- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Quantification of Bacteria Residing in Caenorhabditis elegans Intestine

Published: Vol 10, Iss 9, May 5, 2020 DOI: 10.21769/BioProtoc.3605 Views: 5806
Reviewed by: Juan Facundo Rodriguez AyalaNeelanjan BoseAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Oct 2017
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Quantification of intestinal colonization by pathogenic or commensal bacteria constitute a critical part of the analysis to understand host-microbe interactions during different time points of their interplay. Here we detail a method to isolate non-pathogenic and pathogenic bacteria from C. elegans intestines, and classify gut phenotypes induced by bacterial pathogens using fluorescently-tagged bacteria. Furthermore, these methods can be used to isolate and identify new culturable bacterial species from natural microbiomes of wild nematodes.
Keywords: Host-Pathogen interactionsBackground
In the wild, nematodes are exposed to a wide variety of bacterial and fungal communities (Frézal and Félix, 2015). Under laboratory conditions, the nematode C. elegans has been historically maintained in a single food source (Brenner, 1974). However, the worm has been challenged with various pathogens and an increasing number of non-pathogenic-bacteria of diverse nutritious quality (Garsin et al., 2003; Gracida and Eckmann, 2013; Dirksen et al., 2016; MacNeil et al., 2013; Tan et al., 1999). C. elegans embryos can be extracted from gravid hermaphrodites by using hypochlorite treatment. This procedure eliminates bacteria allowing the new generation to be exposed anew to a microbe. This advantage provides a unique framework to study host-microbe interactions. Genetic tractability of nematodes and bacteria allows to study eukaryotic (Garsin et al., 2003) and prokaryotic (Gallagher and Manoil, 2001) gene function at different time points on this dynamic interplay.
The response of C. elegans to different bacteria depends on both host defenses and bacterial virulence mechanisms (Casadevall and Pirofski, 2003; Hughes and Sperandio, 2008; Casadevall, 2017). Quantifiable physiological outputs (MacNeil et al., 2013; Samuel et al., 2016) and behavioral responses (Zhang et al., 2005; Jin et al., 2016; Palominos et al., 2017) to a variety of microbes have been reported. An important component of the study of bacteria-worm interaction is the quantification of the intestinal bacterial load as well as the ability of bacteria to colonize the animal’s gut. Rodriguez Ayala et al. (2017) explains how to isolate vegetative and resistant spores of Bacillus subtilis from worm’s guts. Palominos et al. (2017) described two independent methods to accurately quantify bacterial colonization proficiency in C. elegans. Here we expand on the methodology used in the latter.
In this protocol we first describe a method to isolate bacteria from guts of worms grown on non-pathogenic (E. coli OP50 or OP50-GFP) and pathogenic (Salmonella Typhimurium 14028, MST1 or MST1-GFP) bacteria. We then quantify the colony forming units (CFU) as measure of individual colony number present in the intestines of animals. Second, green fluorescent protein (GFP)-expressing bacteria are used to qualify the degree of intestinal colonization. All of these constitute reliable methods to measure presence of intact bacteria and degrees of colonization of C. elegans intestine. Moreover, this protocol is a simple method to measure colonization by any bacteria that is culturable or tagged with fluorescent markers (e.g., beneficial or natural bacterial cohabitants of wild nematodes). This may lead to proper identification of food sources to grow other non-cultured nematodes, as well as to study in a Petri dish how nematodes relates to their natural commensals. Finally, these methods constitute a way to study in-vivo interactions between bacteria and its living host for several generations.
