- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Molecular Size Analysis of Recombinant Importin-histone Complexes Using Analytical Ultracentrifugation
Published: Vol 10, Iss 10, May 20, 2020 DOI: 10.21769/BioProtoc.3625 Views: 4559
Reviewed by: Gal HaimovichAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Mar 2019
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Histones constitute the protein components of nucleosomes. Despite their small sizes, histones do not diffuse through the nuclear pore complex. Instead, they are transported to the nucleus by importins, either alone or in complex with histone chaperones. Determining the molecular size of the importin-histone complexes is key to understanding the mechanism of histone transport and also the potential roles of importins as histone chaperones and in the assembly of nucleosomes. Here we report a simple and reproducible sedimentation-velocity based method to determine the molecular sizes of importin-histone complexes using analytical ultracentrifugation. The method does not use any reporter tags or interaction with column resin thereby analyzing the interactions of the native proteins.
Keywords: ImportinBackground
Nucleosomes are the most basic structural and functional units of the eukaryotic chromatin. Histone proteins H2A, H2B, H3 and H4 are the protein components of the nucleosomes. Each nucleosome consists of 147 base pairs of DNA wrapped around an H3-H4 tetramer and two copies of the H2A-H2B dimer (Luger et al., 1997a). Histones, like other proteins in the cell, are synthesized in the cytoplasm. The nucleosomes, however, are assembled in the nucleus. Despite their small size (monomers are 10-15 kDa), histones do not diffuse through the nuclear pore complex and instead are transported either alone or in complex with histone chaperones by the importins (Johnson-Saliba et al., 2000; Baake et al., 2001; Mosammaparast et al., 2001, 2002a and 2002b; Muhlhausser et al., 2001; Jakel et al., 2002).
Analysis of the histone-importin complexes helps elucidate the mechanism of the transport of histones. Histone proteins are highly basic proteins and have non-specific interactions with most column resins, and thus chromatography-based experiments require careful optimization and analyses. Here we describe an alternate solution-based sedimentation-velocity method to accurately determine the sedimentation coefficient of the various possible complexes. This Analytical Ultracentrifugation (AUC) method is highly reproducible and requires very little protein. Furthermore, it does not use any reporter tags, enabling experimentation with native or native-like macromolecules. The method can easily be adopted to study other importin-histone complexes and histone-chaperone complexes not only expressed in bacteria but also from other native sources.
Materials and Reagents
- Culture flask (1,000 ml) (VWR, catalog number: 29136-106 )
- Parafilm (Sigma-Aldrich, catalog number: BR701605 )
- FisherbrandTM Regenerated cellulose dialysis tubing (6,000-8,000D) (Fisher Scientific, catalog number: 21-152-4 )
- Slide-A-LyzerTM MINI Dialysis Device, 7K MWCO, 0.1 ml (Thermo Fisher Scientific, catalog number: 69560 )
- Glass Econo-Column (Bio-Rad, catalog number: 7374156 )
- Amicon Ultra-15 Centrifugal Filter Unit, 3kDa (Millipore Sigma, catalog number: UFC900324 )
- Amicon Ultra-15 Centrifugal Filter Unit,10kDa (Millipore Sigma, catalog number: UFC901024 )
- Amicon Ultra-15 Centrifugal Filter Unit, 50kDa (Millipore Sigma, catalog number: UFC905024 )
- E. coli BL21 DE3 plysS cells (Thermo Fisher Scientific, catalog number: C602003 )
- pET-3a plasmid (Sigma-Aldrich, catalog number: 69418 )
- pET-22b plasmid (Sigma-Aldrich, catalog number: 69744 )
- pGEX-4T3 plasmid (GE Healthcare, catalog number: 28954552 )
- Ampicillin (Goldbio, catalog number: A-301-5 )
- Quick start Bradford Protein Assay Kit (Bio-Rad, catalog number: 5000201 )
- Chloramphenicol (Goldbio, catalog number: C-105-5 )
- Complete, EDTA-free protease inhibitor (Sigma-Aldrich, catalog number: 11873580001 )
- Adenosine 5′-triphosphate disodium salt hydrate (Sigma-Aldrich, catalog number: A26209 )
- Dimethyl sulfoxide (DMSO) (Sigma-Aldrich, catalog number: 276855 )
- DTT (RPI, catalog number: D110000-25.0 )
