- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

HIV-CRISPR: A CRISPR/Cas9 Screening Method to Identify Genes Affecting HIV Replication
Published: Vol 10, Iss 9, May 5, 2020 DOI: 10.21769/BioProtoc.3614 Views: 10229
Reviewed by: Imre GáspárVinay PanwarKirsten A. CoprenAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Dec 2018
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Screening with CRISPR/Cas9 technology has already led to significant discoveries in the fields of cancer biology, cell biology and virology. Because of the relatively low false discovery rates and the ability to perform high-throughput, pooled approaches, it has rapidly become the assay of choice for screening studies, including whole-genome screens. Here, we describe a CRISPR screening protocol that allows for efficient screening of the entire life cycle of HIV-1 through packaging of the HIV-CRISPR lentiviral genomes by infecting HIV-1 virus in trans.
Keywords: HIVBackground
Genetic screens are powerful tools to identify new genes affecting the replication of viruses, including Human Immunodeficiency Virus (HIV). Identification of host genes important in HIV infection allows for enhanced understanding of HIV replication, evolution, transmission and pathogenesis, key interests of the HIV field. In particular, the discovery of HIV restriction factors, which are often Interferon (IFN) Stimulated Genes (ISGs), has been a crucial development in the field of retrovirology in the last decade. High-throughput approaches to discover HIV restriction factors have focused on over-expression screens to identify broadly acting antiviral ISGs (Schoggins et al., 2011) or acting specifically against HIV (Kane et al., 2016). One whole-genome screen for HIV restriction factors was performed by transfecting pools of siRNA pools in target cells (Liu et al., 2011). However, these methods lacked the robustness, versatility, and high-throughput aspects of CRISPR/Cas9 genetic screens. Thus, we developed an innovative approach using the CRISPR/Cas9 technology and relying on the ability of HIV to cross-package lentiviral genomes expressed in cells transduced with CRISPR/Cas9 libraries (OhAinle et al., 2018). This protocol was used originally to discover ISGs that block HIV-1 infection in the monocytic cell line THP-1, using a library of 2000 ISGs we designed. However, it can be used with any single guide RNA (sgRNA) library (whole-genome or subsets of genes) that is synthesized and assembled into the HIV-CRISPR lentiviral backbone. HIV-CRISPR was constructed by replacing the U3-deleted Self-Inactivating (SIN) LTR in the lentiCRISPRv2 vector with the full-length LTR from HIV-1LAI. This modification allows for HIV-CRISPR genomic RNA to be transcribed and available for packaging in trans into HIV virions. It can be used with any human cell line permissive for lentiviral transduction and editing, and can be used to study a variety of HIV isolates, mutants or even other closely-related lentiviruses that can package the HIV-CRISPR lentiviral genomes in trans. Importantly, this protocol assesses key factors important to the life cycle of HIV, including late steps such as assembly or budding, which may not be examined by other approaches relying on the use of reporter viruses.
Materials and Reagents
- 6-well and 12-well tissue culture plates
- 10 cm tissue culture dishes
- T-75 tissue culture flasks
- 0.22 μm filter flasks [such as MilliporeSigmaTM StericupTM Quick Release-HV Vacuum Filtration System (0.45 pore)] (Fisher, catalog number: SCHVU02RE )
- 0.22 μm filters (such as MilliporeSigmaTM MillexTM-GP Sterile Syringe Filters with PES Membrane) (Fisher, catalog number: SLGP033RS )
- Steriflip 50 ml conical tubes (MilliporeSigmaTM SteriflipTM Sterile Disposable Vacuum Filter Units) (Fisher, catalog number: SCGP00525 )
- 50 ml conical tubes
- Filter/barrier and non-filter/barrier tips
- Serological pipettes
- 293T cells (ATCC, catalog number: CRL-3216 ), grow in complete DMEM: DMEM 10% FBS, 1% Pen/Strep
- THP-1 cells (NIH AIDS reagent program, catalog number: 9942 ; RRID, catalog number: CVCL_0006 ), grow in complete RPMI: RPMI 10% FBS, 1% Pen/Strep, 10 mM HEPES, Glutamax
- TZM-bl cells (NIH AIDS reagent program, catalog number: 8129 ), grow in complete DMEM: DMEM 10%FBS, 1% Pen/Strep
- HIV-CRISPR plasmid (NIH AIDS reagent program, catalog number: 13567 )
- HIV-CRISPR Library (PIKA-HIV library, NIH AIDS reagent program, catalog number: 13566 )
- HIV vector packaging and envelope plasmids: pMD2.G plasmid (Addgene, catalog number: 12259 ) and psPAX2 plasmid (Addgene, catalog number: 12260 )
- Fetal Bovine Serum, heat-inactivated (Peak Serum, catalog number: PS-FB2 or equivalent)
- DMEM (Gibco, catalog number: 11965-092 )
- RPMI (Gibco, catalog number: 11875-093 )
- Penicillin/streptomycin (Gibco, catalog number: 15140-122 or equivalent)
- HEPES (Gibco, catalog number: 15630-080 )
- Glutamax (Gibco, catalog number: 35050-061 )
- Mirus Bio TransIT-LT1 Transfection Reagent (Fisher Scientific, catalog number: MIR2305 )
- Bovine Serum Albumin (Sigma, catalog number: A1933 )
- Puromycin dihydrochloride (Sigma, catalog number: P8833 )
- HIV viral stock, such as HIVLAI (BRU) Viral Stock (NIH AIDS reagent Program, catalog number: 2522 )
- DEAE-Dextran hydrochloride (Sigma, catalog number: D9885 )
