oligomeric properties of a bacterial core protein translocase. Embo J 20(10): 2462-2471.
- Eisner, G., Koch, H. G., Beck, K., Brunner, J. and Muller, M. (2003). Ligand crowding at a nascent signal sequence. J Cell Biol 163(1): 35-44.
- Hussain, M., Ichihara, S. and Mizushima, S. (1980). Accumulation of glyceride-containing precursor of the outer membrane lipoprotein in the cytoplasmic membrane of Escherichia coli treated with globomycin. J Biol Chem 255(8): 3707-3712.
- Kawashima, Y., Miyazaki, E., Muller, M., Tokuda, H. and Nishiyama, K. (2008). Diacylglycerol specifically blocks spontaneous integration of membrane proteins and allows detection of a factor-assisted integration. J Biol Chem 283(36): 24489-24496.
- Kiefer, D. and Kuhn, A. (1999). Hydrophobic forces drive spontaneous membrane insertion of the bacteriophage Pf3 coat protein without topological control. EMBO J 18(22): 6299-6306.
- Koch, H. G., Hengelage, T., Neumann-Haefelin, C., MacFarlane, J., Hoffschulte, H. K., Schimz, K. L., Mechler, B. and Muller, M. (1999). In vitro studies with purified components reveal signal recognition particle (SRP) and SecA/SecB as constituents of two independent protein-targeting pathways of Escherichia coli. Mol Biol Cell 10(7): 2163-2173.
- Koch, H. G. and Muller, M. (2000). Dissecting the translocase and integrase functions of the Escherichia coli SecYEG translocon. J Cell Biol 150(3): 689-694.
- Kuruma, Y., Nishiyama, K., Shimizu, Y., Muller, M. and Ueda, T. (2005). Development of a minimal cell-free translation system for the synthesis of presecretory and integral membrane proteins. Biotechnol Prog 21(4): 1243-1251.
- Laemmli, U. K. (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227(5259): 680-685.
- Moser, M., Nagamori, S., Huber, M., Tokuda, H. and Nishiyama, K. (2013). Glycolipozyme MPIase is essential for topology inversion of SecG during preprotein translocation. Proc Natl Acad Sci U S A 110(24): 9734-9739.
- Newitt, J. A. and Bernstein, H. D. (1998). A mutation in the Escherichia coli secY gene that produces distinct effects on inner membrane protein insertion and protein export. J Biol Chem 273(20): 12451-12456.
- Nishikawa, H., Sasaki, M. and Nishiyama, K. (2017). Membrane insertion of F0 c subunit of F0F1 ATPase depends on glycolipozyme MPIase and is stimulated by YidC. Biochem Biophys Res Commun 487(2): 477-482.
- Nishiyama, K., Ikegami, A., Moser, M., Schiltz, E., Tokuda, H. and Muller, M. (2006). A derivative of lipid A is involved in signal recognition particle/SecYEG-dependent and -independent membrane integrations. J Biol Chem 281(47): 35667-35676.
- Nishiyama, K., Maeda, M., Abe, M., Kanamori, T., Shimamoto, K., Kusumoto, S., Ueda, T. and Tokuda, H. (2010). A novel complete reconstitution system for membrane integration of the simplest membrane protein. Biochem Biophys Res Commun 394(3): 733-736.
- Nishiyama, K., Maeda, M., Yanagisawa, K., Nagase, R., Komura, H., Iwashita, T., Yamagaki, T., Kusumoto, S., Tokuda, H. and Shimamoto, K. (2012). MPIase is a glycolipozyme essential for membrane protein integration. Nat Commun 3: 1260.
- Samuelson, J. C., Chen, M., Jiang, F., Moller, I., Wiedmann, M., Kuhn, A., Phillips, G. J. and Dalbey, R. E. (2000). YidC mediates membrane protein insertion in bacteria. Nature 406(6796): 637-641.
- Sasaki, M., Nishikawa, H., Suzuki, S., Moser, M., Huber, M., Sawasato, K., Matsubayashi, H. T., Kumazaki, K., Tsukazaki, T., Kuruma, Y., Nureki, O., Ueda, T. and Nishiyama, K. I. (2019). The bacterial protein YidC accelerates MPIase-dependent integration of membrane proteins. J Biol Chem 294(49): 18898-18908.
- Serek, J., Bauer-Manz, G., Struhalla, G., van den Berg, L., Kiefer, D., Dalbey, R. and Kuhn, A. (2004). Escherichia coli YidC is a membrane insertase for Sec-independent proteins. EMBO J 23(2): 294-301.
- Shimizu, Y., Inoue, A., Tomari, Y., Suzuki, T., Yokogawa, T., Nishikawa, K. and Ueda, T. (2001). Cell-free translation reconstituted with purified components. Nat Biotechnol 19(8): 751-755.
- Stiegler, N., Dalbey, R. E. and Kuhn, A. (2011). M13 procoat protein insertion into YidC and SecYEG proteoliposomes and liposomes. J Mol Biol 406(3): 362-370.
- Ulbrandt, N. D., Newitt, J. A. and Bernstein, H. D. (1997). The E. coli signal recognition particle is required for the insertion of a subset of inner membrane proteins. Cell 88(2): 187-196.
- Welte, T., Kudva, R., Kuhn, P., Sturm, L., Braig, D., Muller, M., Warscheid, B., Drepper, F. and Koch, H. G. (2012). Promiscuous targeting of polytopic membrane proteins to SecYEG or YidC by the Escherichia coli signal recognition particle. Mol Biol Cell 23(3): 464-479.
Article Information
Copyright
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Nishikawa, H., Sasaki, M. and Nishiyama, K. (2020). In vitro Assay for Bacterial Membrane Protein Integration into Proteoliposomes. Bio-protocol 10(10): e3626. DOI: 10.21769/BioProtoc.3626.
- Sasaki, M., Nishikawa, H., Suzuki, S., Moser, M., Huber, M., Sawasato, K., Matsubayashi, H. T., Kumazaki, K., Tsukazaki, T., Kuruma, Y., Nureki, O., Ueda, T. and Nishiyama, K. I. (2019). The bacterial protein YidC accelerates MPIase-dependent integration of membrane proteins. J Biol Chem 294(49): 18898-18908.
Category
Biochemistry > Protein > Synthesis
Molecular Biology > Protein > Activity
Biochemistry > Protein > Interaction
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.