- EDTA (RPI, catalog number: E57020-500.0 )
- Glycerol (Fisher, catalog number: 633-4 )
- Tryptone (Sigma-Aldrich, catalog number: T7293 )
- Tris HCl pH 7.5 (Sigma-Aldrich, catalog number: T5941 )
- β-mercaptoethanol (Sigma-Aldrich, catalog number: M6250 )
- Potassium acetate (Sigma-Aldrich, catalog number: P1190 )
- Magnesium acetate (Sigma-Aldrich, catalog number: M5661 )
- Imidazole (Sigma-Aldrich, catalog number: I5513 )
- Guanosine 5'-Triphosphate Sodium (Sigma-Aldrich, catalog number: G8877 )
- Glutathione sepharose 4B (GE Healthcare, catalog number: 17075601 )
- Guanidine HCl (RPI, catalog number: G49000-100 )
- HEPES (RPI, catalog number: H75030-500.0 )
- IPTG (Gold Biotechnology, catalog number: I24816100 )
- Magnesium chloride (MgCl2) (Sigma-Aldrich, catalog number: M8266 )
- Ni-NTA Agarose (Qiagen, catalog number: 30320 )
- Sodium acetate (Sigma-Aldrich, catalog number: S8750 )
- Sodium chloride (NaCl) (RPI, catalog number: S23020-5000.0 )
- TCEP (Gold Biotechnology, catalog number: TCEP25 )
- Tris hydroxymethyl aminomethane (RPI, catalog number: T60040-5000.0 )
- Triton X-100 (Sigma-Aldrich, catalog number: T878-100ML )
- Urea (RPI, catalog number: U20200 )
- β-mercapto-ethanol (Sigma-Aldrich, catalog number: M6250 )
- Yeast extract (Sigma-Aldrich, catalog number: Y1625 )
- LB media (1 L) (see Recipes)
- 2x YPT Media (1 L) (see Recipes)
- Wash buffer (see Recipes)
- Unfolding buffer (see Recipes)
- Refolding buffer (see Recipes)
- Sodium acetate urea buffer 200 (SAU 200) (see Recipes)
- Sodium acetate urea buffer 600 (SAU 600) (see Recipes)
- AUC buffer (see Recipes)
- Imp9-lysis buffer (see Recipes)
- Imp9-wash buffer (see Recipes)
- Sodium chloride Q buffer (see Recipes)
- Imp9-SEC buffer (see Recipes)
- Ran-lysis buffer (see Recipes)
- Ran-wash buffer (see Recipes)
- Ran-elution buffer (see Recipes)
- Sodium chloride SP buffer (see Recipes)
- Ran GTP exchange buffer (see Recipes)
Equipment
- Q-500 Sonicator (Q Sonica, catalog number: Q500-110 )
- Emulsiflex–C5 cell homogenizer (Avestin, catalog number: Emulsiflex C5 )
- Oakridge tubes–50 ml tubes (Thermo Fisher Scientific, catalog number: 3119-0050 )
- AKTA pure (chromatography system) (GE Healthcare, catalog number: 29046665 )
- HiTrap Q HP (GE Healthcare, catalog number: 17115301 )
- HiTrap SP HP (GE Healthcare, catalog number: 17115201 )
- Superdex 200 Increase 10/300 GL (GE Healthcare, catalog number: 28990944 )
- NanoDrop (Thermo Fisher Scientific, catalog number: ND-2000C )
- Lyophilizer (Labconco, catalog number: 7740020 )
- Avanti J-301 High-Performance Centrifuge (Beckman Coulter, catalog number: 363118 )
- JA-20 Beckman rotor (Beckman Coulter, catalog number: 334831 )
- Eight-hole An-50Ti rotor (Beckman Coulter, catalog number: 363782 )
- AUC (Analytical Ultracentrifuge) centerpiece assemblies, including charcoal-filled Epon centerpieces, sapphire windows, aluminum housings, and fill-port plugs (Beckman Coulter, catalog number: A37299 )
- Beckman-Coulter Optima XL-1 Analytical Ultracentrifuge (AUC) (Beckman Coulter, catalog number: B86437 )
Software
- SEDNTERP 1 (http://www.jphilo.mailway.com/download.htm)
- SEDFIT (http://www.analytical.ultracentrifugation.com)
- REDATE (http://biophysics.swmed.edu/MBR/software.html)
- GUSSI (http://biophysics.swmed.edu/MBR/software.html)
Procedure
Note: Histone purification and H2A-H2B complex assembly protocol is adapted from a previously published protocol (Luger et al., 1997b and 1999). The protocol has been altered slightly to suit the requirements of the current experiment. This protocol describes a methodology for analysis of Imp9–H2A-H2B complexes and can be used to study not only other Importin–histone complexes but also various histone chaperone complexes. A flow chart of the protocol is shown in Figure 1.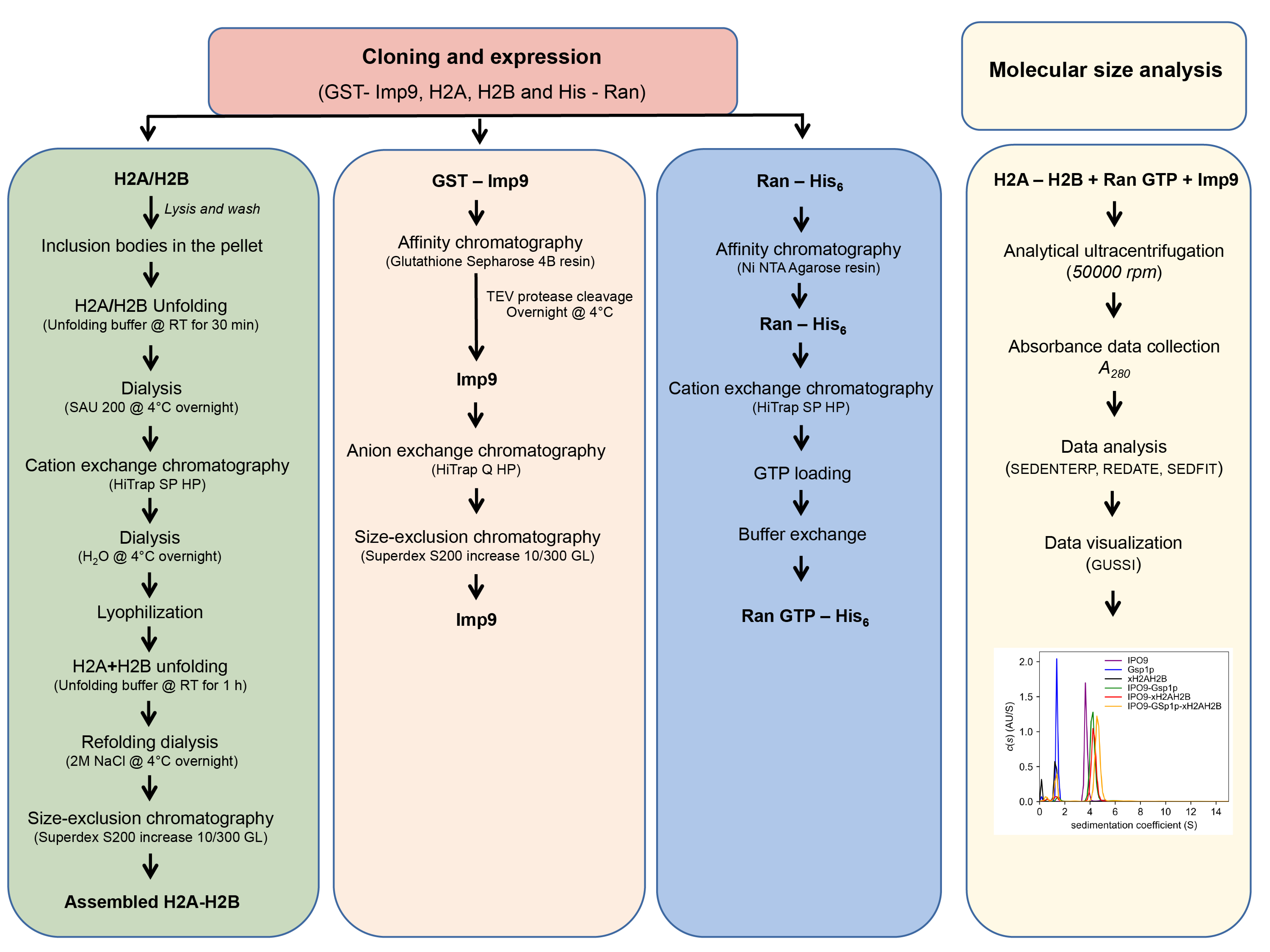
Figure 1. Flow chart depicting various steps of protein purification and analytical ultracentrifugation
- Expression of histones H2A/H2B
- Transform E. coli BL21 DE3 plysS cells with histone expression plasmid (pET-3a containing H2A/H2B gene) and plate on 2x YPT agar plates containing appropriate antibiotics (ampicillin and chloramphenicol). Incubate at 37 °C overnight (12-15 h). Fresh transformation will ensure better protein expression.
