- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Confocal and Super-resolution Imaging of RNA in Live Bacteria Using a Fluorogenic Silicon Rhodamine-binding Aptamer



Published: Vol 10, Iss 9, May 5, 2020 DOI: 10.21769/BioProtoc.3603 Views: 8479
Reviewed by: Imre GáspárRosario Gomez-GarciaAnonymous reviewer(s)
Original research article
The authors used this protocol in:

May 2019
Advertisement





Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Genetically encoded light-up RNA aptamers have been shown to be promising tools for the visualization of RNAs in living cells, helping us to advance our understanding of the broad and complex life of RNA. Although a handful of light-up aptamers spanning the visible wavelength region have been developed, none of them have yet been reported to be compatible with advanced super-resolution techniques, mainly due to poor photophysical properties of their small-molecule fluorogens. Here, we describe a detailed protocol for fluorescence microscopy of mRNA in live bacteria using the recently reported fluorogenic silicon rhodamine binding aptamer (SiRA) featuring excellent photophysical properties. Notably, with SiRA, we demonstrated the first aptamer-based RNA visualization using super-resolution (STED) microscopy. This imaging method can be especially valuable for visualization of RNA in prokaryotes since the size of a bacterium is only a few times greater than the optical resolution of a conventional microscope.
Background
The visualization of specific RNA molecules by fluorescence microscopy has become invaluable during the past two decades to extend our knowledge of RNA function within cells in a spatiotemporal manner (Tyagi, 2009; Xia et al., 2017). Due to the lack of inherently fluorescent RNAs, development of fluorogenic RNA labeling tools for live-cell imaging and their adaptation to state-of-the-art microscopes–especially to super-resolution microscopes–is imperative. Super-resolution microscopy (SRM) is particularly attractive for imaging RNAs in prokaryotic systems since a bacterium is very small (~2.5 μm long, ~0.5-1 μm wide) and the resolution of a standard fluorescence microscope is restricted to ~200-300 nm due to the diffraction limit of light (Reshes et al., 2008; Nienhaus and Nienhaus, 2016). Although a plethora of tools have been developed to fluorescently label an RNA of interest (ROI), their live-cell visualization using SRM (resolution ~20-50 nm) is still challenging (Alexander and Devaraj, 2017), mainly due to the stringent requirements on the photophysical properties of the fluorophores needed for live-cell SRM. These properties include high photostability and brightness, cellular membrane permeability, water solubility, absorption and emission wavelengths preferentially in the far-red and near-infrared (NIR) region, on-off switching capability and a sufficient signal-to-background ratio during imaging (Wang et al., 2019). Thus, there is a strong need for novel RNA reporter systems that are compatible with SRM.
An especially attractive approach to visualize RNAs in live cells is the use of genetically encoded fluorescence light-up aptamers, which are short oligonucleotides that bind small, conditionally fluorescent probes with high affinity and are developed using systematic evolution of ligands by exponential enrichment (SELEX) (Holeman et al., 1998; Ouellet, 2016). Tagged to the ROI, the aptamers can be visualized due to a fluorescence increase upon small molecule binding (Figure 1). Several fluorescence enhancing RNA aptamers spanning the visible region of the spectrum have recently been developed and proven to be powerful tools for RNA labeling (Bouhedda et al., 2017; Neubacher and Hennig, 2019).
The famous Spinach, Broccoli, Mango, Corn aptamers and their improved variants bind to fluorophores with low fluorescence quantum yield in solution and become fluorescent due to the restriction of intramolecular movements upon aptamer binding (Figure 1A) (Paige et al., 2011; Filonov et al., 2014; Dolgosheina et al., 2014; Song et al., 2017). Other prominent examples are SRB-2, DNB, BHQ and Riboglow aptamers that disrupt a quencher-dye conjugate upon binding to either the dye or the quencher moiety, thereby generating fluorescence enhancement (Figure 1B) (Murata et al., 2011; Sunbul and Jäschke, 2013 and 2018; Arora et al., 2015; Braselmann et al., 2018). Most of these aptamer-ligand pairs, however, do not absorb and emit in the far-red or NIR region. Although MG, Riboglow, Mango TO-3 and the recently reported DIR-pro (which has not been reported for RNA labeling) are excited in the far-red, their fluorophores show either low photostability and brightness, phototoxicity or are hardly cell permeable (Tan et al., 2017; Autour et al., 2018; Yerramilli and Kim, 2018).