Materials and Reagents
- Laboratory Labeling Tape
- Disposable Kontes® Pellet Pestle® Grinders (VWR, catalog number: KT749520-0000 )
- Corning® 0.2 μm syringe filters (Sigma-Aldrich, catalog number: CLS431215 )
- 15 ml Falcon tubes (Fisher, catalog number: 14-959-49B )
- 50 ml Falcon tubes (Fisher, catalog number: 14-432-22 )
- 1.5 ml microcentrifuge tubes (FisherbrandTM, catalog number: 05-408-129 )
- Weighing boats (FisherbrandTM, catalog number: 08-732-112 )
- Pipet tips (Fisher, catalog numbers: 02-707-401 , 02-707-415 , 02-707-436 )
- 90-mm Petri dishes (Nunc®, catalog number: Z717223-320EA )
- Microscope slides (Fisher, catalog number: 12-518-100B )
- 22 x 22 Microscope slide coverslips (VWR, catalog number: 470145-876 )
- Pasteur glass pipette (Fisher Scientific, catalog number: 13-678-20A )
- Plastic disposable graduated pipettes (FisherbrandTM, catalog number: 13-711-9BM )
- Borosilicate glass disposable rimless culture tubes (Thomas Scientific, catalog number: 99445-10 )
- C. elegans strains; for wild type use Bristol N2 strain from Caenorhabditis Genetics Center (CGC)
- E. coli OP50 and OP50-GFP can be obtained from the CGC (https://cgc.umn.edu/)
- Salmonella MST1-GFP is available upon request
- Glycerol ≥ 99.5% (Sigma-Aldrich, catalog number: G9012 )
- NaCl (Merckmillipore, catalog number 106404 )
- BD BactoTM Peptone (Fisher, catalog number: S71604 )
- GibcoTM BactoTM Tryptone (Fisher, catalog number: DF0123-17-3 )
- Streptomycin Sulfate (Thermo Fisher Scientific, catalog number: 11860038 )
- Gentamicin sulfate (Sigma-Aldrich, catalog number: G4918 )
- Ampicillin anhydrous basis (Sigma-Aldrich, catalog number: A9393 )
- BD BactoTM Agar (VWR, catalog number: 90000-760 )
- CaCl2·2H2O (Sigma-Aldrich, catalog number: C7902 )
- Cholesterol (Sigma-Aldrich, catalog number: C75209 )
- Ethyl alcohol pure ≥ 99.5% (Sigma-Aldrich, catalog number: 459836 )
- Levamisole hydrochloride ≥ 99% (Sigma-Aldrich, catalog number: 1359302 )
- K2HPO4 (Sigma-Aldrich, catalog number: RES20765 )
- KH2PO4 (Sigma-Aldrich, catalog number: 1551139 )
- MgSO4 (Sigma-Aldrich, catalog number: 230391 )
- Yeast extract (GibcoTM, catalog number: 212710 )
- Nematode Growth Medium (NGM) plates (see Recipes)
- Solid Luria-Bertani (LB) (see Recipes)
- Liquid LB (see Recipes)
- Sterile 1 M MgSO4 solution (see Recipes)
- Sterile 1 M CaCl2 (see Recipes)
- Sterile Phosphate buffer (see Recipes)
- M9 buffer (see Recipes)
- Levamisole 250 mM (stock) in M9 (see Recipes)
- M9 + Lev (M9 with 25 mM levamisole) (see Recipes)
- M9 + Lev + Ab (M9 with 25 mM levamisole with antibiotics) (see Recipes)
- 1 mM levamisole for microscopy (see Recipes)
- 87% glycerol for bacterial stocks (see Recipes)
- 2% agar for microscopy (see Recipes)
Equipment
- Hand Tally Counter (Humboldt, catalog number: H-9700 )
- Pipetman P10, P200 P1000 (Gilson, catalog numbers: F144802 , F123601 , F123602 )
- Platinum pick made as described in Wollenberg et al. (2013)
- Bunsen burner
- Autoclave
- Stirring hotplate
- Thermal block
- Refrigerated Centrifuge (Eppendorf)
- Laminar flow cabinet
- Incubator for stable temperature
- Incubator for liquid culture
- Dissecting stereoscope with fluorescence
- Freezer (-20 °C)
- 500 ml glass beaker
- 0.5 and 1 L DURAN® Original glass bottles (www.duran-bottle-system.com)
- Stirrer
- Inverted fluorescence microscope with x40 and x60 magnification, and Nomarski filters (Nikon, model: Eclipse Ti-5 )
Software
- FIJI (Schindelin et al., 2012) (FIJI is just ImageJ) Version 2.0, available at http://imagej.net(Fiji/Downloads)
- Microsoft® Excel 2015
- GraphPad Prism 6 (©Graphpad Software)
Procedure
Note: All microbiological techniques should be carried out in a clean bench, next to a Bunsen burner.
- Bacteria and nematode growth
- Bacteria were store in 50% glycerol stocks at -20 °C. Stocks were prepared by mixing 287 μl of sterile 87% glycerol and 213 μl of cultured bacteria. Mix by inversion.
- Streak E. coli OP50-GFP and Salmonella Typhimurium MST1-GFP from glycerol stocks onto individual Luria-Bertani (LB) plates and grow them overnight at 37 °C. LB plates for these two bacteria should contain ampicillin (50 μg/ml).
- Next day, pick a single colony and grow it on 10 ml of liquid LB supplemented with 50 μg/ml ampicillin at 37 °C for 6 h on a 50 ml Falcon tube. The OD600 should range between 1.5 and 2. Colonies can be picked by using a sterile 200 μl pippet tip, or a smear loop (prior sterilization with fire). Tube lid should be with loose, but secured by lab tape. Shake at 200 rpm.
- Seed 3 ml of each bacterial culture onto 90-mm NGM agar plates. Allow it to dry and use them next day. Keep at room temperature.