- Pick single colonies and start 3-5 ml pre-culture in 2x YPT media with appropriate antibiotic. Grow them at 37 °C overnight.
- Inoculate 100 ml 2x YPT media with antibiotics with 100 µl turbid pre-cultures and incubate until the OD reaches 0.4-0.5.
- Inoculate six 1 L 2x YPT media containing antibiotics evenly with the culture from Step A3 and grow at 37 °C until the OD reaches 0.4-0.5 and induce with 250 μM IPTG for 4 h at 37 °C.
- After 4 h, harvest the cells by centrifugation at 4,000 x g for 20 min at 4 °C.
- Resuspend the cell pellets in 35 ml/L (cell culture) of wash buffer.
- Flash freeze the cell suspension in liquid nitrogen and store at -80 °C. The frozen cell suspension can be stored for a week at -80 °C.
- Histone purification from inclusion bodies
- Thaw the cell suspension at room temperature (25 °C) and place the tube containing the cell suspension in an ice-beaker with the sonicator probe in the tube.
- Set up the sonicator on pulse mode (1 s on and 1 s off). Lyse the cells on pulse mode at 50% amplitude for 3 min, repeating it 3-4 times or until the cell suspension becomes less viscous. The pulse mode and ice-beaker help prevent over-heating of the sample.
- Transfer the lysate into Oakridge tubes and spin at 25,000 x g for 20 min at 4 °C. Discard the supernatant and resuspend the pellet with 35 ml/L (cell culture) of wash buffer containing 1% (v/v) Triton X-100.
- Spin at 25,000 x g for 20 min at 4 °C. Discard the wash buffer.
- Repeat the wash described above twice with wash buffer without Triton X-100. Discard the wash buffer after the spin (25,000 x g for 20 min at 4 °C). Collect the final pellet. The pellet contains the protein (H2A/H2B) inclusion bodies.
- The inclusion body pellet can be stored at -20 °C for a week.
- Thaw the inclusion body pellet at room temperature first by dissolving the pellet in 1ml of dimethyl sulfoxide (DMSO) and then suspending it well in 20 ml of unfolding buffer. Incubate it on a rocker gently (10-15 rpm) at room temperature for 30 min.
- Spin the suspension at 25,000 x g for 20 min at 4 °C to remove any particulates.
- Transfer the supernatant from the previous step carefully to the dialysis tubing (6-8 kDa cutoff).
- Dialyze the sample overnight in SAU-200 (4 x 1 L), changing the buffer every hour for the first three rounds and leaving the last one overnight. All dialysis steps are carried out at 4 °C.
- Carefully transfer the dialyzed sample to Oakridge tubes and spin at 25,000 x g for 20 min at 4 °C to remove any particulates.
- Connect HiTrap SP HP (Cation exchange chromatography) column to the FPLC system (AKTA pure) and equilibrate with SAU-200. Set up a FPLC program to inject the sample and run a step gradient from 0% SAU 600 (100% SAU 200) to 40% SAU 600 (60% SAU 200) in 10 column volumes and 40% SAU 600 (60% SAU 200) to 100% SAU 600 (0% SAU 200) in 10 column volumes. 200 ml each of SAU200 and SAU600 should be enough to form the step gradient in a 5 ml HiTrap SP HP column.
- Inject the dialyzed sample into HiTrap SP HP column pre-equilibrated with SAU-200.
- Elute histones bound to the HiTrap SP HP column using the step gradient setup in Step B12.
- H2A/H2B elutes at about 38-42% SAU 600. Pool the peak fractions from HiTrap SP HP column and transfer carefully to a dialysis tubing (6-8 kDa cutoff) and dialyze in ice-cold water (Water + 5 mM β-mercapto-ethanol) overnight (4 × 4 L), changing the water every hour for the first three rounds and leaving the last one overnight.
- Determine the concentration of histones in the dialyzed sample by measuring the absorbance at 280 nm against water using a NanoDrop and applying the known extinction coefficient and path length in Beer’s Law and aliquot the dialyzed samples into cryovials at approximately 1 mg total protein per aliquot. The theoretical molar extinction coefficients can be obtained using expasy protoparam tool (http://ca.expasy.org/tools/protparam.html).
- Prepare an icebox with dry ice and fill it with ice-cold ethanol.
- Transfer the cryovials from Step B16 into the icebox to flash freeze.
- Remove the cap of the cryovials and seal with parafilm and puncture the parafilm.
- Turn on the Lyophilizer and close the ballast. Wait for the vacuum to reach < 100 mT and condenser temperature to -40 °C.
- Load samples into the Lyophilizer bottles. Open the vacuum valve and let it run overnight.
- After the run, turn off the vacuum and take out the samples. Remove the parafilm and replace with cap.
- Store at -80 °C until ready to assemble the complex. Lyophilization extends the shelf life of histone proteins by several months.
- Histone dimer H2A-H2B complex assembly
- Dissolve lyophilized histone aliquots to a concentration of approximately 2 mg/ml in unfolding buffer and incubate at room temperature for at least 30 min but not more than one hour. Determine the concentration of unfolded histones by measuring the absorbance at 280 nm against the unfolding buffer using a NanoDrop and applying the known extinction coefficient and path length in Beer’s Law.