Prompted by these shortcomings, we recently developed a novel light-up RNA aptamer that binds to 3-carboxy silicon rhodamine (SiR) (Wirth et al., 2019). SiRs are an exciting new fluorophore class in the field of protein labeling for SRM because they are photostable, NIR-emitting fluorophores that change their equilibrium between the non-colored spirolactone and the fluorescent zwitterion in response to their environment (Figure 1C) (Lukinavičius et al., 2013). This property is responsible for their high cell permeability and fluorogenic behavior and contributes to the popularity of SiR dyes and their derivatives in the last decade (Wang et al., 2019). Applying SELEX on a combinatorial RNA library with a diversity of ~2 x 1015, we discovered a 50 nt-minimal aptamer, SiRA, which binds the target SiR 1 with a KD of 430 nM and shows a significant fluorescence turn-on upon dye binding (Figure 2). This finding introduces a new concept of fluorogenicity to the field of aptamer-based RNA imaging, namely environment-dependent intramolecular spirocyclization (Figure 1C). SiRA is remarkably resistant to photobleaching and constitutes the brightest far-red light up aptamer system known to date (quantum yield 0.98; extinction coefficient 86,000 M-1·cm-1). Using the SiRA system, we visualized the expression of RNAs in bacteria in no-wash live-cell imaging experiments (Wirth et al., 2019). The innovation in this protocol is that we can use SiRA’s excellent photophysical properties for stimulated emission depletion (STED) SRM of mRNA in live bacteria. To date, SiRA is the only light-up aptamer system proven to work in SRM applications.
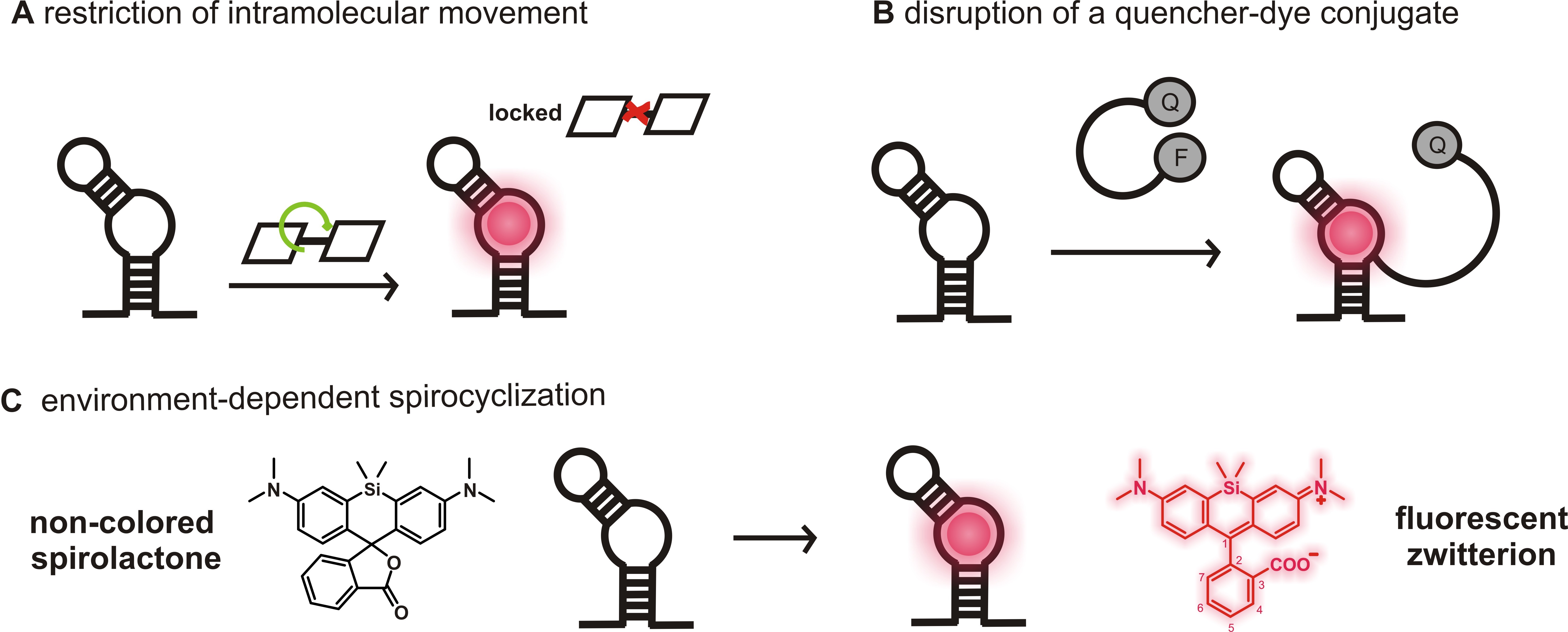