- Pick 5 L4 worms (genetic background of interest) onto each plate seeded with E. coli OP50-GFP and S. Typhimurium MST1-GFP. Experiment should be carried out in triplicates.
Allow worms to grow until desired developmental stage (Figure 1). In our case, we allow worms to grown for 2 days and we selected L4 grown on each bacterial lawn, because it is easy to recognize as a specific developmental stage.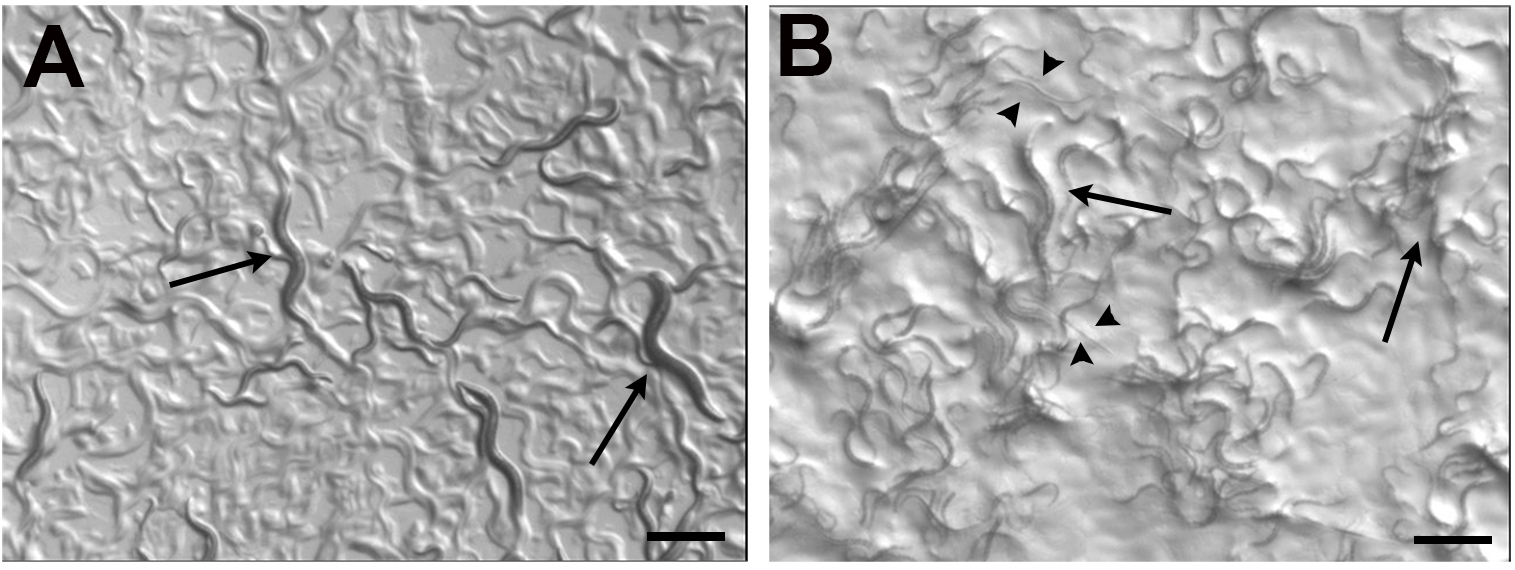
Figure 1. Wild type (N2) worms growing on E. coli OP50 and Salmonella Typhimurium MST1. As previously described (Palominos et al., 2017), the exposure to bacterial pathogens for two consecutive generations cause that a percentage of C. elegans population enter diapause, forming the dauer larvae (arrowheads). Adults can be seen in both conditions (arrows). Scale bars = 500 μm.
- Isolate bacteria from C. elegans intestines: Homogenization and serial dilution
Note: This procedure should be carried out in a sterile area. Ideally, under a laminar flow cabinet. All solutions should be autoclaved prior use.- Prepare levamisole (25 mM) in M9 solution (M9 + Lev). This will cause worms to paralyze and stop the pharyngeal pumping, avoiding the solution to enter to the worm’s interior. Keep on ice.
- Select 30 worms of the desired stage grown on each bacterium lawn and pass them with a platinum pick onto 1.5 ml microcentrifuge tubes containing 1 ml of M 9+ Lev. Three tubes should be prepared per bacteria with worms from each replica (Step A4) This will be biological replicate number 1 (replicates number 2 and 3 should be carried out in a different day).
- Centrifuge worms for 2 min at 376 x g. Discard supernatant.
- Fill with 1 ml of M9 + Lev.
- Repeat Steps B3-B4, twice.
- Discard M9+Lev and resuspend worms in 1 ml of M9 + Lev supplemented with Antibiotics (M9 + Lev + Ab).