- Qubit dsDNA HS Assay Kit (Thermo Fisher, catalog number: Q32854 )
- QIAamp Viral RNA Mini Kit (Qiagen, catalog number: 52904 )
- QIAamp DNA Blood Midi Kit (Qiagen, catalog number: 51185 )
- QIAquick PCR Purification Kit (Qiagen, catalog number: 28104 )
- RNAse AWAY Decontamination Reagent (Thermo Fisher, catalog number: 10328011 )
- SuperScript II Reverse Transcriptase (Thermo Fisher, catalog number: 18064014 )
- Herculase II Fusion DNA Polymerase (Agilent, catalog number: 600679 )
- PCR oligos (see Supplemental)
Oligos for the PCR1 reaction are custom DNA oligos from IDT, with standard purification/desalting (or equivalent). The PCR2 reaction oligos (R2_R, R2_Indexes) are HPLC-purified ultramers from IDT (or equivalent). - dNTPs (Thermo Fisher, catalog number: R0192 )
- DMSO (Sigma, catalog number: 472301 )
- Nuclease-free H2O
- Agarose
- Agencourt AMPure XP Beads (Beckman Coulter Life Sciences, catalog number: A63880 )
- HiSeq Rapid SBS Kit v2 (Illumina, catalog number: FC-402-4022 )
- HiSeq Rapid Cluster Kit v2 (Illumina, catalog number: GF-402-4002 )
- MiSeq Reagent Kit v2 (Illumina, catalog number: MS-102-2001 )
- PhiX Control V3 Kit (Illumina, catalog number: FC-110-3001 )
- Tris base (Sigma, catalog number: 10708976001 )
- Boric acid (Sigma, catalog number: B6768 )
- 0.5 M EDTA (Sigma, catalog number: E9884 )
- 10x TBE running buffer (see Recipes)
Optional:
- Human IFN Alpha Hybrid (Universal Type I Interferon) (PBL assay science, catalog number: 11200-1 ). Dilute in PBS 0.1% BSA and keep aliquots at -80 °C
- Sucrose
- NaCl
- 20% sucrose solution (see Recipes)
Equipment
- SW28 Beckman Polyallomer ultracentrifuge tubes (Beckman Coulter, catalog number: 326823 )
- Microcentrifuge (Eppendorf, model: 5425R or equivalent)
- Ultracentrifuge (Beckman Coulter, model: Optima L-90K or equivalent)
- SW28 (Eppendorf, catalog number: 342207 ) or SW32Ti (Beckman Coulter, catalog number: 369694 ) Ultracentrifugation rotor (or equivalent)
- Eppendorf 5810 tabletop centrifuge (Eppendorf, catalog number: 0 22625004 )
- Swinging-bucket rotor (Rotor S-4-104) (Eppendorf, catalog number: 5820755008 )
- Swinging-bucket rotor plate adapters (Eppendorf, catalog number: 0 22638866 )
- Vortex
- Water baths
- Nanodrop
- Applied Biosystems ProFlex Thermal Cycler (or equivalent)
- Magnetic rack (96-well)
- Qubit 4 Fluorometer (Thermo Fisher)
- Illumina MiSeq, HiSeq or equivalent
- CO2 tissue culture incubator
- -80 °C freezer
Software
- MAGeCK: https://sourceforge.net/p/mageck/wiki/Home/
- Bowtie: http://bowtie-bio.sourceforge.net/index.shtml
- FASTX Toolkit Barcode Splitter: http://hannonlab.cshl.edu/fastx_toolkit/
Procedure
Overview of the procedure (Figure 1):
•Preparation of HIV-CRISPR Library Lentiviral Vector Stock (A)
•Quantification of the HIV-CRISPR Lentiviral Vector Stock Titer (B)
•HIV-CRISPR Library Transduction (C)
•HIV-CRISPR Library Cell Infection (D)
•HIV-CRISPR Cellular Genomic DNA Extraction (E)
•HIV-CRISPR Viral RNA Extraction (F)
•Cellular Genomic DNA HIV-CRISPR Library Amplification (G)
•Viral RNA HIV-CRISPR Library Amplification (H)
•Library Clean-Up: Double-Sided SPRI (I)
•Quantification of Libraries (J)
•Pooling libraries for High-Throughput Sequencing (K)
•High-Throughput Sequencing (HTS) of HIV-CRISPR Libraries (L)
•HIV-CRISPR Screen Analysis (M)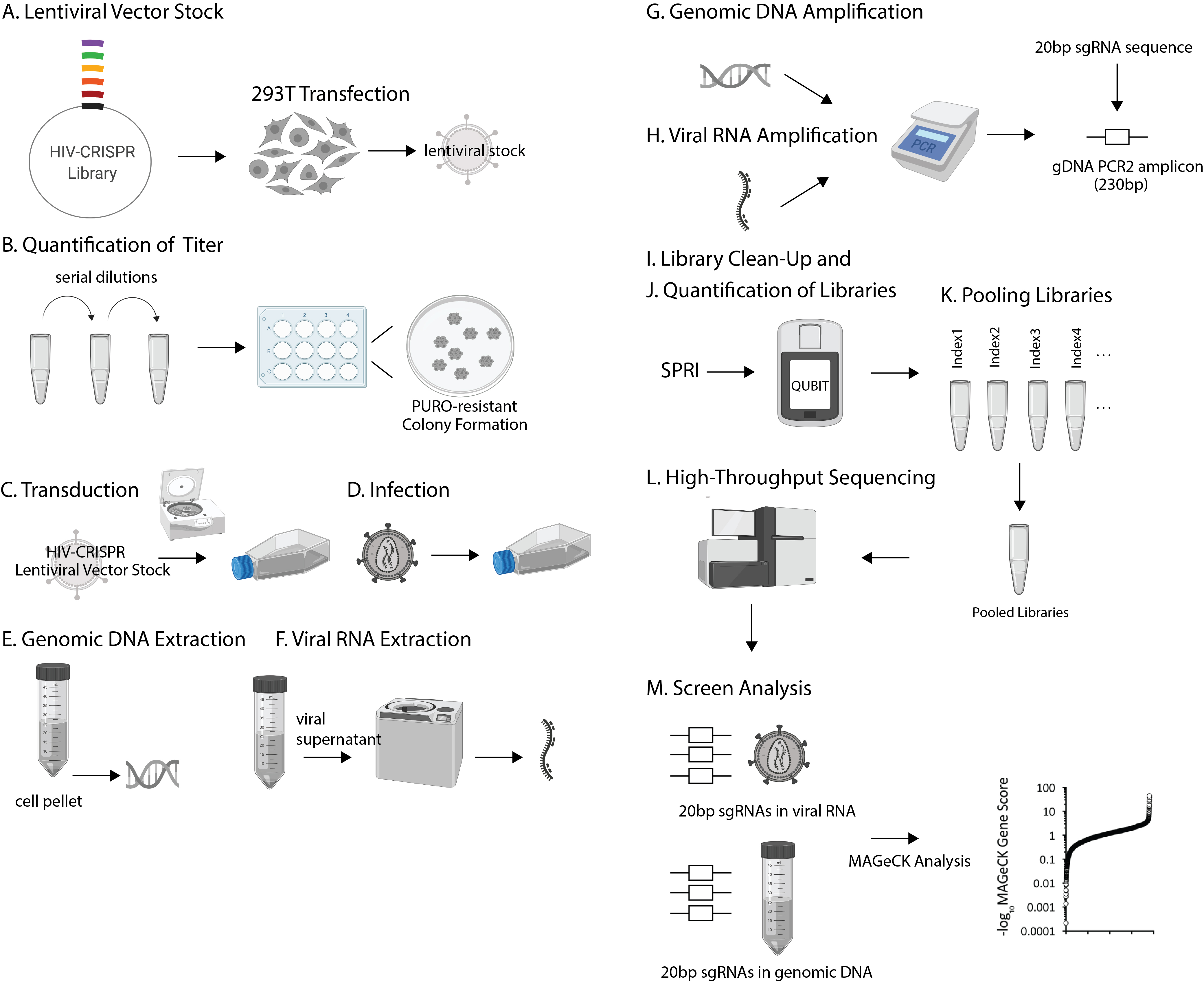
Figure 1. Schematic of the overall HIV-CRISPR Screening procedure
- Preparation of HIV-CRISPR Library Lentiviral Vector Stock
The goal of Procedure A is to generate a library-scale HIV-CRISPR lentiviral vector stock to be used to create HIV-CRISPR cells (Procedure C).- Day 0: plating 293T cells
One day prior to transfection seed 293T cells at a concentration of 2 x 105 cells/ml in 2 ml/well in 6-well plates. Use low passage 293T cells maintained below confluency for several passages in order to increase lentiviral titers. Typically, 20 6-well plates are prepared at a time and are usually enough for several transductions of a ~15,000 sgRNA library or one transduction with a ~100,000 sgRNA whole-genome library. - Day 1: transfection
- Check the confluency of cells: ideally, they should be at 50% confluency at the time of transfection.
- For each well of a 6-well plate, mix 667 ng of the HIV-CRISPR Library plasmid, 500 ng of the psPAX2 plasmid (HIV GagPol) and 333 ng of the pMD2.G plasmid (VSV-G).
- Add all DNA to 200 μl of serum-free medium (SFM-DMEM) and vortex for 5 s.
- Bring the TransIT-LT1 transfection reagent to room temperature and vortex for 5 s, making sure that no precipitate is visible.
- Add 4.5 μl TransIT-LT1 (ratio of 3 μl MIRUS to 1 μg of total plasmid DNA) directly to SFM media/DNA and vortex for 10 s.
- Incubate the DNA/TransIT-LT1 mix for 20 min to form complexes.
- Add transfection mix slowly to each well of 293T cells and gently swirl the plate to distribute.
- Day 2: replace transfection media
Remove the media and replace it with 1.5 ml of complete DMEM per well. - Day 3: collect viral supernatant
- Remove viral supernatant from cells.
Optional: replace media with 1.5 ml of complete DMEM per well for a second collection the following day. - Clarify supernatant by centrifuging cell debris for 5 min at 1,000 x g at room temperature.
- Collect cleared viral supernatant in a new 50 ml Falcon tube.