Figure 1. Fluorogenicity concepts of light-up RNA aptamers used for RNA imaging. A. A fluorophore with low fluorescence quantum yield due to molecular motions in solution (OFF) becomes highly fluorescent upon aptamer binding due to the restriction of intramolecular movements (ON). B. The contact-quenched fluorophore-quencher conjugates (OFF) light up upon binding to either the fluorophore or the quencher moiety (ON). Here, an example of a light-up system using a fluorophore binding aptamer was shown. C. In solution, SiRs can favor the non-colored spirolactone (OFF). The change in environment upon aptamer binding causes a shift of the equilibrium to the fluorescent zwitterion (ON).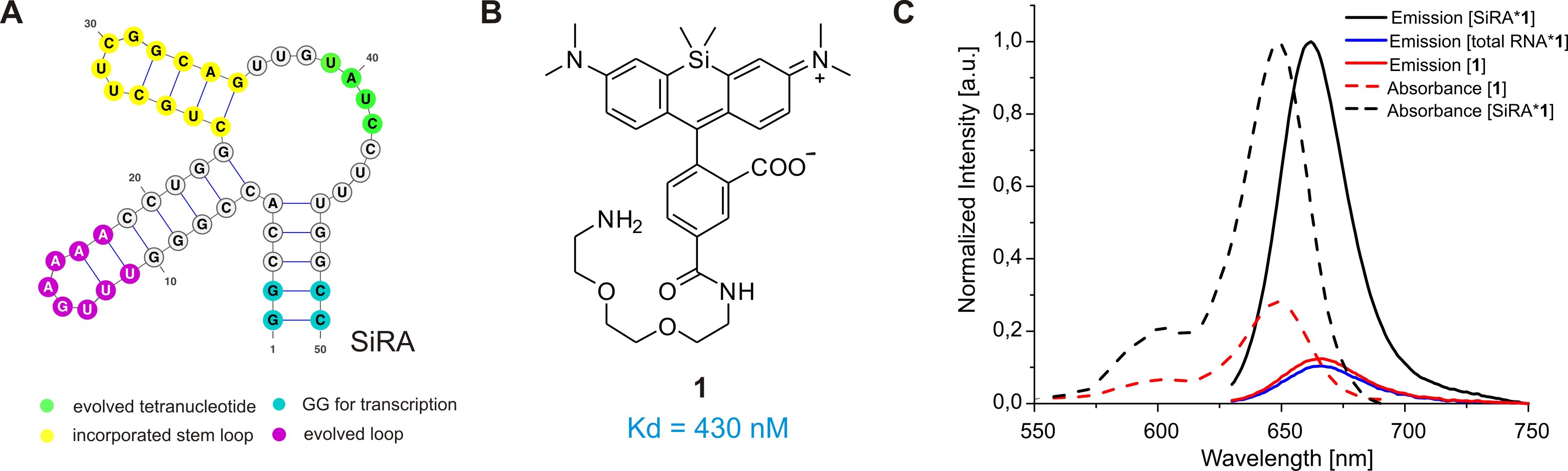
Figure 2. The SiRA aptamer and its properties. A. The predicted secondary structure of the 50-nt SiRA aptamer evolved during the selection process (important features are marked in color). B. Structure of the fluorogenic dye 1 used in this protocol, which binds to SiRA with nanomolar affinity. C. Normalized absorption and emission spectra of free SiR 1 and the [SiRA*1] complex.
In the following protocol, we describe the preparation of E. coli bacteria for live-cell confocal and STED imaging of GFP-mRNA in detail. We herein use a plasmid-based expression system and tag the GFP-mRNA with tandem repeats of the novel SiRA aptamer (SiRA5) at the 3′ untranslated region (UTR). This protocol can be adapted to visualize any plasmid expressed target RNA; however, the expression levels and the half-life of RNAs need to be kept in mind. Further optimization of the protocol might be required for specific applications.