- Mix resuspended equimolar mixtures of H2A and H2B and dilute the mixture to 1 mg/ml in unfolding buffer and incubate them on a room temperature rocker at 10-15 rpm for 1 h.
- Centrifuge the sample at 25,000 x g for 10 min at 4 °C to remove any precipitates and transfer the supernatant to a dialysis tubing (6-8 kDa cutoff).
- Dialyze in refolding buffer overnight at 4 °C, changing the buffer at least four times (4 × 2 L), every hour for the first three rounds and leaving the last one overnight.
- Centrifuge the dialyzed sample at 25,000 x g for 10 min at 4 °C to remove any particulates and concentrate the sample in a 10 kDa Amicon centrifugal concentrator to an appropriate injection volume (A maximum load of 0.5 ml of 10 mg total protein is recommended per injection for good resolution on the S200 Superdex increase column).
- Inject the sample onto Superdex S200 increase column pre-equilibrated with refolding buffer. H2A-H2B dimer elutes at an elution volume of approximately 16.5 ml in a 24 ml Superdex S200 increase column.
- Pool the peak fractions, concentrate in a 10 kDa Amicon centrifugal concentrator to approximately 10 mg/ml.
- Aliquot into 100 μl aliquots, flash freeze and store them at -80 °C.
- Expression of Imp9
- Transform E. coli BL21 DE3 cells with GST-Imp9 expression plasmid (modified pGEX-4T3 (thrombin site replaced with TEV protease cleavage site [Chook and Blobel, 1999]) containing Imp9 gene) (Padavannil et al., 2019) and plate on LB agar plates containing ampicillin. Incubate at 37 °C overnight. Fresh transformation will ensure better protein expression.
- Pick single colonies and start 3-5 ml pre-culture in LB media with ampicillin. Grow them at 37 °C overnight.
- Inoculate 100 ml LB media with ampicillin with 0.5-1 ml turbid pre-cultures and incubate until the OD reaches 0.4-0.5.
- Inoculate 4 x 1 L LB media containing ampicillin evenly with the culture from Step D3 and grow at 37 °C until the OD reaches 0.6 and induce with 500 μM IPTG for 12 h at 20 °C.
- Harvest the cells by centrifugation at 4,000 x g for 20 min at 4 °C.
- Purification of Imp9
- Suspend the harvested cells in Imp9-lysis buffer.
- Lyse the cells in Emulsiflex–C5 cell homogenizer.
- Transfer the lysate into Oakridge tubes and spin at 40,000 x g for 30 min at 4 °C and collect the supernatant.
- Set up a gravity flow in a Glass Econo-Column column in the cold room (4 °C) and equilibrate 1.5 ml (per 1 liter of cell culture) of Glutathione Sepharose 4B resin with Imp9-lysis buffer.
- Elute out the equilibration lysis buffer from Step E4 and add the supernatant from Step E3. Pass the supernatant through the resin by gravity flow a couple of times.
- Wash the GST–Imp9 bound resin with the Imp9-wash buffer twice.
- Wash the GST–Imp9 bound resin with the Imp9-ATP wash buffer once (Imp9-ATP wash buffer is lysis buffer with 5 mM ATP).
- Wash the GST–Imp9 bound resin with an additional Imp9-wash buffer.
- Check the concentration (a rough estimate using Bradford reagent should suffice) of the fusion protein on beads to determine the amount of TEV protease to be added.
- Cleave GST tag on column by incubating GST–Imp9 bound resin with TEV protease containing Im9-wash buffer overnight at 4 °C (add 100 μl of TEV protease (100 μM) for every 50 mg of fusion protein). Gently mix it once and incubate at 4 °C. Do not rock the column after addition of TEV protease.
- Elute Imp9 from the column. GST tag stays bound to the resin.
- Connect HiTrap Q HP (Anion exchange chromatography) column to the FPLC system (AKTA pure) and equilibrate with 100 mM sodium chloride Q buffer. Set up an FPLC program to inject the sample and run a linear gradient from 100% 100 mM sodium chloride Q-buffer to 100% 1 M sodium chloride Q-buffer in 20 column volumes. 150 ml of each buffer should be enough to form the linear gradient in a 5 ml HiTrap Q HP column.
- Inject Imp9 from Step E11 to HiTrap Q HP column pre-equilibrated with 100 mM sodium chloride Q buffer.
- Elute the protein from the column using the set linear gradient of 100% 100 mM sodium chloride Q-buffer to 100% 1 M sodium chloride Q-buffer (Imp9 elutes at approximately 22% B [78% A and 22% B]. The peak is quite distinct and the fractions within the peak are pooled).
- Pool the fractions containing Imp9 and concentrate the protein using 50 kDa Amicon centrifugal concentrator to an appropriate injection volume (A maximum load of 0.5 ml of 10 mg total protein is recommended per injection for good resolution on the S200 Superdex increase column).
- Inject the protein from Step E15 to an S200 Superdex increase column pre-equilibrated with Size-exclusion buffer (Imp9-SEC buffer). Imp9 elutes at an elution volume of approximately 13 ml in a 24 ml column.
- Pool the peak fractions from the column and concentrate the protein using 50 kDa Amicon centrifugal concentrator to the required concentration (Imp9 can be concentrated to up to 20 mg/ml. The concentrated sample can be flash frozen in liquid nitrogen and stored at -80 °C until ready to use).
- Expression of Ran [yeast Gsp1 (1-179, Q71L)]
- Transform E. coli BL21 DE3 cells with Ran [Gsp1 (1-179, Q71L)] expression plasmid (pET-22b containing yeast Ran [Gsp1 (1-179, Q71L) gene] and plate on LB agar plates containing ampicillin. Incubate at 37 °C overnight. Fresh transformation will ensure better protein expression.
- Pick single colonies and start 3-5 ml pre-culture in LB media with ampicillin. Grow them at 37 °C overnight.
- Inoculate 100 ml LB media with ampicillin with 0.5-1 ml turbid pre-cultures and incubate until the OD reaches 0.4-0.5.
- Inoculate 4 x 1 liter LB media containing ampicillin evenly with the culture from Step F3 and grow at 37 °C until the OD reaches 0.6 and induce with 300 μM IPTG for 12 h at 20 °C. Ran expresses as Ran–His6. The His6-tag is not cleaved during purification.
- Harvest the cells by centrifugation at 4,000 x g for 20 min at 4 °C.