- (Optional) Day 4: Collect the viral supernatant, similarly as on Day 3. Pool the supernatants from collection 1 and 2 together.
- Remove viral supernatant from cells.
- Filtering: Filter viral supernatant with a 0.22 μm filter flask.
Note: Supernatants can be stored at 4 °C overnight or for several days without significant loss of infectivity. - Virus concentration: Add 33 ml of viral supernatant to SW28 Beckman Polyallomer ultracentrifuge tubes. If less than 33 ml viral supernatant is available, fill with DMEM media. Carefully add 4 ml of 20% sucrose solution to each SW28 Beckman Polyallomer ultracentrifuge tube with a 5 ml pipet. The tube should be filled up to 0.5 cm from the top to avoid collapsing during ultracentrifugation. Finish filling the tube with complete DMEM media as needed.
- Load the SW28 ultracentrifuge tubes into a pre-chilled SW28 or SW32Ti rotor. Spin at 70,000 x g for 1 h at 4 °C.
- Decant clarified viral supernatants and let tubes dry inverted on a paper towel for several minutes. Put each tube into a 50 ml conical tube and resuspend the (non-visible) pellets in the desired volume (from 200 μl to 1 ml) and incubate at 4 °C.
- After 15-60 min of incubation, vortex gently for 5 s. Incubate at 4 °C for several hours (or ideally overnight) with occasional vortexing.
- Gently resuspend viral pellets in DMEM media using a 1 ml pipet (avoid generating bubbles), combine all supernatants, mix, aliquot and freeze at -80 °C. Small aliquots (20-50 μl) can be made for titering in TZM-bl or other cells.
- Scale up the transfection as needed, depending on the amount of lentivector needed.
- Lentiviral vector production is more efficient in 6-well plates rather than 10 cm or other dishes.
- All waste should be handled according to institutional biosafety rules.
- Day 0: plating 293T cells
- Quantification of the HIV-CRISPR Lentiviral Vector Stock Titer
The aim of this step is to quantify the titer of the lentiviral vector stock produced in Procedure A. The Colony Forming Assay used here will allow calculating the volume of lentiviral vector stock necessary to achieve the desired Multiplicity of Infection (MOI). Multiplicity of Infection (MOI) is a ratio of infectious particles to the number of cells and can be used to deliver a specific number of transducing lentiviral vectors on average to cells in culture.- Day 0: cell plating
Plate TZM-bl cells at 1.5 x 105 cells/ml in a 12-well plate (1 ml per well). - Day 1: virus infections
- Replace the media with 300 μl of complete DMEM, with a final concentration of 20 μg/ml DEAE-Dextran.
- Thaw an aliquot of the HIV-CRISPR lentivector preparation.
- Prepare the following HIV-CRISPR lentivector dilutions in duplicate in complete DMEM: 1:10, 1:100, 1:1,000, 1:3,000, 1:10,000 (for example, dilute 20 μl to 180 μl for a 1:10 dilution, serial dilute from there)
- Add 20 μl of diluted lentivector per well of a 12-well plate in duplicate.
- Infect cells by spin-infection (spinoculation) for 30 min at 1,100 x g at room temperature, using a tabletop centrifuge with swinging-bucket rotors/plate adapters.
- Add 200 μl media to well to fill to 500 μl per well. Incubate cells overnight at 37 °C in a CO2 incubator.
- Day 2: puromycin selection
Replace media with complete DMEM with 0.5 μg/ml puromycin (1 ml per well).
Note: Puromycin concentration is cell-type dependent and can be determined with a drug kill curve (see Step C2c). - Day 3-Day 11: cell killing
- Replace the media with fresh Puromycin-containing DMEM media as needed to clear off dead cells until colonies of cells are visible and can be manually counted (typically in the 1: 1,000-1: 3,000 range).
- Calculate the titer (Infectious Units/ml of lentiviral supernatant). For example, if 40 colonies are counted (on average) at the 1:1,000 dilution, this would be equivalent to 40,000 IU for 20 μl of inoculum used in the titering assay. This is equal to 2 x 106 IU/ml in the lentiviral supernatant.
- Day 0: cell plating
- HIV-CRISPR Library Transduction
This step consists in the generation of a pool of cells stably expressing the Cas9 enzyme and the sgRNA library. Upon transduction with the lentiviral stock produced in Procedure A and quantified in Procedure B, each target cell will carry an integrated lentiviral HIV-CRISPR lentiviral genome encoding for Puromycin resistance, the Cas9 enzyme, and a specific sgRNA from the library.- Day 0: cell plating and transduction
- Plate the cells to be transduced in a 12-well plate. For adherent cells, cells should be at approximately 75% confluency. For suspension cells, use a concentration of 2 x 106 cells per well in 500 μl in a 12-well.
Note: Plate enough cells to ensure 500x coverage of the library, meaning that each sgRNA in the library is sampled a minimum of 500 times. To calculate the number of cells to transduce, multiply the number of individual sgRNAs in the library by 500. For example, the GeCKOv2 whole genome library that contains 123,411 sgRNAs: therefore, 62 x 106 transduced cells should be targeted. - Add DEAE-Dextran to the media to make a final concentration of 20 μg/ml to enhance transduction efficiency.
Note: This concentration of DEAE-Dextran is optimal to enhance transduction of THP-1 cells. For other cell types, optimize this concentration with a dose response to ensure optimal transduction rates while minimizing cell toxicity. - Add the appropriate volume of HIV-CRISPR library lentiviral vector to reach an MOI of 0.5-0.75, thereby allowing for a cell population post-selection in which only one sgRNA (and therefore gene knockout) is present in most cells in the population.
- Infect cells by spin-infection (spinoculation) for 30 min at 1,100 x g at room temperature in a tabletop centrifuge with swing bucket rotors/plate adapters. Add 500 μl of media to each well and incubate overnight at 37 °C in a CO2 incubator.
- The amount of lentiviral stock to use in order to achieve an effective MOI of 0.5-0.75 may differ significantly in other cell types, as the rate of lentiviral transduction efficiency varies. Optimization of this step can be performed for each cell line used. Testing different amounts of DEAE-dextran and various dilutions of the lentiviral stock allows for cell type-specific determination of optimal transduction conditions.
- Leftover lentivector stock not used at this step may be frozen back once without major loss of titer.
- Plate the cells to be transduced in a 12-well plate. For adherent cells, cells should be at approximately 75% confluency. For suspension cells, use a concentration of 2 x 106 cells per well in 500 μl in a 12-well.
- Day 1
- For suspension cells, combine all wells of the transduced cells in one tube by resuspending the cells with a 1 ml pipet (pipet up and down to fully resuspend all cells from each well) and collecting in a Falcon tube. Make sure that no cells stay attached to the bottom of the well. For adherent cells, trypsinize the cells to remove the cells from the plate.
- Pellet cells for 5 min at 1,000 x g.
- Resuspend cell pellet at a final concentration of 5 x 105 cells/ml in complete RPMI (or other media as appropriate) with 0.5 μg/ml Puromycin (or other appropriate concentration).
Note: This concentration of Puromycin was optimized for selection in THP-1 cells, and other concentration as appropriate for each cell type. We recommend using puromycin concentrations previously published in the literature for each cell type of interest, or as determined by a kill curve prior to the transduction. The kill curve is performed by adding increasing concentrations of puromycin on non-transduced cells, which allows determination of the minimal concentration necessary to kill 100% of cells after several days of puromycin selection in culture.
- Day 2-Day 14: HIV-CRISPR cell library selection
- Continue puromycin selection for 10-14 days to select for transduced cells and to allow for CRISPR/Cas9-mediated gene knockout to occur.
- Replace the media every 2-3 days with complete media containing puromycin to expand the cells, always maintaining the cells under selection. Split cells when appropriate, always maintaining enough cells to ensure good coverage of the library (> 500x).
- When selection is complete, use cells for the experiment and freeze leftover cells in 90% FBS and 10% DMSO for future experiments.