Materials and Reagents
- Pipette tips (Sarstedt, catalog numbers: 70.113 0, 70.760.002 , 70.762 )
- Microcentrifuge tubes 1.5 ml (Sarstedt, catalog number: 72.690.001 )
- 15 μ-Slide 8-well uncoated, glass bottom #1.5 polymer coverslip (Ibidi GmbH, catalog number: 80827 )
- High clarity PP conical centrifuge tube 15 ml (Falcon®, catalog number: 352096 )
- High clarity PP conical centrifuge tube 50 ml (Falcon®, catalog number: 352070 )
- Medical Millex-GS syringe filter unit, 0.22 μm (EDM Merck, catalog number: SLGSM33SS )
- BD Discardit disposable syringes 10 ml (Becton Dickinson, catalog number: 300296 )
- PYREX® Media bottles, graduated, Corning®, 1,000 ml (VWR, PYREX®, catalog number: 1395-10L )
- FluoSpheresTM Carboxylate-Modified Microspheres, 0.04 μm, dark red fluorescent (660/680), 5% solids, azide free (Thermo Fisher Scientific, catalog number: F8789 )
- Gold beads (GC80, BB International, catalog number: EM. GC80 )
- E. coli BL21 StarTM (DE3) Invitrogen competent E. coli strains (Thermo Fisher, catalog number: C601003 ), store at -80 °C
- pET-GFP expression plasmid (pET His6 GFP TEV LIC cloning vector, 1 GFP) (Addgene, Plasmid number: 29663), store at -20 °C
- pET-GFP-SiRA5 expression plasmid, store at -20 °C
The pET-GFP-SiRA5 plasmid used in this protocol is not commercially available. Generally tandem repeats of an aptamer of choice (depending on the target RNA and expression level), can directly be fused to the ROI (in this case GFP mRNA) using proper restriction sites in the 3′ UTR (here: SalI and XhoI fast digest restriction enzymes, Thermo ScientificTM, catalog numbers: FD0644 and FD0695 , respectively) as previously described in the literature (Sunbul et al., 2018). In this study, we inserted five repeats of the SiRA aptamer with interspersed linker nucleotides according to Figure 3.
Figure 3. Excerpt of the pET-GFP-SiRA5 bacterial expression plasmid used for the visualization of GFP mRNA in this protocol. lacO: lac operator, rbs: ribosomal binding site, ROI: RNA of interest, P: promoter, T: terminator, SalI and XhoI: Restriction sites, stop: Stop codon for RNA translation.
The following sequences show an excerpt of the cloned expression vector (pET-GFP-SiRA5) as well as the pET-GFP used in this study.pET-GFP-SiRA5:black SiRA (3-nt stem loop)
...CGAGGAAACCTGTACTTCCAATCCAATATTGGAAGTGGATAACGGATCCGAATTCGAGCGCCGTCGACAGGCCCACCGGGTTTGAAAACCTGGCTGCTTCGGCAGTTGTATCCTTTGGGCCTAAGAGAGACCACCGGGTTTGAAAACCTGGCTGCTTCGGCAGTTGTATCCTTTGGTCTCAATAACCAGCCACCGGGTTTGAAAACCTGGCTGCTTCGGCAGTTGTATCCTTTGGGCCTAAGAGAGACCACCGGGTTTGAAAACCTGGCTGCTTCGGCAGTTGTATCCTTTGGTCTCAATAACCAGCCACCGGGTTTGAAAACCTGGCTGCTTCGGCAGTTGTATCCTTTGGCTGGCTCGAGCACCACCACCACCACCACTGAGATCCGGCTGCTAACAAAGCCCGAAAGGAAGCTGAGTTGGCTGCTGCCACCGCTGAGCAATAACTAGCATAACCCCTTGGGGCCTCTAAACGGGTCTTGAGGGGTTTTTTGCTGAAAGGAGGAACTATATCCGGATTGGCGAATGGGACGCGCCCTGTAGCGGCGCATTAAGCGCGGCGGGTG…
pET-GFP:
...CGAGGAAACCTGTACTTCCAATCCAATATTGGAAGTGGATAACGGATCCGAATTCGAGCGCCGTCGACAAGCTTGCGGCCGCACTCGAGCACCACCACCACCACCACTGAGATCCGGCTGCTAACAAAGCCCGAAAGGAAGCTGAGTTGGCTGCTGCCACCGCTGAGCAATAACTAGCATAACCCCTTGGGGCCTCTAAACGGGTCTTGAGGGGTTTTTTGCTGAAAGGAGGAACTATATCCGGATTGGCGAATGGGACGCGCCCTGTAGCGGCGCATTAAGCGCGGCGGGTG…
grey Variable Stem Loop or Spacer Region
blue SalI Restriction Site
pink XhoI Restriction Site - Autoclaved LB medium, Lennox (Carl Roth®, catalog number: X964.4 )