- Purification and GTP loading of Ran
- Suspend the harvested cells in Ran-lysis buffer.
- Lyse the cells in Emulsiflex–C5 cell homogenizer.
- Transfer the lysate into Oakridge tubes and spin at 40,000 x g for 30 min at 4 °C and collect the supernatant.
- Set up a gravity flow in a Glass Econo-Column column in the cold room (4 °C) and equilibrate 1.5 ml (per 1 liter of cell culture) of Ni-NTA agarose resin with Ran-lysis buffer.
- Elute out the equilibration lysis buffer from Step G4 and add the supernatant from Step G3. Pass the supernatant through the resin by gravity flow a couple of times.
- Wash the Ran bound resin with the Ran-wash buffer twice.
- Elute the Ran from the column with Ran-elution buffer.
- Load the eluted Ran with GTP by incubating it on ice for 30 min with 40 mM GTP (final concentration) (add GTP to required concentration from 100 mM GTP stock solution).
- Connect HiTrap SP HP (Cation exchange chromatography) column to the FPLC system (AKTA pure) and equilibrate with 50 mM sodium chloride SP-buffer. Set up an FPLC program to inject the sample and run a linear gradient from 100% 50 mM sodium chloride SP buffer to 100% 1 M sodium chloride SP buffer in 20 column volumes. 150 ml of each buffer should be enough to form the linear gradient in a 5 ml HiTrap SP HP column.
- Inject the GTP-loaded Ran to pre-equilibrated HiTrap SP HP column.
- Elute the protein using the set linear gradient of 100% of 50 mM sodium chloride SP-buffer to 100% 1 M sodium chloride SP-buffer [Ran elutes at 40% SP-buffer B (40% B and 60% A) The peak is quite distinct and the fractions within the peak are pooled].
- Pool the fractions containing Ran-GTP and concentrate the protein to 10 mg/ml using 3-kDa Amicon centrifugal concentrator. Aliquot the concentrated protein to 100 μl aliquots and store at -80 °C.
- Sample preparation for AUC
- Dialyze preassembled histones sequentially at 4 °C in 1 L of 1 M NaCl refolding buffer (2 h), 1 L of 500 mM NaCl refolding buffer (2 h), and 2 L of AUC buffer (overnight). Histone dimers tend to dissociate and aggregate on sudden exposure to low salt. Sequential dilution over time helps to maintain the dimer. Histone tetramers and histone octamer probably behave the same and should be treated similarly.
- Dialyze purified Imp9 at 4 °C in AUC buffer overnight.
- Dialyze purified Ran GTP at 4 °C in AUC buffer overnight.
- Inject the proteins (H2A-H2B dimer, Imp9 and Ran GTP separately) into Superdex S200 increase column pre-equilibrated with AUC buffer. Save the AUC buffer from the run to make dilutions and to use as a reference buffer in the AUC.
- Histone dimer (H2A-H2B) and Imp9 have an elution volume of 16 ml and 13 ml in a 24 ml Superdex S200 increase column respectively. Ran GTP elutes at an elution volume of 18 ml in a 24 ml Superdex S200 increase column. Pool the peak fractions and concentrate the proteins to approximately 10 mg/ml using Amicon centrifugal filter units. The proteins can be stored at -80 °C for a week. Also store the AUC buffer used to run the Superdex S200 column at -80 °C to avoid any buffer mismatch.
- Sample loading to the AUC cell
- Calculate the concentration of each protein required for the AUC run based on their molar extinction coefficients.
- Mix the dialyzed samples to the final volume of 450 μl for the sedimentation velocity experiment. 1) 450 μl Imp9 alone (3 μM), 2) 450 μl RanGTP alone (10 μM), 3) 450 μl H2A-H2B (10 μM), 4) 3 μM Imp9 + 3 μM RanGTP in a total volume of 450 μl, 5) 3 μM Imp9 +3 μM H2A-H2B in a total volume of 450 μl, 6) 3 μM Imp9 + 3 μM H2A-H2B + 10 μM RanGTP in a total volume of 450 μl. Incubate the proteins at 4 °C overnight the day before the AUC run to ensure proper equilibration of the complexes.
- Assemble standard Epon-filled centerpieces (Balbo et al., 2009) (Figure 2).

Figure 2. Assembly of Epon-filled centerpieces. (a) An unassembled cell. The parts are (A) window liners, (B) window housings (with window cushions installed), (C) sapphire windows, (D) cell housing, (E) screw ring, (F) screw-ring gasket, (G) centerpiece, (H) fill-port plugs, and (I) fill-port gaskets. The window liners are placed into the window housings such that the gap in the liner is opposite the registration groove of the respective housing (seen at the top of both housings in this view. The windows are inserted into the window housings. One window is placed into the cell housing face up, then the centerpiece is inserted, then the second window (face down). On this, the screw-ring gasket is positioned, followed by the screw ring. The screw ring is torqued to between 120 and 140 in-lbs. The cell is filled with solutions through external fill ports, and then the fill port gaskets followed by the fill-port plugs are installed. Details are in Balbo et al. (2009). (b) The filled and assembled cell, viewed from the “top” of the cell. - Set up the Beckman-Coulter Optima XL-1 Analytical Ultracentrifuge (AUC) for a sedimentation velocity experiment (Balbo et al., 2008).
- Load 450 μl of the samples into the sample sectors and load 450 μl of the reference buffer (AUC buffer) into the reference sectors of double-sector centerpieces and place them into an eight-hole An-50Ti rotor. Position the rotor in the centrifuge, and incubate under vacuum at 20 °C for 2.5 h. Commence centrifugation at 50,000 rpm.
Monitor the sedimentation using absorbance at 280 nm (A280). Collect scans as rapidly as possible. Centrifugation may be ceased when all evidence of sedimentation is absent.