- Day 0: cell plating and transduction
- HIV-CRISPR Library Cell Infection
Cells generated in Procedure C are infected by HIV in order to assess the effect of each single gene knock-out in the library of cells on HIV replication. Typically, a screen for each condition (i.e., virus) is done in duplicate to enhance screen results.- Day 0
Count and plate cells at 2 x 106 cells/ml in 500 μl in 12-well dishes.
Note: Cells can be pretreated one day prior with 1000 U/ml of recombinant Universal Type I IFN alpha for 24 h. - Day 1
- Infect cells in a 12-well plate with 20 μg/ml DEAE-Dextran. Different MOIs may be used depending on the aim of the experiment (identification of cofactors or restriction factors; sensitivity of the phenotype to saturation, low infection levels, etc.). Total volume for infection can vary, smaller overall volumes give higher rates of infection.
- Infect cells by spin-infection (spinoculation) for 30 min at 1,100 x g at room temperature, using a tabletop centrifuge with swinging-bucket rotors/plate adapters.
- Add 500 μl of media in each well and incubate overnight at 37 °C in a CO2 incubator.
- Day 2
Spin cells for 5 min at 1,000 x g and resuspend them in media (with or without IFN as appropriate). - Day 4
Collect cells and supernatants for nucleic extraction, amplification and sequencing.
- Depending on the cell line or the desired outcome, different MOIs or time of collection can be used.
- Important for remaining protocol steps: contamination with plasmid DNA, genomic DNA or amplified libraries can be a problem and steps should be taken to reduce the likelihood of contamination. Use clean pipets with filter tips, freshly opened kits and reagents, and dedicated hoods and benches for each step as possible.
- Day 0
- HIV-CRISPR Cellular Genomic DNA Extraction
Cellular genomic DNA is extracted from cells to allow subsequent PCR amplification (Procedure G) of the HIV-CRISPR lentiviral genomes (including the sgRNAs) integrated in the cell population.- Pellet cells by spinning for 5 min at 1,000 x g to separate cells and virus-containing supernatant. Save supernatants for viral RNA extraction (Procedure F). The pellets can be stored at 4 °C for a few days (or frozen at -80 °C although lysis becomes more difficult) before DNA extraction.
- Extract genomic DNA (gDNA) using the QIAamp DNA Blood Midi Kit as per the protocol, with the following modifications:
- Ideally use a maximum of 5 x 106 cells per extraction (scale up to multiple extractions as required).
- Perform the extra centrifugation step to remove as much ethanol as possible before eluting (ethanol can inhibit downstream amplification steps).
- Elute genomic DNA in 250 μl H2O instead of the buffer provided in the DNA Blood Midi kit (which contains EDTA and can inhibit downstream amplification steps).
- Perform two successive 250 μl elutions. Keep Elution 1 and Elution 2 separate, and quantify DNA using a nanodrop (typical yield is 100-300 ng/μl). The second elution is usually less concentrated, but more efficiently amplified because of reduced ethanol carry-over.
- Freeze gDNA at -20 °C or -80 °C.
- HIV-CRISPR Viral RNA Extraction
HIV virions are collected from the supernatant and purified. Viral RNA from virions is extracted to allow subsequent PCR amplification of the sgRNA encoded by the HIV-CRISPR viral genomic RNA.- Collect viral supernatant when separating cells in Procedure E above. Viral supernatant can be stored for up to a week at 4 °C before or after filtering (see Procedure A or long-term at -80 °C).
- Filter supernatants with a 0.22 μm filter or with a Steriflip 50 ml conical tube.
- Concentrate viral particles over a 20% sucrose solution, as described in Step A6. The centrifugation is carried out at 70,000 x g for 1 h at 4 °C.
- Slowly resuspend the viral pellets in 140 μl of PBS, and incubate overnight at 4 °C, occasionally vortexing.
Note: This step can be performed after several hours at 4 °C. - Transfer to a 1.5 ml Eppendorf tube. Store at 4 °C for several days or freeze at -80 °C if not used right away for RNA extraction.
- Extract viral RNA using the QIAamp Viral RNA Mini Kit as per the protocol, with the following modifications:
- This step should be carried on a dedicated RNA bench. Clean the bench and pipettes thoroughly with RNase away. Use filter/barrier tips.
- The carrier RNA is not added to Buffer AVL during lysis.
- Viral RNA (vRNA) is eluted in 40 μl of buffer AVE to facilitate downstream reverse transcription (RT) reactions.
- Freeze vRNA at -80 °C and avoid multiple freeze/thaw cycles.
- Cellular Genomic DNA HIV-CRISPR Library Amplification
sgRNA sequences present in the integrated HIV-CRISPR vectors in the pool of cells are amplified by PCR from cellular genomic DNA (Procedure E). Primers for amplification flank the variable sgRNA sequences, resulting in unbiased amplification of all sgRNA sequences present in the HIV-CRISPR Cell Library. The library representation of sgRNA sequences in the cellular genomic DNA is used to normalize the data during Screen Data Analysis (Procedure L).- Thaw isolated genomic DNA (gDNA) preps (from Procedure E) and PCR reagents on ice.
- Calculate the amount of gDNA to amplify to ensure approximately 500x coverage of the library (using an estimate of 6.6 pg of total genomic DNA per cell on average) and the number of PCR reactions to perform, using a maximum of 2 μg of genomic DNA for each PCR reaction to ensure most efficient amplification. For example, for the GeCKO v2 library containing 123,411 sgRNAs, a 500x coverage corresponds to 6.17 x 107 cells, or 410 μg of DNA. In this case, perform 205 PCR reactions.
- Perform PCR on the HIV-CRISPR lentiviral genome, to obtain a PCR1 product of 445 bp (Figure 2). PCR reactions are set up as follows (Table 1):
Notes:- The list of primers used is available in Supplemental.
- Preparation of PCR reactions is done in a dedicated, DNA-free clean area or hood with dedicated, plasmid-free pipettes to avoid contamination during amplification.
Table 1. First-Round PCR (PCR1)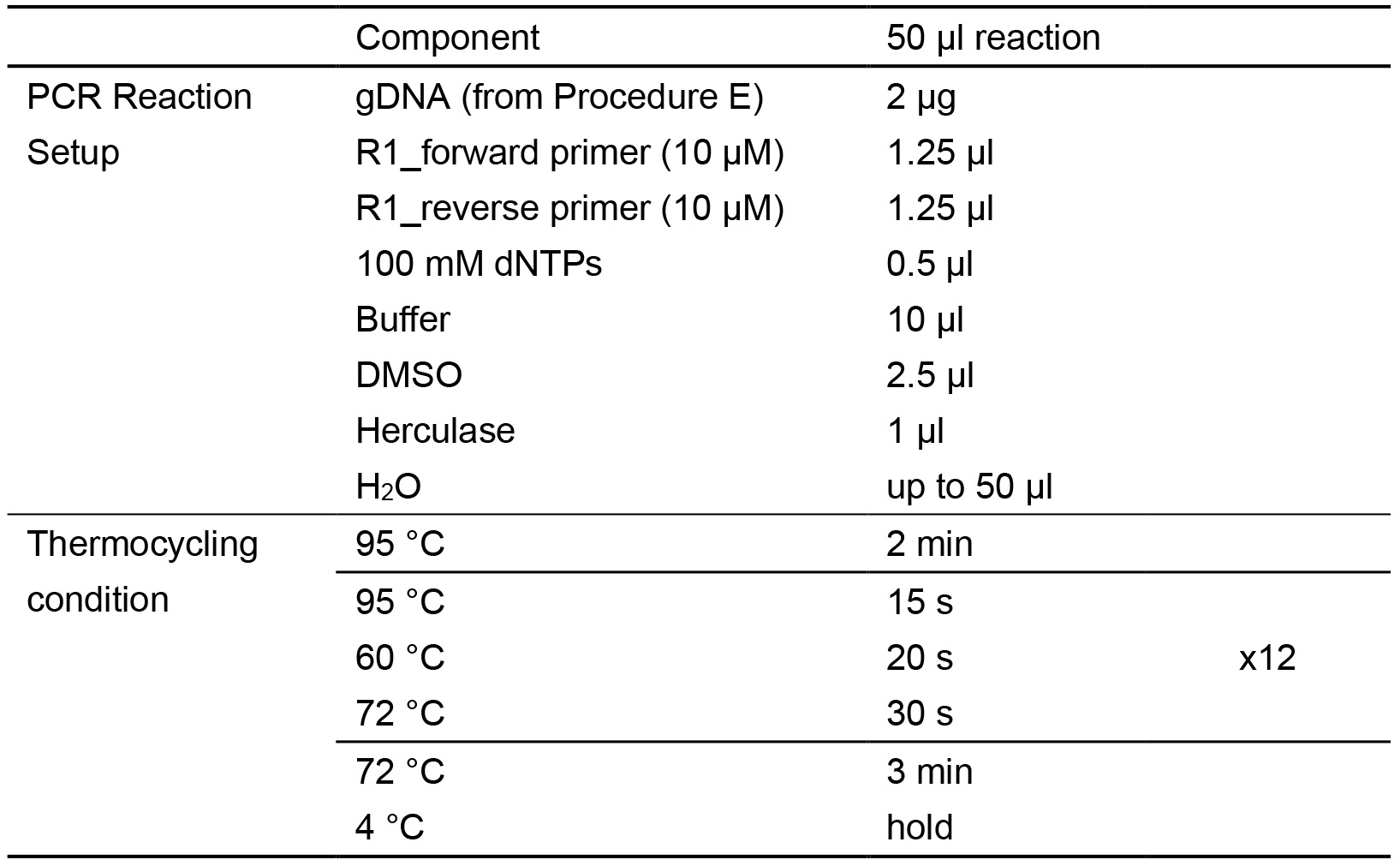
- Combine together all 50 μl PCR1 reactions in a single tube.