- LB Broth with agar, Lennox (Sigma-Aldrich, catalog number: L2897 )
- Kanamycin sulfate from Streptomyces kanamyceticus (Sigma-Aldrich, catalog number: K1377 ), prepare 1 M aqueous solution, filter-sterilize and store at -20 °C
- Isopropyl-β-D-thio-galactopyranoside (IPTG) (Sigma-Aldrich, catalog number: I5502 ), prepare 1 M aqueous solution, filter-sterilize and store at -20 °C
- Potassium hydroxide pellets EMPLURA® (Sigma-Aldrich, catalog number: 1050121000 ), prepare 10 M aqueous solution
- Poly-D-lysine hydrobromide (Sigma-Aldrich, catalog number: P7886 ), prepare 2.5 mg/ml in DPBS, store at 4 °C
- Dulbecco's Phosphate Buffered Saline (DPBS) (Sigma-Aldrich, catalog number: D8537 )
- MilliQ water
- Silicon rhodamine based fluorogenic dye, 100 μM solution in DMSO
The fluorogenic dye used in this protocol is not commercially available. Therefore, it has to be synthesized and purified by HPLC as described in the literature (Wirth et al., 2019).
Note: The HPLC purification has to be carried out with great care, since the fluorescence of remaining impurities can decrease the signal-to-background ratio during the imaging experiment. - M9 Broth (Fluka Sigma-Aldrich, catalog number: 63011 )
- Magnesium sulfate monohydrate (Sigma-Aldrich, catalog number: 434183 ), prepare 1 M aqueous solution and autoclave
- D-(+)-Glucose (Sigma-Aldrich, catalog number: G8270 ), prepare 40% (w/v) aqueous solution and filter-sterilize
- Magnesium chloride hexahydrate (Sigma-Aldrich, catalog number M2670 ), prepare 1 M aqueous solution and autoclave
- Calcium chloride dihydrate (Sigma-Aldrich: catalog number: 1725801000 ), prepare 1 M aqueous solution and autoclave
- Live-cell imaging solution (Thermo Fisher Scientific, catalog number: A14291DJ )
- M9 imaging solution (see Recipes)
Equipment
- Research® plus pipettes, variable volume 200 and 1,000 μl (Sigma-Aldrich, Eppendorf®, catalog numbers: Z683817 and Z683825 )
- Steam Sterilizer (e.g., Varioklav®, model: 155S )
- Thermomixer comfort 24 x 1.5 ml (Eppendorf®)
- Incubator and shaker (Thermo Fisher Scientific, type 496)
- BioPhotometer (Eppendorf®, type 6131)
- High speed Micro Centrifuge (Neuation Technology Pvt. Ltd., catalog number: iFuge M12 )
- Freezer
- Refrigerator
- Confocal microscope (Nikon, model: A1R )
For this protocol, a point scanning confocal microscope with hybrid scanner (galvano/resonant) was used, equipped with a Nikon N Apo 60x NA 1.4 λs OI (WD 0.14 mm, FOV 0.21 x 0.21 mm) objective. For fluorescence excitation, 488 nm (green channel) and 640 nm (red channel) lasers were used. For detection, a high-sensitive GaAsP detector combined with 525/50 nm and 700/75 nm (center wavelength/bandwidth) emission filters for green and red channels, respectively, was used. - STED microscope
The STED microscope used in this protocol is home-built and has been described in detail (Gao et al., 2017). In addition, a 650-nm long-pass dichroic mirror (ET700SP; Chroma, Bellows Falls, VT) and a time-correlated single photon counting (TCSPC) card (SPC-150, Becker & Hickl GmbH, Berlin, Germany) are needed.
Note: The experiment in the protocol was carried out with 640 nm excitation and 779 nm depletion; only the vortex phase plate for plain 2D STED imaging was used.