Data analysis
The end result of the data-analysis method detailed below is the c(s) distribution (Schuck, 2000). The result thus takes the form of a two-dimensional distribution, with the single population of species presented as a function of their respective sedimentation coefficients. Larger proteins or assemblies will have larger sedimentation coefficients. The method is based on the concept of scaling solutions to the Lamm Equation (Lamm, 1929) directly to the data (a(r,t)) according to![]()
where r is the radius (in cm) from the center of rotation, s is the sedimentation coefficient, t is time in seconds since the beginning of the centrifugation, and D is the translational diffusion coefficient, and L depicts the Lamm Equation. This allows the AUC data to be directly fitted. Noise in the data can cause unrealistic, high-frequency fluctuations in the c(s) distribution, and thus it is regularized along lines discussed by Provencher (Provencher, 1982, Schuck, 2000, Schuck et al., 2002). Systematic noise elements in the data can easily be detected and removed, resulting in higher-quality fits (Schuck and Demeler, 1999). The sedimentation coefficients in the distributions, coupled with the refined frictional ratios from the analysis, can be used to determine molar masses for species and complexes, but in the latter case, these masses should only be relied on in situations when the complex is expected to be fully occupied for the entirety of the SV experiment and the refined frictional ratio can be safely assumed to represent that of the complex (i.e., most of the signal comes from the complex or all species detected have similar frictional ratios).
Note: Screen shots of various steps of data analysis are provided as a supplement to the article (Figure S1).
Sedimentation velocity data analysis
- Calculate the buffer density and viscosity from the buffer composition using SEDNTERP.
- Calculate the partial-specific volume of the protein using SEDNTERP (Laue et al., 1992) or SEDFIT (Zhao et al., 2011).
- Use REDATE to change the time stamps in the Beckman data using the algorithm suggested by the Schuck laboratory (Zhao et al., 2013).
Note: REDATE optionally makes folders for each wavelength of data acquisition. - Start SEDFIT
- Choose “Data → Load New Files”; load only data scans that show evidence of sedimentation, i.e., late scans with no evidence of sedimentation should be excluded. 50-150 scans are sufficient, and every “nth” scan may be loaded to observe this limit.
- Use the mouse to define the positions of the meniscus (red line), the sector bottom (blue line), and the data-analysis boundaries (green lines).
- Choose “Model → Continuous c(s) distribution” (Schuck, 2000).
- Choose “Parameters”:
- resolution 50/100
- s min 0
- s max 15/10
- refine (activate using the respective the check box)–frictional ratio (Proteins 1.2-2.0)
- refine–Baseline
- check–Fit time-independent noise
- check–Fit RI noise only if analyzing interferometric data
- refine–Meniscus
- confidence level (F-ratio)–set to 0.68 for 1 sigma of regularization.
- Input the partial-specific volume, solution density, and solution viscosity in their proper places.
- From the Main Menu, choose “Run” (i.e., refine all linear parameters).
- Adjust parameters if significant data/fit mismatches are in evidence. The most common culprits at this stage are frictional ratio and meniscus. Redo the Run until the fit lines reasonably resemble the data.
- From the Main Menu, choose “Fit” (i.e., iteratively refine all parameters).
- Assess the quality of the fit. The root-mean-square deviation (rmsd) should be low (usually less than 0.01 signal units), and minimal systematicity should be evidenced in the residual plots. Poor values/appearances at this stage may indicate data-acquisition problems, turbulence, or convection, and it may be necessary to redo the experiment if the data are so compromised. The default fitting option is the Simplex algorithm; change it to Marquardt Levenberg (“Options → Fitting Options → Marquardt Levenberg”), note the rmsd value, and fit the data again. Continue alternating between Simplex and Marquardt Levenberg until the rmsd values no longer change upon fitting.