Note: The total number of PCR1 reactions to be pooled at this step will vary, depending on the total amount of genomic DNA amplified (as determined in Step G2). - Clean up the PCR1 pool with the QIAquick PCR clean up kit, following the kit instructions:
http://2012.igem.org/wiki/images/a/a3/QIAquick_PCR-purification.pdf.
Note: We typically pool up to 12 PCR reactions on a single column and elute in 50 μl of H2O. This allows for concentration of the PCR1 product, while reducing the number of individual columns needed and avoiding saturation of the columns (that can bind up to 10 μg of DNA according to the manufacturer’s instructions). - Combine the cleaned up PCR1 reactions to create a cleaned-up PCR1 Pool.
- Perform the PCR2 reaction, leading to amplification of a PCR2 product (size = 230 bp). PCR reactions are set up as follows (Table 2):
Notes:- Four PCR2 reactions are performed to amplify sufficient product for clean -up and pooling for HTS while also avoiding PCR bias that could be introduced in a single PCR reaction.
- Each sample (genomic DNA replicate) must be amplified with a unique barcoded Indexing Primer (these are the “R2_IndexX” primer set). Extra care should be taken to avoid cross-contamination of Indexing Primers as this would result in mis-assignment of reads during barcode demultiplexing. The list of primers used is available in Supplemental. Other custom primer sets can be designed and used if desired.
Table 2. Second-Round PCR (PCR2)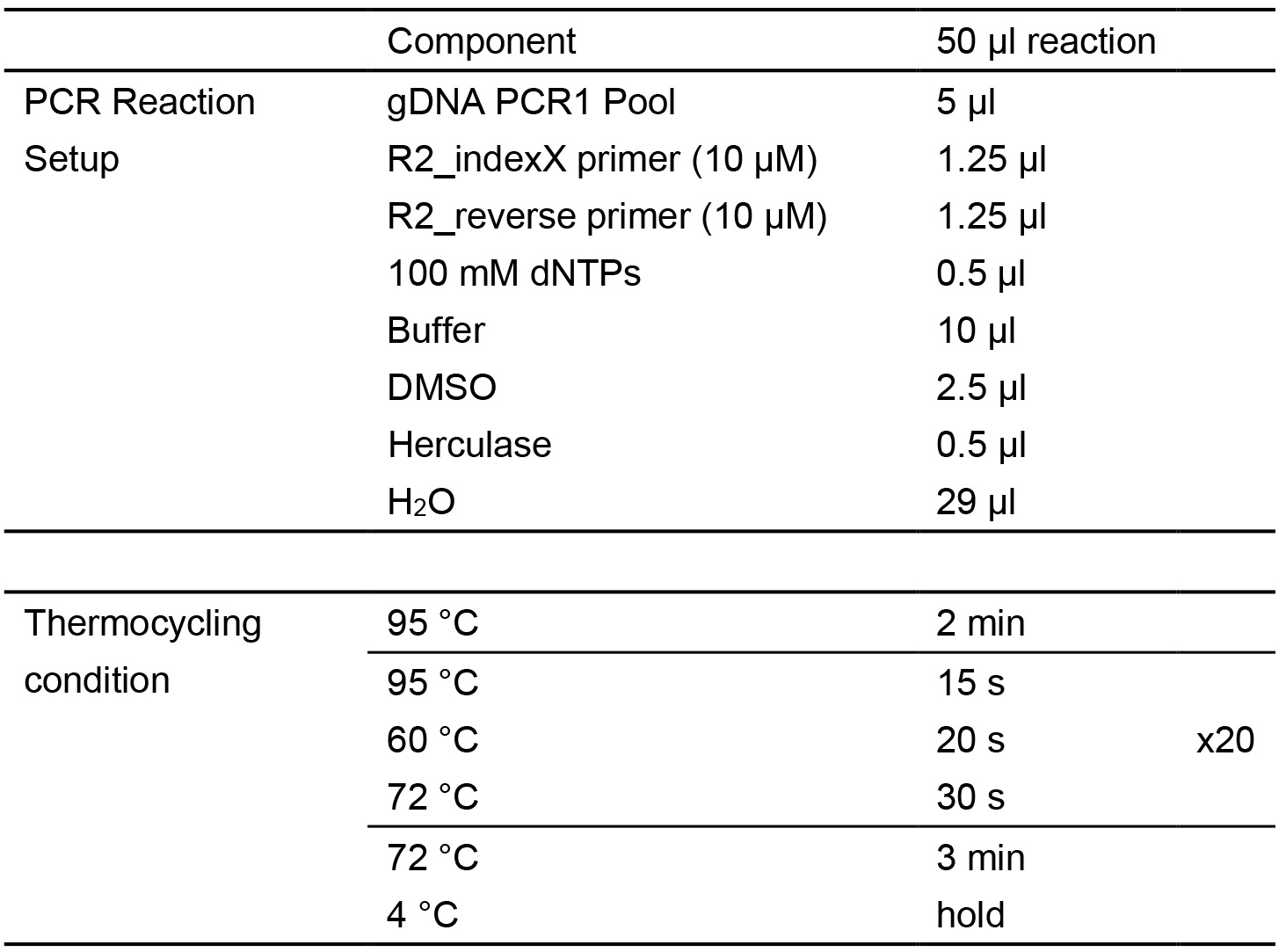
- PCR amplification can be verified by running a small amount of the PCR2 reactions on a 2% analytical TBE gel (230 bp product).
Notes:- Carried-through PCR1 product (the 445 bp product amplified in the first round of PCR) may remain visible but should not affect downstream pooling and HTS as most of this product is removed during the Library Clean Up (Procedure I). Carried through product could affect accurate quantification but does not contain sequences necessary for HTS and, therefore, do not interfere with sequencing (Procedure K).
- TBE (as opposed to TAE) allows for easier visualization of the 230 bp amplicons but should not be used for gel purification.