Software
- ImageJ Fiji, version 2.0.0. with Java 1.6.0_24
- Software confocal microscope: NIS-Elements image acquisition and analysis software (Nikon)
- Software STED microscope: Imspector (Max-Planck-Innovation GmhH, Muenchen, Germany)
- Matlab (MathWorks, Natick, MA)
- OriginPro 2015 32 bit (OriginLab, Northampton, MA)
Procedure
- E. coli transformation (Day 1: ~2.5 h followed by over-night incubation)
Note: We recommend a fresh transformation prior to each imaging experiment to ensure proper mRNA-SiRA expression.- Add 50 ng of the plasmids (pET-GFP and pET-GFP-SiRA5) into each autoclaved microcentrifuge tube and put them on ice.
- Thaw chemically competent E. coli BL21 StarTM (DE3) cells for 15 min on ice.
- Add 40 μl of E. coli BL21 StarTM (DE3) cells to each tube and incubate for an additional 30 min on ice.
- Heat the tubes for 45 s at 42 °C followed by incubation on ice for 2 min.
- Add 250 μl of pre-warmed (37 °C) LB medium to each tube and shake them at 650 rpm for 60 min.
- For the selection of bacteria carrying the plasmid, streak 20 μl of the bacteria on LB agar plates containing 30 μg/ml of kanamycin.
- Incubate the LB agar plates overnight at 37 °C.
Note: In the morning, place the agar plates at 4 °C and keep them there throughout the day.
- Induction of mRNA expression in bacteria (Day 2: 15 min followed by over-night incubation; Day 3: ~ 5 h)
- Add 5 ml of LB medium supplemented with 30 μg/ml kanamycin into a 15 ml Falcon and start an overnight culture using a single colony picked from each LB agar plate using a sterile pipette tip.
- Incubate the Falcon at 37 °C with vigorous shaking at 170 rpm overnight.
- The next morning, measure the optical density (OD600) for a 1:10 dilution of each overnight culture.
- Generate a starter culture from the overnight culture. For this, add 10 ml of LB medium supplemented with 30 μg/ml kanamycin in a 50 ml Falcon and add the overnight culture to give an OD600 of 0.05.
- Shake the culture at 37 °C and 170 rpm till the OD600 reaches 0.35.
Note: This usually takes 1.5-2 h. - Induce the expression of GFP mRNA by adding 10 μl of IPTG (1 mM final concentration) and shake the culture for an additional 3 h at 37 °C.
Note: Currently the SiRA system has only been used for imaging highly overexpressed GFP mRNAs. In order to visualize lower copy numbers, a larger number of tandem repeats can be designed and further optimization of the protocol (e.g., dye concentration) might be necessary.
- Preparation of poly-D-lysine coated glass chamber slides (Day 3: ~3 h, in parallel to Steps B5-B6)
Note: The poly-D-lysine solution for coating needs to be prepared freshly every time just before adding it to the well. The poly-D-lysine coated glass chamber slides, however, can be prepared up to 24 h in advance and stored in the refrigerator at 6 °C. Warming up to RT before use is recommended.- Remove the 8-well ibidi glass slides from the packaging and incubate each well with 200 μl of a 10 M aqueous KOH solution for 5 min.
- Remove the KOH solution from the wells very carefully, wash them thoroughly multiple times with 400 μl sterile MilliQ water and air-dry them for 60 min.
- Mix 1,000 μl of water with 20 μl of the poly-D-lysine stock (~50 μg/μl final concentration) and add 250 μl of the solution quickly into each well (recipe for 4 wells).
- Incubate the wells for 45 min at room temperature.
- Wash each well twice with 300 μl sterile MilliQ water and air-dry for at least 30 min prior to the immobilization of the bacteria.
- Preparation of E. coli samples for imaging (Day 3: ~1 h)
Note: For live-cell imaging experiments, we recommend to image up to 2 wells at a time, due to the short mRNA half-life and bacterial metabolism.- Transfer a 200 μl aliquot of each culture (Step B6) to an autoclaved microcentrifuge tube and pellet the cells at 5,600 x g for 2 min at room temperature.
- Remove the LB medium and suspend the bacteria in 1 ml of M9 imaging solution (Recipe 1).
- Add 200 μl of the cell suspension into each well of the coated 8-well ibidi slide.
- Incubate the bacteria for 35 min at room temperature to allow for adhesion to the surface.
- Gently wash each well with 400 μl of M9 imaging solution.