- Under the conditions that (a) the species of interest dominates the signal or (b) all species may be assumed to have the same frictional ratio, the molar masses of species may be estimated by choosing “Display → Show peak Mw in c(s)” and pressing the button that appears in the c(s) distribution plot.
- Before plotting, it may be desirable to increase the resolution of the distribution (in the Parameters window) to 150 and redo the fit.
- Choose “Plot → GUSSI c(s) plot” for the first distribution. For subsequent distributions that are to be overlaid on the first, choose “Copy → Copy Distribution” (thereby placing it on the clipboard) and then paste it (“Distributions → Paste a Distribution”) into the GUSSI instance that contains previous distributions. A typical GUSSI output is shown in Figure 3.
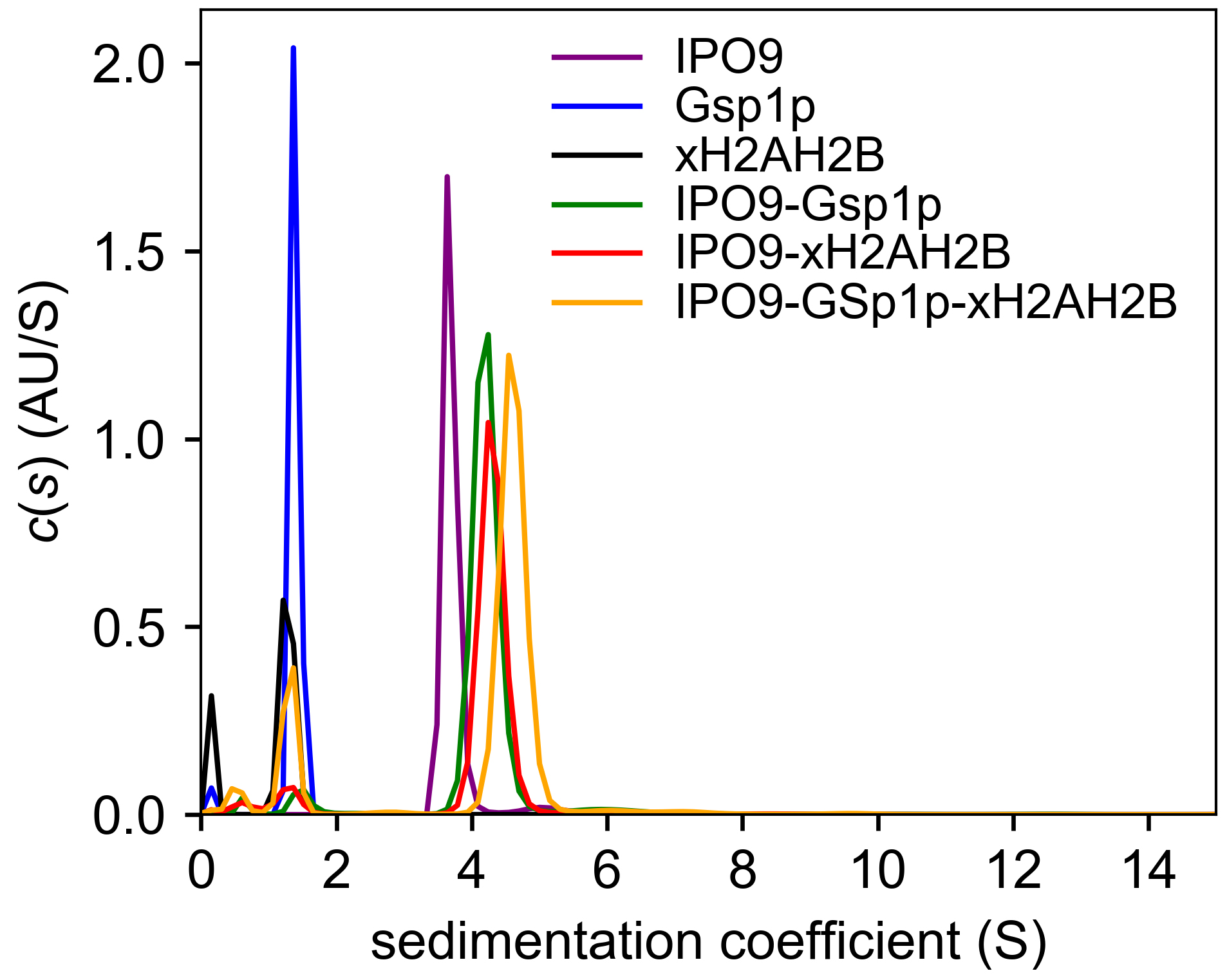
Figure 3. A typical GUSSI output. Analytical ultracentrifugation produced sedimentation profiles for Imp9, H2A-H2B, RanGTP, the 1:1 molar ratio mix of Imp9 and H2A-H2B dimer, the 1:1 molar ratio mix of Imp9 and RanGTP, and the 1:1:3 molar ratio mix of Imp9, H2A-H2B dimer and RanGTP. - In GUSSI, choose “Integrate → Integrate All” to obtain weighted s-values for all species simultaneously (this can be done individually in SEDFIT as well by using the integration function, which is summoned by pressing Ctrl-I).
- Confidence intervals for weighted s-values (if necessary) (Schuck, 2016).
- In SEDFIT, after an optimized analysis, define the integration limits and note down the values.
- Note the optimized meniscus value.
- Choose “Statistics → Calculate variance ratio (F-statistics)” for a 68.3% confidence level and note down the target rmsd value.
- Choose “Parameters”: Fix the meniscus to a lower value (e.g., lower it by 0.01 cm) and perform a fit; observe the rmsd to see if it finishes above the target value. If it does not, lower the fixed meniscus value and repeat until the rmsd exceeds the target value.
- Keeping the meniscus fixed at this new value, choose “Statistics → Monte-Carlo for integrated weight → average s values” and perform a minimum of 1,000 iterations at a 68.3% confidence level.
- Note the confidence interval returned by the program (two values).
- Repeat, fixing the meniscus to values higher than the optimal one.
- Choose as the confidence interval the highest and lowest of the four values that were returned by the program.
Recipes
- LB media (1 L)
10 g Tryptone
10 g NaCl
5 g Yeast extract - 2x YPT Media (1 L)
16 g Bacto Tryptone
10 g Yeast extract
5 g NaCl - Wash buffer
10 mM Tris HCl pH 7.5
1 mM EDTA
5 mM β-mercaptoethanol - Unfolding buffer
7 M Guanidinium HCl
20 mM Tris HCl pH 7.5
10 mM DTT - Refolding buffer
2 M NaCl
10 mM Tris HCl
1 mM EDTA
5 mM β-mercaptoethanol - Sodium acetate urea buffer 200 (SAU 200)
7 M Urea
20 mM Sodium Acetate, pH 5.2
200 mM NaCl
1 mM EDTA
5 mM β-mercaptoethanol - Sodium acetate urea buffer 600 (SAU 600)
7 M Urea
20 mM Sodium Acetate pH 5.2
600 mM NaCl
1 mM EDTA
5 mM β-mercaptoethanol - AUC buffer
20 mM HEPES pH 7.3
200 mM sodium chloride
2 mM magnesium chloride
2 mM TCEP
8% glycerol - Imp9-lysis buffer
50 mM Tris-HCl pH 7.5
100 mM NaCl
1 mM EDTA
2 mM DTT
20% Glycerol
Complete, EDTA-free protease inhibitor - Imp9-wash buffer
50 mM Tris-HCl pH 7.5
100 mM NaCl
1 mM EDTA
2 mM DTT
20% Glycerol - Sodium chloride Q buffer
20 mM Tris-HCl pH 7.5
100 mM NaCl/1 M NaCl
1 mM EDTA
2 mM DTT
20% Glycerol - Imp9-SEC buffer
20 mM HEPES pH 7.3
110 mM potassium acetate
2 mM magnesium acetate
2 mM DTT
15% Glycerol - Ran-lysis buffer
50 mM HEPES pH 8.0
200 mM NaCl
10% Glycerol
2 mM magnesium acetate
2 mM β-mercaptoethanol
5 mM Imidazole
Complete, EDTA-free protease inhibitor - Ran-wash buffer
20 mM HEPES pH 8.0
200 mM NaCl
10% Glycerol
2 mM magnesium acetate
2 mM β-mercaptoethanol
40 mM imidazole - Ran-elution buffer
20 mM HEPES pH 7.5
50 mM NaCl
10% glycerol
2 mM magnesium acetate
2 mM β-mercaptoethanol
300 mM imidazole - Sodium chloride SP buffer
20 mM HEPES pH 7.5
50 mM NaCl/1 M NaCl
4 mM magnesium acetate
1 mM DTT
10% glycerol - Ran GTP exchange buffer
20 mM HEPES pH 7.5
100 mM NaCl
4 mM magnesium acetate
1 mM DTT
10% glycerol
Note: The buffers are made using standard procedures. Weigh the components into a volume less than the final volume, adjust the pH while constantly stirring the buffer and after the pH has been adjusted, add water to achieve the final volume. Adjust the pH of the media to 7.0 before adding water to achieve the final volume.