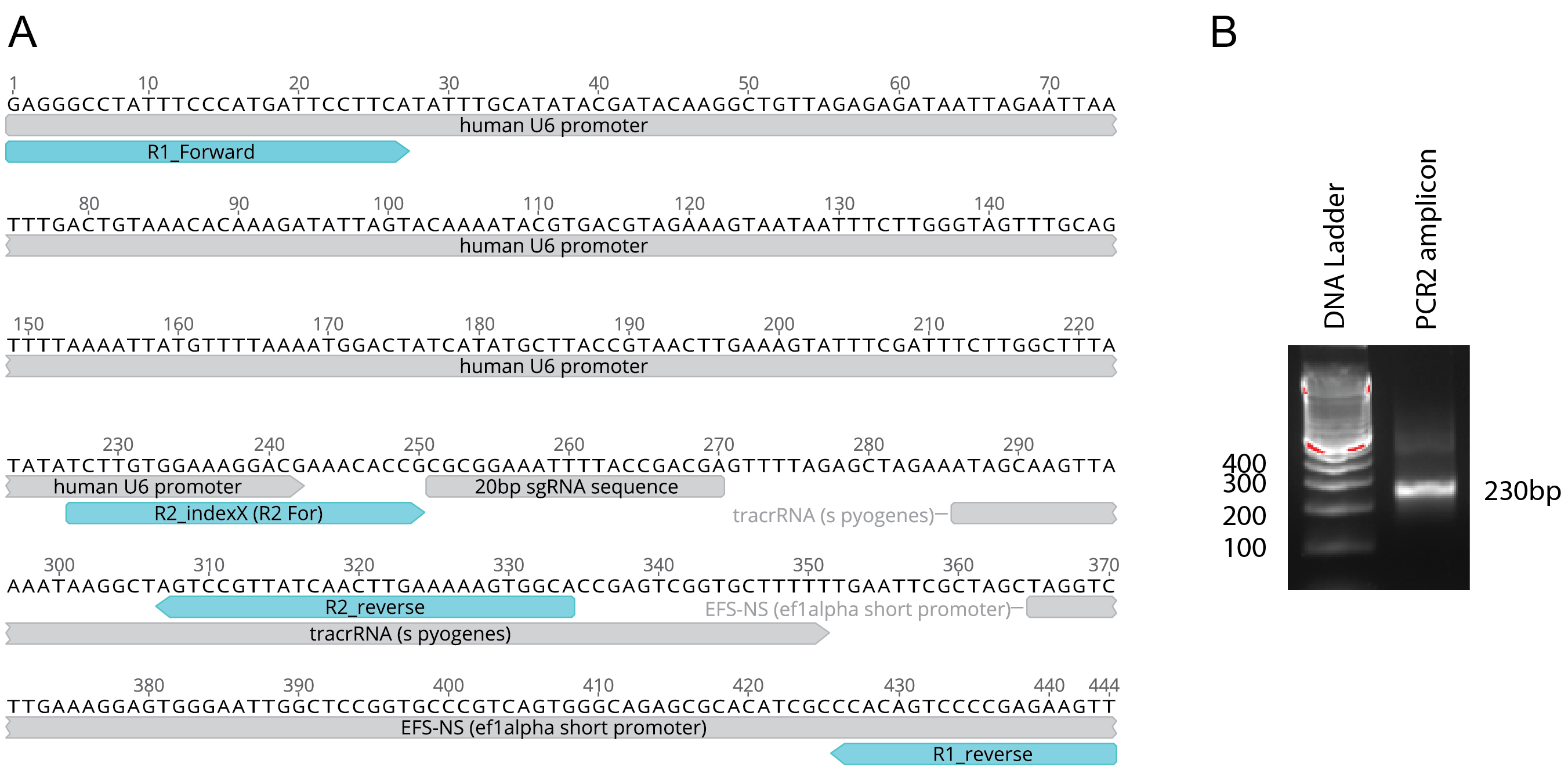
Figure 2. Round 1 (PCR1) and Round 2 (PCR2) amplicons. A. A zoomed view of the HIV-CRISPR sequence is shown with the R1 (PCR1) and R2 (PCR2) primer binding sites mapped. An example 20 bp sgRNA sequence is displayed in the HIV-CRISPR vector backbone. B. Example gel showing expected PCR2 amplicon (230 bp). Note that some 445 bp PCR1 amplicon product is also visible.
- Combine together all PCR2 reactions to create the gDNA PCR2 Pool. This includes all four PCR2 reactions, as indicated in Step G7.
- Freeze the gDNA PCR2 Pool at -20 °C or at 80 °C.
- Viral RNA HIV-CRISPR Library Amplification
The sgRNAs encoded in the packaged HIV-CRISPR viral genomes (packaged in trans by HIV) are amplified by PCR. This allows for the relative frequency of each sgRNA in the virions to be measured.
Note: Preparation of PCR reactions are done in a dedicated, DNA-free clean area or hood with dedicated, plasmid-free pipettes to avoid contamination during amplification.- Thaw viral RNA (vRNA from Procedure F), the SuperScript II Reverse Transcriptase kit and Herculase PCR reagents on ice.
- Perform reverse transcription of viral RNA as follows (Table 3):
Note: The list of primers used is available in Supplemental.
Table 3. RT
- Incubate RT reactions for 5 min at 65 °C in a Thermal Cycler, then place on ice.
- Add 1 μl of the Superscript II enzyme to each RT reaction.
Note: Mix well by pipetting up and down carefully to avoid bubbles. This step is key to ensure adequate mixing of reagents for maximum-efficiency amplification of all HIV-CRISPR viral RNA templates. - Incubate in a Thermal Cycler as follows:
42 °C 50 min
70 °C 15 min - Pool together all RT reactions to create the vRNA RT Pool.
- Next, PCR1 amplification of the reverse-transcribed viral RNA cDNAs is performed similar to genomic DNA amplification (Table 4).
Table 4. First-Round PCR for RT (PCR1)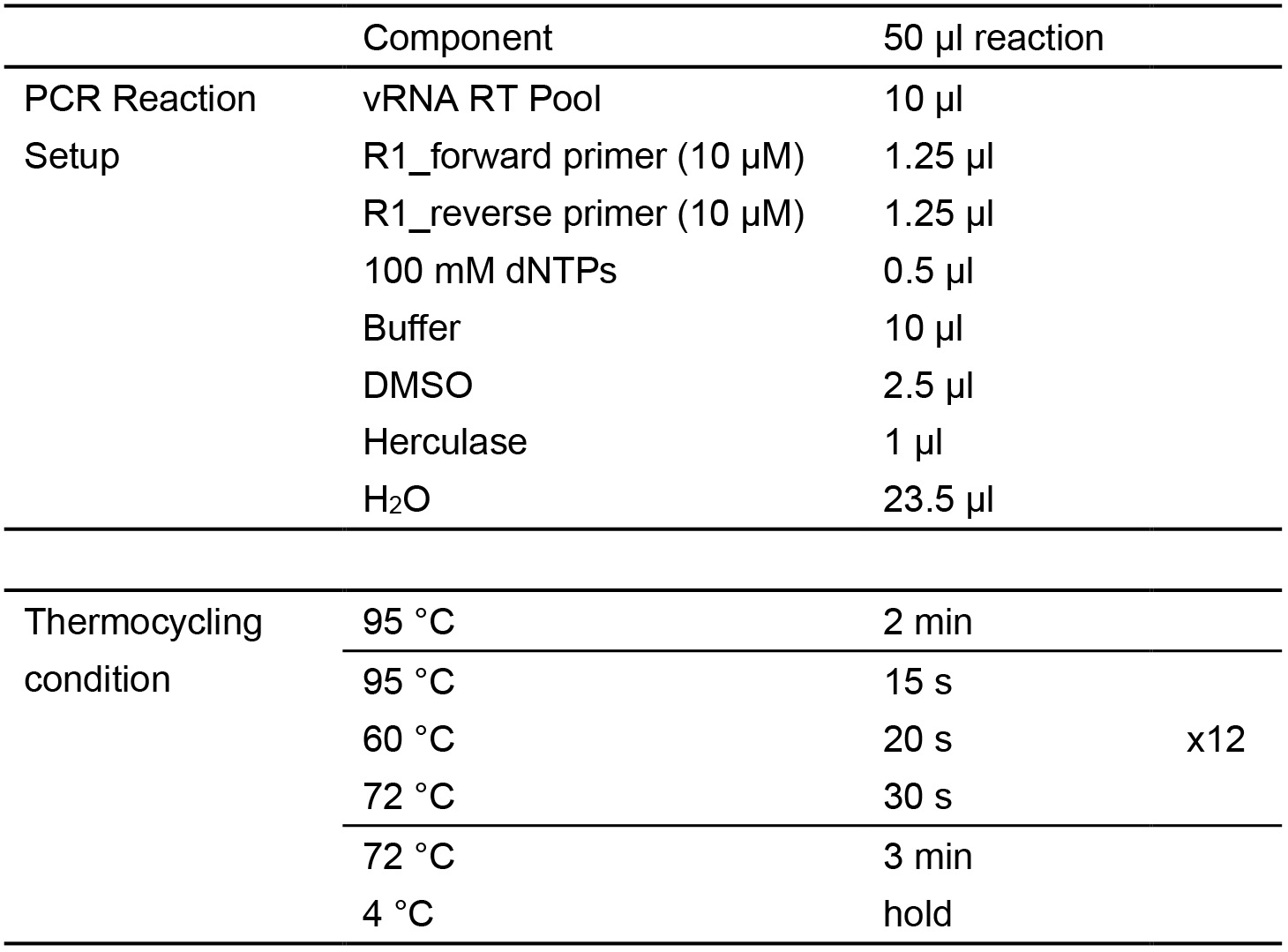
- Up to 8 PCR1 reactions are pooled together and cleaned up using a single column of the QIAquick PCR clean up kit, with a 50 μl final elution volume (vRNA PCR1 Pool).