- Add 300 μl of M9 imaging solution containing 500 nM of the silicon rhodamine fluorogenic dye and directly prepare for imaging.
Note: Permeation of the dye into the bacterium is quite fast (5-10 min) at 37 °C; therefore, the time for microscope adjustment is sufficient for incubation. If permeation issues occur, prewarming of the dye/M9 imaging solution to 37 °C is recommended.
- Imaging GFP-SiRA5 mRNA using a confocal microscope (Day 3: ~1-2 h)
- Turn on the 488 nm (green channel) and 640 nm (red channel) lasers for fluorescence illumination of the point scanning confocal microscope and use the 525/50 nm and 700/75 nm emission filters for green and red channel, respectively.
- Attach the temperature incubation chamber to the microscope, prewarm to 37 °C and place the 8-well slide with adherent E. coli into the chamber.
- Use the bright-field illumination to focus on the immobilized E. coli with the Nikon N Apo 60x NA 1.4 λs OI (WD 0.14 mm, FOV 0.21 x 0.21 mm) objective, starting with the pET-GFP-SiRA5 transformed cells.
- Determine a suitable exposure time for the GFP and red channel as well as the correct z-axis. Choose the settings such that you do not saturate the pixels but still obtain the highest fluorescent signal.
- Acquire the brightfield and fluorescence images of all channels (Figure 4).
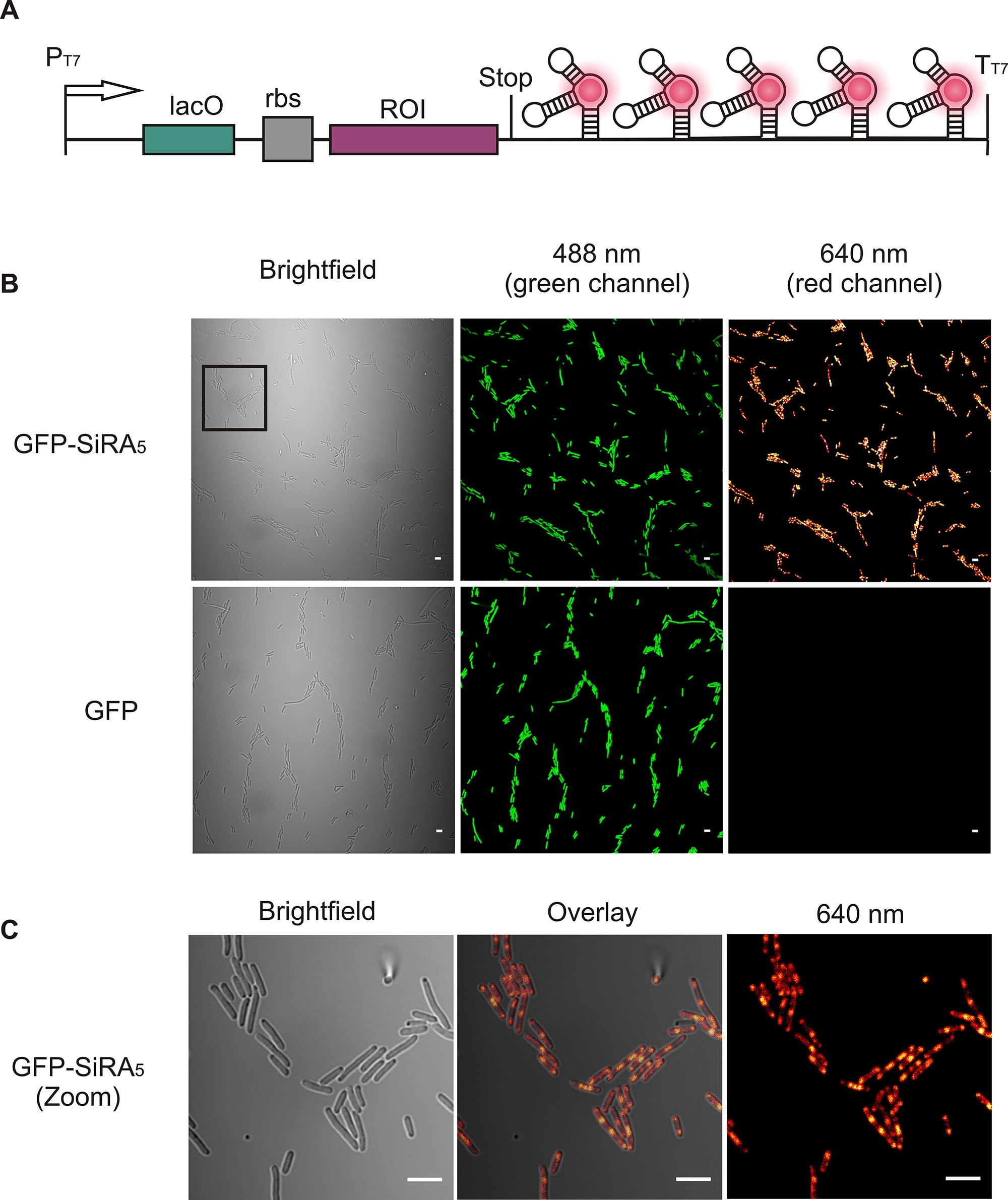
Figure 4. Example of GFP mRNA imaging in live E. coli using a confocal microscope. A. Schematic of the plasmid used in the imaging experiment. Binding of the fluorophore to the SiRA aptamers generates a fluorescence signal in the red detection channel of the microscope. B. No-wash live-cell imaging of GFP mRNA in live E. coli cells. The cells were transformed with either the GFP plasmid or the GFP-SiRA5 plasmid, and the images taken with the bright field, green (protein expression) and red channel (mRNA detection). Scale bars = 5 µm. C. Zoom-in of the images depicted in Figure 4B. Scale bars = 5 µm. - Repeat Steps E3-E5 with the E. coli cells carrying the control plasmid pET-GFP using the same microscope settings.
- For background correction, we use Fiji/Image J image analysis software. Manually pick a surface area without attached E. coli cells and subtract its mean fluorescence signal from the whole image.
- (Optional) Adjustment of the STED microscope
- Ensure spatial and temporal overlap of the 640-nm excitation and 779-nm depletion beam foci by scanning a gold bead (GC80) with detection in both color channels.