- Repeat Steps B3-B5, but with M9 + Lev + Ab, instead of M9 + Lev.
- Discard supernatant, resuspend pelleted worms in 1 ml of M9 + Lev + Ab and incubate for one hour.
- Repeat Steps B3-B5.
Note: Use just M9 + Lev. - Discard M9 + Lev as much as possible and lyse worms with a sterile pestle (could be motorized if available).
Note: Worms on each tube should be lysed with a sterile pestle. Lyse worms for 1 min, or until the worm pellet is completely dissolved. - Resuspend lysed worms in 500 μl of M9.
Note: Serial dilutions consist of series of successive measured dilutions that are prepared in order to reduce the concentration of bacteria and obtain a known number of colony forming units per sample. - Dilute 1:10 the worm lysate, in seven serial dilutions (dilution #1 to #7) in M9. For example, take 1 ml of worm lysate into 9 ml of M9 buffer. This will be dilution #1. Then, take 1 ml of the dilution #1 into 9 ml of M9 buffer, making dilution #2. Repeat sequentially until dilution #7.
- Take 200 μl of dilution #5 (10-5), #6 (10-6), and #7 (10-7), and plate them on solid LB with antibiotics. Streptomycin is used to select E. coli OP50, ampicillin is used to select fluorescent Salmonella Typhimurium MST1-GFP and E. coli OP50-GFP strains.
- Incubate plates overnight at 37 °C.
- Quantification of bacterial colonization by calculating Colony Forming Units (CFU) in C. elegans intestines (or simple plating)
Notes:- Next day, check for undesired bacterial or fungi contamination ensuring you will count colonies of bacteria with the reported morphology. For example, E. coli OP50 colonies are tiny, round, creamy white in color and with defined borders. In contrast, Salmonella colonies are shiny, with dense center and round margins.
- You will have 9 plates of isolated colonies from each bacterium, in this case, 18 plates in total (3 from E. coli replicates, and other three replicates from Salmonella, per each 10-5, 10-6, 10-7 dilution).
- Count the number of colonies with a Hand Tally Counter (Humboldt). Digital counters, or image-based counters can also be used.
- Register each value in an MS® Excel sheet.
- Calculate the CFU per worm using the formula:
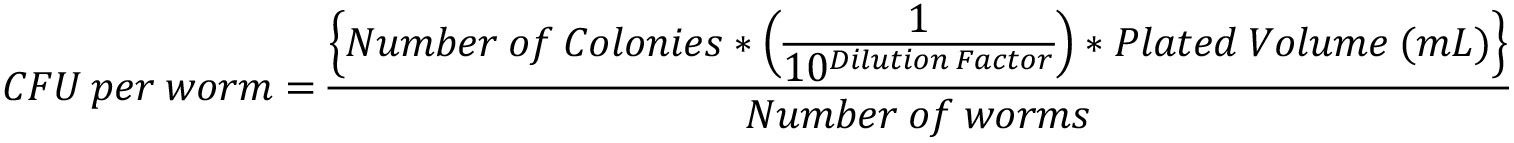
- Quantification of the degree of bacterial colonization in the gut using fluorescence microscopy
- Prepare agar pads as described by Monica Driscoll (www.wormatlas.org/agarpad.htm) or in WormBook. Instead of a 5% agar solution, we use 2%.
- Pick 15 worms per bacterial condition to individual agar pads containing a drop of 1 mM levamisole hydrochloride using a platinum pick. Small amounts of playdough can be placed at each corner of a 22 x 22 coverslip before covering the preparation to avoid pressuring the worm.
- Classify worms according to the presence of GFP positive bacteria in their guts. As described in Palominos et al. (2017), worms with no detectable fluorescence or discrete bacteria in the pharynx are classified as “undetectable” (Figure 2A’). Worms with one-third of the intestine with fluorescent bacteria are classified as “partial”, and “full” when GFP tagged bacteria is found along the whole intestine (Figure 2B’).
- Quantify the number of animals with different phenotypes. Save raw data.
- Take representative images of each phenotype.