- 12.5 μl of the vRNA PCR1 Pool is used as template in four PCR2 reactions (Table 5):
Notes:- Each sample (vRNA replicate) must be amplified with a unique barcoded Indexing Primer (these are the “R2_IndexX” primer set). Extra care should be taken to avoid cross-contamination of Indexing Primers as this would result in mis-assignment of reads during barcode demultiplexing. The list of primers used is available in Supplemental. Other custom primer sets can be designed and used if desired.
- Four PCR2 reactions are performed to amplify sufficient product for clean -up and pooling for HTS while also avoiding PCR bias that could be introduced in a single PCR reaction.
Table 5. Second-Round PCR (PCR2)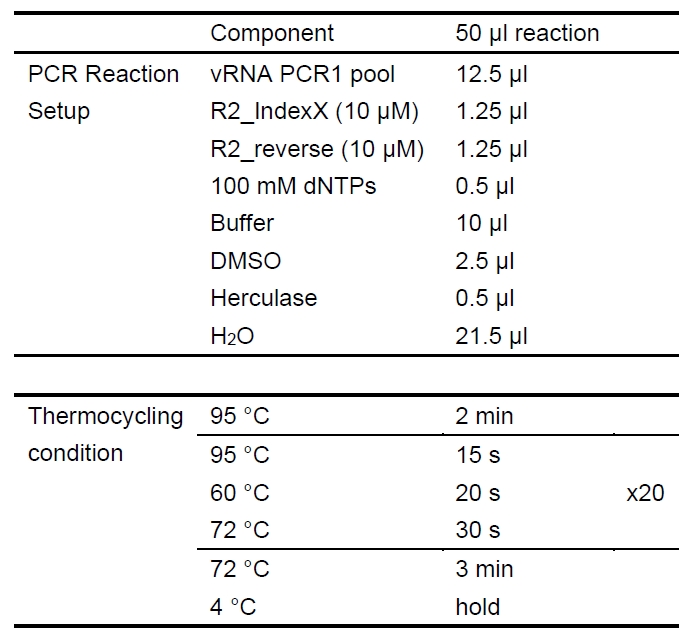
- PCR amplification can be verified by running a small amount (i.e., 5 μl) of the PCR2 reactions on a 2% analytical TBE gel (see Figure 2: 230 bp product).
Note: Carried-through PCR1 product (445 bp product) may remain visible but should not affect downstream pooling and HTS as most of this product is removed during the Library Clean Up (Procedure I). Carried-through product could affect accurate quantification but does not contain sequences necessary for HTS and, therefore, do not interfere with sequencing (Procedure K). - Combine together all PCR2 reactions to create a vRNA PCR2 Pool.
- Freeze the vRNA PCR2 Pool at -20 °C or at 80 °C.
- Library Clean-Up: Double-Sided SPRI
Individual libraries are cleaned up prior to sequencing in order to remove excess primer and other nonspecific products (including PCR1 product that may be carried through). Double-sided Solid Phase Reversible Immobilization (SPRI) accomplishes this but other approaches, such as Gel Purification can also be used. The bead:DNA ratio used here results in sufficient isolation of the 230 bp amplicons used in sequencing, but other ratios may also be appropriate.- Vortex an appropriate volume of AMPureXP beads thoroughly and bring them to room temperature.
- Mix PCR2 Pool with the beads at a 0.8x ratio (e.g., 40 μl of DNA for 32 μl of beads) and vortex for 5 s.
- Incubate for 5 min at room temperature.
- Place tubes on a magnetic rack and incubate for 5 min.
- Transfer the supernatant to a new tube, without disturbing the beads.
- Incubate the supernatant from the previous step with beads at a 1.5x ratio (e.g., 50 μl of supernatant with 75 μl of beads). Vortex for 5 s and incubate for 5 min at room temperature.
- Place tube on the magnetic rack for 5 min and carefully remove and discard the supernatant by pipetting, without disturbing the beads.
- Add 200 μl of 80% ethanol (make fresh), keeping the tube on the magnetic stand. Incubate for 30 s and remove and discard the supernantant.
- Optional: Repeat Step I7 one more time.
- Air dry the beads on the magnet for 5 min but no longer (to avoid overdrying the DNA/beads mixture).
- Remove the tube from the magnet and resuspend the beads in 45 μl of nuclease-free H2O.
- Vortex DNA/Bead mix for 5 s and incubate at room temperature for 2 min.
- Place on the magnetic rack for 3 min. Carefully transfer 35 μl of supernatant to a new tube without carrying over beads. The supernatant should appear clear; if not, repeat this step to get rid of leftover beads.
- Store cleaned-up libraries at -20 °C or -80 °C (or for at least several weeks at 4 °C).
- Quantification of Libraries
The amplicon libraries need to be precisely quantified prior to pooling and sequencing to maximize sequencing output of adapter- and sgRNA-containing amplicon DNA during High-Throughput Sequencing (HTS) (Procedure L). Accurate quantification of library DNA is done with the Qubit dsDNA HS Kit and will be enhanced by calibrating the fluorometer with the provided calibration standards each time the assay is performed. Qubit dsDNA HS Assay Kit Manufacturer’s protocol:
assets.thermofisher.com/TFS-Assets/LSG/manuals/Qubit_dsDNA_HS_Assay_UG.pdf.- Quantify the dsDNA present in each cleaned-up PCR2 library sample, following the instructions for the Qubit dsDNA HS Kit as follows:
- Prepare the Qubit solution by diluting Qubit reagent 1:200 in the provided buffer.
- For each sample, add 2 μl of sample to 198 μl of the diluted buffer.
- Read out quantification of dsDNA in each sample on the Qubit and record.
- Quantify the dsDNA present in each cleaned-up PCR2 library sample, following the instructions for the Qubit dsDNA HS Kit as follows:
- Pooling libraries for High-Throughput Sequencing
In this step, the amplicon libraries previously prepared are sequenced by High-Throughput Sequencing (HTS). Libraries are typically sequenced using Illumina miSeq or HiSeq, but other HTS methods may also be appropriate.- Calculate the concentration of all cleaned-up libraries and convert to nM.