Acknowledgments
We thank Bing Li for plasmids expressing H2A and H2B. This work was funded by NIGMS of NIH under Awards R01GM069909 (YMC), U01GM98256-01 (YMC), the Welch Foundation Grants I-1532 (YMC), the Leukemia and Lymphoma Society Scholar Award (YMC) and the University of Texas Southwestern Endowed Scholars Program (YMC). The histone purification and assembly protocol is a modified version of histone purification and assembly protocol from Karolin Luger’s published work.
Competing interests
The authors declare no conflicts of interest or competing interests.
References
- Baake, M., Bauerle, M., Doenecke, D. and Albig, W. (2001). Core histones and linker histones are imported into the nucleus by different pathways. Eur J Cell Biol 80(11): 669-677.
- Balbo, A., Brown, P. H. and Schuck, P. (2008). Experimental Protocol for Sedimentation Velocity Analytical Ultracentrifugation.
- Balbo, A., Zhao, H., Brown, P. H. and Schuck, P. (2009). Assembly, loading, and alignment of an analytical ultracentrifuge sample cell. J Vis Exp(33). pii: 1530. doi: 10.3791/1530.
- Chook, Y. M. and Blobel, G. (1999). Structure of the nuclear transport complex karyopherin-beta2-Ran x GppNHp. Nature 399(6733): 230-237.
- Jakel, S., Mingot, J. M., Schwarzmaier, P., Hartmann, E. and Gorlich, D. (2002). Importins fulfil a dual function as nuclear import receptors and cytoplasmic chaperones for exposed basic domains. EMBO J 21(3): 377-386.
- Johnson-Saliba, M., Siddon, N. A., Clarkson, M. J., Tremethick, D. J. and Jans, D. A. (2000). Distinct importin recognition properties of histones and chromatin assembly factors. FEBS Lett 467(2-3): 169-174.
- Lamm, O. (1929). Die Differentialgleichung der Ultrazentrifugierung. Arkiv för matematik, astronomi och fysik 21B (2): 1-4.
- Laue, T., Shah, B. D., Rdigeway, R. M. and Pelletier, S. L. (1992). Computer-aided interpretation of analytical sedimentation data for proteins. In: Harding, S. E., Rowe, A. J. and Horton, J. C. (Eds.). Analytical Ultracentrifugation in Biochemistry and Polymer Science. 90-125.
- Luger, K., Mader, A. W., Richmond, R. K., Sargent, D. F. and Richmond, T. J. (1997a). Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389(6648): 251-260.
- Luger, K., Rechsteiner, T. J., Flaus, A. J., Waye, M. M. and Richmond, T. J. (1997b). Characterization of nucleosome core particles containing histone proteins made in bacteria. J Mol Biol 272(3): 301-311.
- Luger, K., Rechsteiner, T. J. and Richmond, T. J. (1999). Expression and purification of recombinant histones and nucleosome reconstitution. Methods Mol Biol 119: 1-16.
- Mosammaparast, N., Ewart, C. S. and Pemberton, L. F. (2002a). A role for nucleosome assembly protein 1 in the nuclear transport of histones H2A and H2B. EMBO J 21(23): 6527-6538.
- Mosammaparast, N., Guo, Y., Shabanowitz, J., Hunt, D. F. and Pemberton, L. F. (2002b). Pathways mediating the nuclear import of histones H3 and H4 in yeast. J Biol Chem 277(1): 862-868.
- Mosammaparast, N., Jackson, K. R., Guo, Y., Brame, C. J., Shabanowitz, J., Hunt, D. F. and Pemberton, L. F. (2001). Nuclear import of histone H2A and H2B is mediated by a network of karyopherins. J Cell Biol 153(2): 251-262.
- Muhlhausser, P., Muller, E. C., Otto, A. and Kutay, U. (2001). Multiple pathways contribute to nuclear import of core histones. EMBO Rep 2(8): 690-696.
- Padavannil, A., Sarkar, P., Kim, S. J., Cagatay, T., Jiou, J., Brautigam, C. A., Tomchick, D. R., Sali, A., D'Arcy, S. and Chook, Y. M. (2019). Importin-9 wraps around the H2A-H2B core to act as nuclear importer and histone chaperone. Elife 8: e43630.
- Provencher, S. W. (1982). CONTIN: A general purpose constrained regularization program for inverting noisy linerar algebraic and integral equations. Comput Phys Commun 27: 229-242.
- Schuck, P. (2000). Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys J 78(3): 1606-1619.
- Schuck, P. (2016). Sedimentation Velocity Analytical Ultracentrifugation: Discrete Species and Size-Distributions for Macromolecules and Particles. ISBN: 9780367878283.
- Schuck, P. and Demeler, B. (1999). Direct sedimentation analysis of interference optical data in analytical ultracentrifugation. Biophys J 76(4): 2288-2296.
- Schuck, P., Perugini, M. A., Gonzales, N. R., Howlett, G. J. and Schubert, D. (2002). Size-distribution analysis of proteins by analytical ultracentrifugation: strategies and application to model systems. Biophys J 82(2): 1096-1111.
- Zhao, H., Brown, P. H. and Schuck, P. (2011). On the distribution of protein refractive index increments. Biophys J 100(9): 2309-2317.
- Zhao, H., Ghirlando, R., Piszczek, G., Curth, U., Brautigam, C. A. and Schuck, P. (2013). Recorded scan times can limit the accuracy of sedimentation coefficients in analytical ultracentrifugation. Anal Biochem 437(1): 104-108.
Article Information
Copyright
![]() Padavannil et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Padavannil et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Padavannil, A., Brautigam, C. A. and Chook, Y. M. (2020). Molecular Size Analysis of Recombinant Importin-histone Complexes Using Analytical Ultracentrifugation. Bio-protocol 10(10): e3625. DOI: 10.21769/BioProtoc.3625.
- Padavannil, A., Sarkar, P., Kim, S. J., Cagatay, T., Jiou, J., Brautigam, C. A., Tomchick, D. R., Sali, A., D'Arcy, S. and Chook, Y. M. (2019). Importin-9 wraps around the H2A-H2B core to act as nuclear importer and histone chaperone. Elife 8: e43630.
Category
Molecular Biology > Protein > Protein-protein interaction
Biochemistry > Protein > Quantification
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.