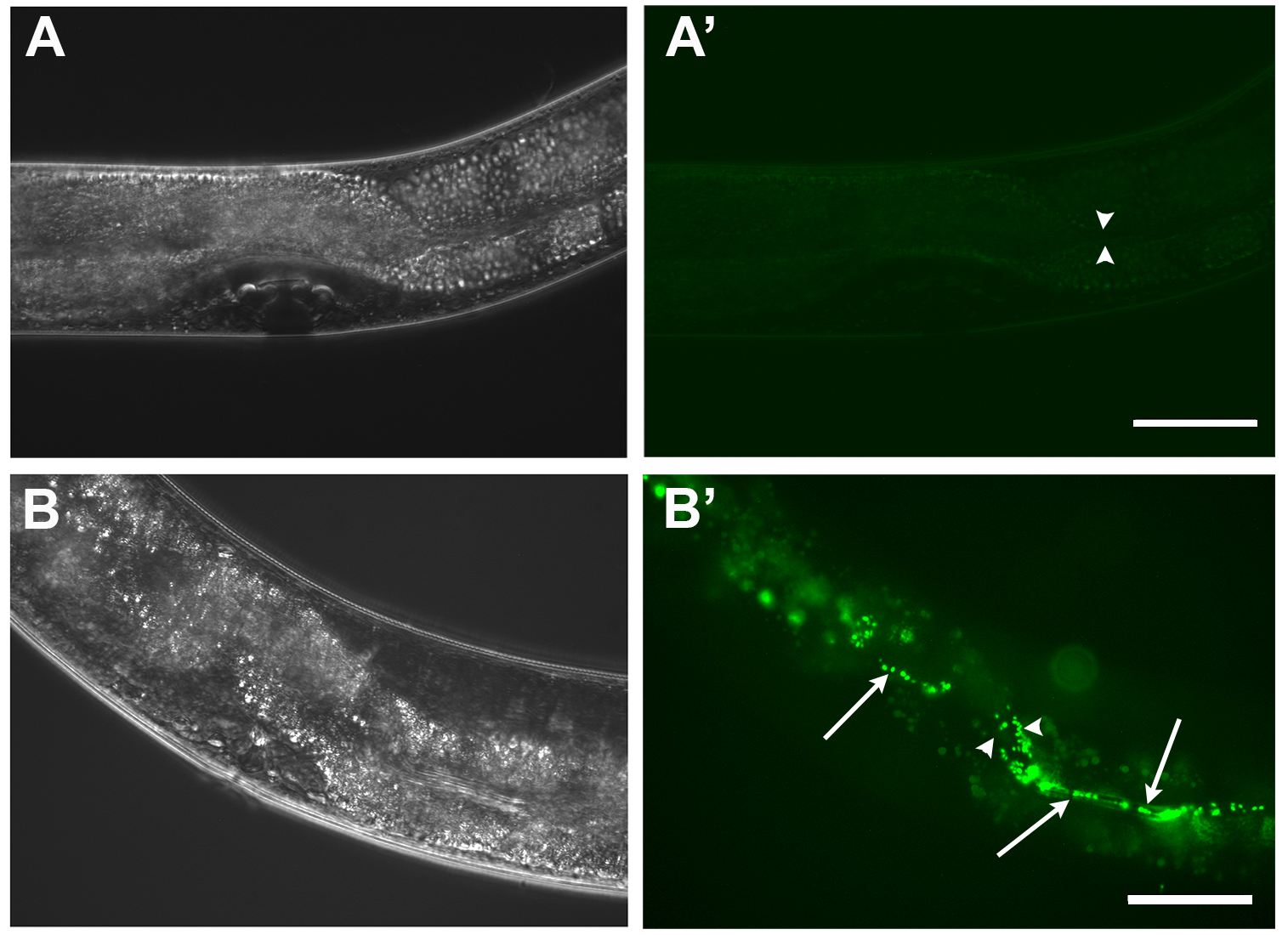
Figure 2. Intestinal colonization of C. elegans gut by fluorescent bacteria. A-A’. L4 wild type worm grown on E. coli OP50-GFP. Most worms on these bacteria possess just scattered bacteria in the pharyngeal grinder. B-B’. L4 wild type worm grown on Salmonella Typhimurium MST1-GFP. Worm fed on Salmonella presenting a “full” colonization phenotype, showing individual and clumped GFP positive bacteria (arrows) along the intestine. Moreover, intestinal expansion can be observed (arrowheads). Scale bars = 50 μm.
Data analysis
- Each experiment should be done at least three times (biological replicas, independent experiments done in different days) and in triplicates (three samples in each replica). See Palominos et al., 2017, Materials and Methods for more information. Each biological replicate will be the numerical value corresponding to the average of the three triplicates.
- One way of plotting the results, is by using Grouped Columns in Prism 6 software (GraphPad). Each column is the average of one biological replica.
- For CFU calculations use Unpaired t test analysis, with 0.05 significance (Figure 3A).
- For Intestinal colonization use two-way ANOVA, Holm-Sidak test, with 0.05 significance (Figure 3B).