For example, for an amplicon size of 230 bp, 1 ng/μl equates to a concentration of 6.59 nM. - Dilute the libraries to a final concentration of 2 nM in nuclease-free H2O or other appropriate dilution for your sequencing method.
- Pool together individual 2 nM libraries in a single tube to create a final 2 nM Pool for sequencing.
- Calculate the concentration of all cleaned-up libraries and convert to nM.
- High-Throughput Sequencing (HTS) of HIV-CRISPR Libraries
In this step, the amplicon libraries previously amplified, cleaned-up and quantified are sequenced by High-Throughput Sequencing (HTS). An Illumina miSeq or HiSeq can be used, but other HTS methods may also be appropriate.
Notes:- The typical output of a miSeq run is usually 20% that of a HiSeq Rapid Run Lane. However, depending on the screen, a miSeq can be adequate. The sequencing depth required will depend on a number of factors, including the precise experimental setup, strength of selection and efficiency of nucleic acid extraction/amplification/clean-up. Typically, we obtain 1-5 million reads per sample, which is more than sufficient. More information on how to determine appropriate sequencing depth can be found on the manufacturer’s website: Illumina.com/science/technology/next-generation-sequencing/plan-experiments/coverage.html.
- Adding PhiX DNA may be necessary to avoid sequence quality issues caused by the low diversity samples, a common problem in amplicon sequencing. A PhiX spike of at least 20% is a useful starting point and can be optimized further in order to gain additional sequencing depth of the samples. Because 50 cycles are sufficient for HIV-CRISPR sequencing with the current amplification protocol/primer sets, we use the HiSeq or miSeq on a Single-Read 50 (SR50) setting.
- HIV-CRISPR Screen Analysis
A variety of data analysis methods can be used to analyze CRISPR screen data. The goal of this protocol is to identify genes affecting HIV replication: therefore, our data analysis approach aims at calculating which gene knock-out leads to increased or reduced HIV infection. To do this, we calculate the relative frequency of each sgRNA sequence in HIV-CRISPR genomes packaged in virions as compared to sgRNA sequences encoded in HIV-CRISPR genomes integrated in cells. The assumption is that sgRNAs targeting antiviral factors will be enriched in the virions and sgRNAs targeting cofactors that are required for or that enhance infection will be depleted in the viral fraction. Statistical methods explained below allow to take into account all the sgRNAs targeting a single gene to assign a score to each gene.
An example analysis pipeline: Raw BCL files are converted to FASTQ files using the bcl2fastq conversion software from Illumina. FATSQ files are then demultiplexed to assign reads to the different samples, using FASTX Toolkit Barcode Splitter. For each sample, reads are then trimmed to the 20 bp variant sgRNA sequence and aligned to the sgRNA library using Bowtie (Langmead et al., 2009). As a negative control, NTC (Non-Targeting Control) sgRNA sequences can be iteratively binned to create an NTC sgRNA gene set as large the set of genes in the sgRNA library itself. Relative enrichment or depletion of sgRNAs and gene scores are determined with the MAGeCK statistical package using default parameters (Li et al., 2014). MAGeCK takes into account the enrichment of all sgRNAs for a gene across replicates to assign a MAGeCK Gene Score for each gene. Examples of analyzed datasets can be found in our original publication (OhAinle et al., 2018).
Note: Replicate CRISPR/Cas9 screens tend to generate reproducible results (r2 values of > 0.9 for gDNA replicates and > 0.8 for vRNA replicates can be achieved). The most robust hits are those hits that are conserved across experiments. Variability in the results can be due to a lack of sampling of the library at various steps in the process: adequate infection of cells, efficient nucleic acid extraction, sequencing depth (i.e., insufficient read depth), or other experimental variables. The correlation between replicates, the degree of enrichment of Non-Targeting Control (NTC) sgRNAs, the overall MAGeCK Gene Scores, p-value and False Discovery Rate (FDR) of top scoring hits (as compared to the “NTC gene set”) are good indicators of the quality of the data generated. However, any hits identified must be validated to determine a true cutoff of signal to noise (i.e., empirically-derived). To avoid the risk of contamination, fresh kits and reagents are used where possible.
Recipes
- 10x Tris/Borate/EDTA (TBE) running buffer (500 ml)
54 g of Tris base
27.5 g of boric acid
20 ml of 0.5 M EDTA
Adjust the pH to 8.3 by adding HCl
Dilute to 1x in water prior to electrophoresis - 20% sucrose solution (200 ml)
40 g sucrose
400 μl 0.5 M EDTA
4 ml 1 M HEPES
5 ml 4 M NaCl
Filter using Steriflip 50 ml conical tubes and keep sterile at 4 °C for up to a year
The EDTA stock is kept at 4 °C
Acknowledgments
This protocol was first described in OhAinle et al. (2018). The work done to develop this protocol was supported by NIH grant R01 AI147877 (M.O.), a CCEH Pilot Grant P30 DK56465 (M.O.) and a UW/FHCRC CFAR New Investigator Award P30 AI027757 (M.O.). Construction of the PIKAHIV library was supported by DP1 DA039543 to Julie Overbaugh, a Belgian American Educational Foundation Fellowship to Jolien Vermeire and R01 AI30927 to Michael Emerman. We thank Emily Hsieh and Vanessa Montoya for help in editing the protocol; Patrick Paddison, Phil Corrin and Lucas Carter for technical advice and the Fred Hutch Genomics Shared Resource and Michael Emerman for support in developing the HIV-CRISPR screening method.
Competing interests
The authors declare no competing interests.
References
- Kane, M., Zang, T. M., Rihn, S. J., Zhang, F., Kueck, T., Alim, M., Schoggins, J., Rice, C. M., Wilson, S. J. and Bieniasz, P. D. (2016). Identification of Interferon-Stimulated Genes with Antiretroviral Activity. Cell Host Microbe 20(3): 392-405.
- Langmead, B., Trapnell, C., Pop, M. and Salzberg, S. L. (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10(3): R25.
- Li, W., Xu, H., Xiao, T., Cong, L., Love, M. I., Zhang, F., Irizarry, R. A., Liu, J. S., Brown, M. and Liu, X. S. (2014). MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol 15(12): 554.
- Liu, L., Oliveira, N. M., Cheney, K. M., Pade, C., Dreja, H., Bergin, A. M., Borgdorff, V., Beach, D. H., Bishop, C. L., Dittmar, M. T. and McKnight, A. (2011). A whole genome screen for HIV restriction factors. Retrovirology 8: 94.
- OhAinle, M., Helms, L., Vermeire, J., Roesch, F., Humes, D., Basom, R., Delrow, J. J., Overbaugh, J. and Emerman, M. (2018). A virus-packageable CRISPR screen identifies host factors mediating interferon inhibition of HIV. Elife 7: e39823.
- Schoggins, J. W., Wilson, S. J., Panis, M., Murphy, M. Y., Jones, C. T., Bieniasz, P. and Rice, C. M. (2011). A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472(7344): 481-485.
Article Information
Copyright
![]() Roesch and OhAinle. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Roesch and OhAinle. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Roesch, F. and OhAinle, M. (2020). HIV-CRISPR: A CRISPR/Cas9 Screening Method to Identify Genes Affecting HIV Replication. Bio-protocol 10(9): e3614. DOI: 10.21769/BioProtoc.3614.
- OhAinle, M., Helms, L., Vermeire, J., Roesch, F., Humes, D., Basom, R., Delrow, J. J., Overbaugh, J. and Emerman, M. (2018). A virus-packageable CRISPR screen identifies host factors mediating interferon inhibition of HIV. Elife 7: e39823.
Category
Immunology > Host defense > Human
Microbiology > Microbe-host interactions > Virus
Systems Biology > Transcriptomics > RNA-seq
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.