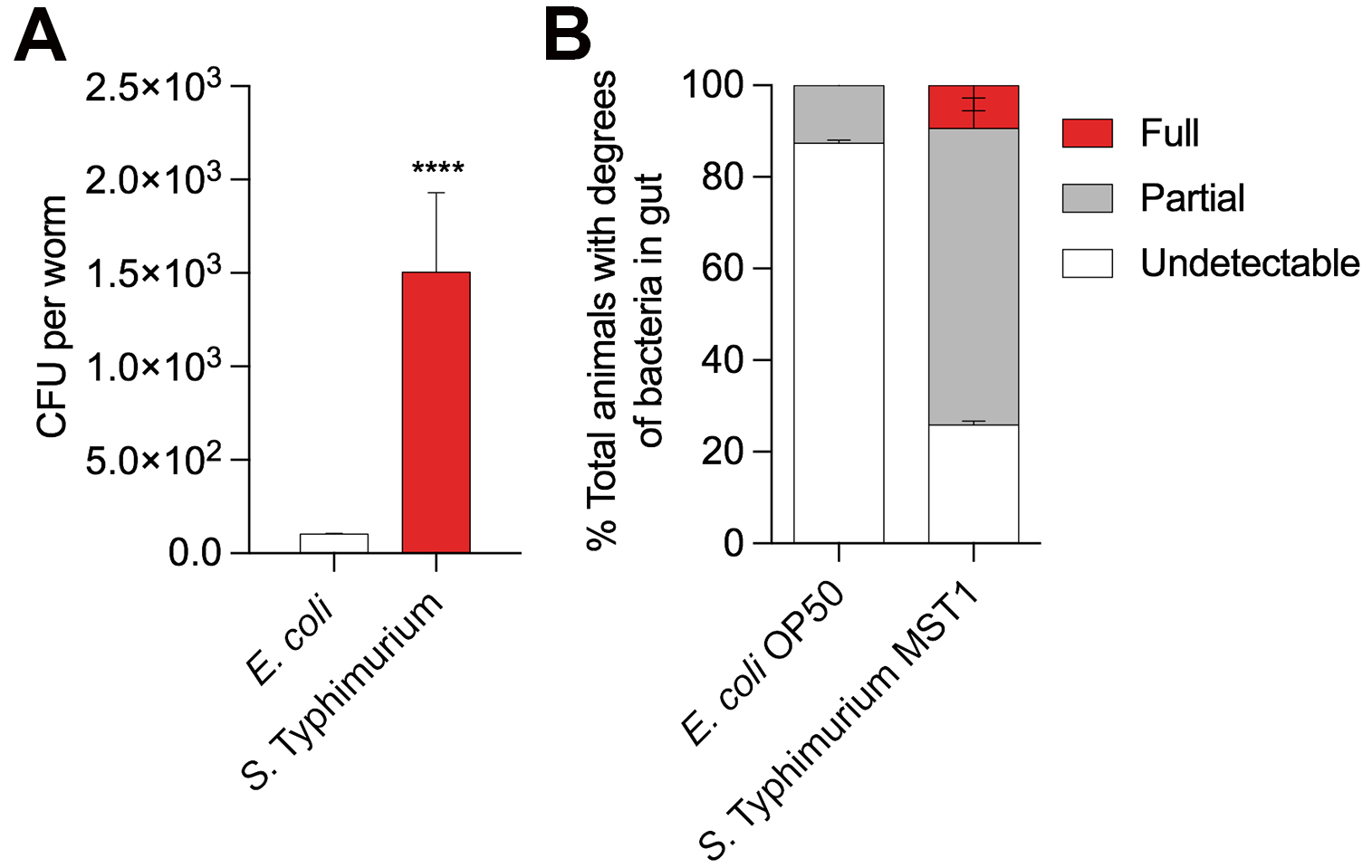
Figure 3. Two different assays to quantify intestinal colonization by bacteria in Caenorhabditis elegans. A. Colony Forming Units (CFU) per worm growing in E. coli OP50 (white) and Salmonella Typhimurium MST1 (red) for two generations. Unpaired t-test, P < 0.0001. N = 90 worms per biological replica, each column contains three replicates. B. Percentage of L4 worms showing different phenotypes of intestinal colonization when grown on E. coli OP50-GFP and S. Typhimurium MST1-GFP. Full, partial and undetectable are defined in Procedure D. Two-way ANOVA, Holm-Sidak test, P < 0.05. N = 45 worms per biological replica.
Notes
- Worms selected for CFU analysis should be healthy. Avoid fungi or other kind of contamination before starting the experiments.
- All solutions and containers should be sterile.
- Intestinal colonization assays can be done using 60 mm plastic Petri dishes seeded with 150 μl of bacterial culture (Procedure A).
- Always select worms in the same developmental stage.
- Autoclave plastic pestles if not sterile.
Recipes
- Nematode Growth Medium (NGM) plates
- Add the following to a 1 L Schott Bottle
3 g NaCl
2.5 g Bacto Peptona
17 g Bacto Agar
Double distilled water (928 ml) - Stir bar
- Autoclave for 20 min at 121 °C
- Place on stir plate, wait until cooled at around 55 °C
- Add the following
1 ml of 1 M CaCl2 sterile
1 ml of 1 M MgSO4 sterile
25 ml of 1 M KH2PO4 pH 6.0 sterile
1 ml of 5 mg/ml cholesterol (prepared in 95% ethanol, and stored at RT) - Pour onto 90 mm sterile plates in a laminar flow cabinet
- Let dry for one night
- Seed with appropriate bacteria, or store at 4 °C
- Add the following to a 1 L Schott Bottle
- Solid Luria-Bertani (LB)
- Dissolve 15 g of Bacto agar, 10 g of Bacto Tryptone, 10 g of NaCl and 5 g of Yeast extract in 1,000 ml dH2O
- Autoclave 20 min at 121 °C
- Swirl it gently to distribute melted agar evenly through the solution. Be careful! Superheated liquids may boil over when swirled
- Once cooled to 55 °C add antibiotics at 50 mg/ml (ampicillin and streptomycin)
- Swirl avoiding bubbles
- Set up a color/mark code (e.g., two red lines for LB-streptomycin, two black lines for LB-ampicillin plates)
- Pour around 30-35 ml per plate
- When medium has hardened completely, invert and store them at 4 °C until needed
- Remove plates for storage 1-2 h before using them
- Liquid LB
- Dissolve 10 g of Bacto Tryptone, 10 g of NaCl and 5 g of Yeast extract in 1,000 ml
- Autoclave 20 min at 121 °C
- Once cooled to 55 °C add antibiotics at 50 mg/ml (ampicillin and streptomycin)
- Sterile 1 M MgSO4 solution
- Dissolve 123.24 g MgSO4·7H2O in 500 ml of pure MilliQ water
- Autoclave at 121 °C for 20 min
- Store at room temperature (RT)
- Sterile 1 M CaCl2
- Dissolve 5.55 g of CaCl2 dehydrate in 50 ml of MilliQ water
- Autoclave
- Store at RT
- Sterile Phosphate buffer
- Dissolve 10.7 g of K2HPO4 and 32.5 g of KH2PO4 to 300 ml of MilliQ water
- Adjust pH to 6.0
- Autoclave
- Store at RT
- M9 buffer
- Dissolve the following in 1 L of pure MilliQ water
3 g KH2PO4
6 g Na2HPO4
5 g NaCl - Autoclave at 121 °C for 20 min, then add 1 ml of sterilized 1 M MgSO4 solution
- Store at RT
- Dissolve the following in 1 L of pure MilliQ water
- Levamisole 250 mM (stock) in M9
- Mix 9.03 g of levamisole hydrochloride in 15 ml of sterile M9
- Stir if necessary
- Aliquot in 1 ml microcentrifuge tubes
- Keep it at -20 °C. Avoid re-thawing
- M9 + Lev (M9 with 25 mM levamisole)
- Mix 5 ml of 250 mM levamisole stock solution with 45 ml of sterile M9
- Keep it on ice
- M9 + Lev + Ab (M9 with 25 mM levamisole with antibiotics)
- Prepare 5 ml of Gentamicin-Ampicilin stock solution (10 mg/ml) by dissolving 50 mg of gentamicin and 50 mg ampicillin in 5 ml of MilliQ water.
- Filter with a 0.2 μm syringe filter. Keep it on ice until finishing aliquoting. Store at -20 °C
- Use 2,000 μl of the stock Gen-Amp (10 mg/ml) and mix with 18 ml of M9 + Lev. Keep it on ice until used
- 1 mM levamisole for microscopy
- Mix 40 μl of M9 + Lev with 960 μl of M9
- Filter with a 0.2 μm syringe filter
- Store at 4 °C. Keep on ice when using
- 87% glycerol for bacterial stocks
- Mix 8.7 ml of 100% glycerol in 1.3 ml of MilliQ water
- Autoclave
- Store at room temperature, covered from light
- 2% agar for microscopy
- Prepare 3 ml of 5% agar/M9 in a 5 ml glass culture tube
- Place the glass culture tube with 2% agarose inside a 500 ml glass beaker, with 150-200 ml of RT water (as imitating a water bath)
- Microwave for 60 s as medium potency
Note: Check homogeneity of the mixture. Resuspend with a plastic pipette if necessary. Be sure that the agarose is completely melted before going to the next step. - Before making the pads keep 2% agar at 60 °C in a heating block or bath
- After use, store it at 4 °C
Acknowledgments
This work has been adapted from Palominos et al. (2017). Some worm strains were provided by the CGC, which is funded by NIH Office of Research Infrastructure Programs (P40OD010440). The development of protocols was funded by Fondecyt 1120209 to F.P.C; Fondecyt 1131038, CONICYT Chile-USA 2013-0041, and the Millenium Initiative to AC. The Centro Interdisciplinario de Neurociencia de Valparaiso is a Millennium Institute supported by the Millennium Scientific Initiative of the Chilean Ministry of Economy, Development, and Tourism (P029-022-F).
Competing interests
The authors declare they have no financial interest.
References
- Brenner, S. (1974). The genetics of Caenorhabditis elegans. Genetics 77(1): 71-94.
- Casadevall, A. (2017). The pathogenic potential of a microbe. mSphere 2(1).
- Casadevall, A. and Pirofski, L. A. (2003). The damage-response framework of microbial pathogenesis. Nat Rev Microbiol 1(1): 17-24.
- Dirksen, P., Marsh, S. A., Braker, I., Heitland, N., Wagner, S., Nakad, R., Mader, S., Petersen, C., Kowallik, V., Rosenstiel, P., Felix, M. A. and Schulenburg, H. (2016). The native microbiome of the nematode Caenorhabditis elegans: gateway to a new host-microbiome model. BMC Biol 14: 38.
- Frézal, L. and Felix, M. A. (2015). C. elegans outside the Petri dish. Elife 4: 05849.
- Gallagher, L. A. and Manoil, C. (2001). Pseudomonas aeruginosa PAO1 kills Caenorhabditis elegans by cyanide poisoning. J Bacteriol 183(21): 6207-6214.
- Garsin, D. A., Villanueva, J. M., Begun, J., Kim, D. H., Sifri, C. D., Calderwood, S. B., Ruvkun, G. and Ausubel, F. M. (2003). Long-lived C. elegans daf-2 mutants are resistant to bacterial pathogens. Science 300(5627): 1921.
- Gracida, X. and Eckmann, C. R. (2013). Fertility and germline stem cell maintenance under different diets requires nhr-114/HNF4 in C. elegans. Curr Biol 23(7): 607-613.
- Hughes, D. T., and Sperandio, V. (2008). Inter-kingdom signalling: communication between bacteria and their hosts. Nat Rev Microbiol 6, 111-120.
- Jin, X., Pokala, N. and Bargmann, C. I. (2016). Distinct circuits for the formation and retrieval of an imprinted olfactory memory. Cell 164(4): 632-643.
- MacNeil, L. T., Watson, E., Arda, H. E., Zhu, L. J. and Walhout, A. J. (2013). Diet-induced developmental acceleration independent of TOR and insulin in C. elegans. Cell 153(1): 240-252.
- Palominos, M. F., Verdugo, L., Gabaldon, C., Pollak, B., Ortíz-Severín, J., Varas, M. A., Chávez, F. P., and Calixto, A. (2017). Transgenerational diapause as an avoidance strategy against bacterial pathogens in Caenorhabditis elegans. MBio 8(5).
- Rodriguez Ayala, F., Cogliati, S., Bauman, C., Leñini, C., Bartolini, M., Villalba, J. M., Argañaraz, F. and Grau, R. (2017). Culturing bacteria from Caenorhabditis elegans gut to assess colonization proficiency. Bio-protocol 7(12): e2345.
- Samuel, B. S., Rowedder, H., Braendle, C., Félix, M. A. and Ruvkun, G. (2016). Caenorhabditis elegans responses to bacteria from its natural habitats. Proc Natl Acad Sci U S A 113(27): E3941-3949.
- Schindelin, J., Arganda-Carreras, I., Frise, E., Kaynig, V., Longair, M., Pietzsch, T., Preibisch, S., Rueden, C., Saalfeld, S., Schmid, B., Tinevez, J. Y., White, D. J., Hartenstein, V., Eliceiri, K., Tomancak, P. and Cardona, A. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9(7): 676-682.
- Tan, M. W., Mahajan-Miklos, S., and Ausubel, F. M. (1999). Killing of Caenorhabditis elegans by Pseudomonas aeruginosa used to model mammalian bacterial pathogenesis. Proc Natl Acad Sci U S A 96(2), 715-720.
- Zhang, Y., Lu, H. and Bargmann, C. I. (2005). Pathogenic bacteria induce aversive olfactory learning in Caenorhabditis elegans. Nature 438(7065): 179-184.
Article Information
Copyright
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Palominos, M. F. and Calixto, A. (2020). Quantification of Bacteria Residing in Caenorhabditis elegans Intestine. Bio-protocol 10(9): e3605. DOI: 10.21769/BioProtoc.3605.
Category
Microbiology > Microbe-host interactions > Bacterium
Cell Biology > Cell isolation and culture > Cell isolation
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.