- At first, separate the reflected/scattered excitation and depletion light beams into two beams by a 650-nm long-pass dichroic mirror. Then, filter the two beams by band-path filters 780/10 nm (center/width) and 640/10 nm until they are detected by the two avalanche photodiodes (APDs).
Use the Imspector software to synchronize the scan signal and photon detection intervals, thus generating two-channel images without spectral crosstalk.
Note: If necessary, the depletion beam needs to be slightly adjusted to ensure that the center of the STED image (with a doughnut-like profile) is precisely overlaid with the excitation image (with a Gaussian profile). - Check the temporal overlap between the excitation and depletion pulses. Therefore, collect the reflected 640-nm and 779-nm light by the gold beads by using a time-correlated single photon counting (TCSPC) card and sort them with respect to the common trigger signal. The delay between the 640 nm excitation and 779 nm pulses can be monitored in the oscilloscope display-module of the TCSPC software.
Note: For optimal depletion, the delay between the excitation and depletion pulses can be adjusted (in our example ~200 ps) by using a delay box, which routes the trigger output of the depletion laser to the trigger input of the excitation laser.
- Imaging dark-red beads (Day 3: ~1 h)
Note: Images of dark-red beads are taken to determine the resolution of the STED microscope and should be carried out before or in parallel to the sample preparation.- Dilute FluosphereTM 40-nm dark-red beads (660/680) from the stock solution into PBS at a volume ratio of 1:104.
- Add 200 μl of the solution to one of the poly-D-lysine coated imaging chambers and incubate for 5 min at room temperature to immobilize the beads on the surface.
- Wash the coverslip twice with 250 μl MilliQ water and finally add 250 μl MilliQ water for imaging.
- Place the coverslip onto the microscope slide holder.
- Adjust the excitation power to 5 μW at the sample and image with the depletion laser at different powers between 0 mW to 150 mW. Scan a rectangular area of 10 x 10 μm2 with 1,024 x 1,024 pixels, covering dozens of individual dark-red beads.
Note: To minimize photo-bleaching upon multiple scans of the same region, it is recommended to use a fresh region for STED imaging at the different STED laser intensities. - Generate a comparable image between confocal and STED modes (Figure 5A). Therefore, image the same area sequentially by confocal and STED microscopy.
Note: By changing the depletion power, the effective resolution can be varied, as shown by images of a bead taken at different powers of the depletion beam (Figure 5B).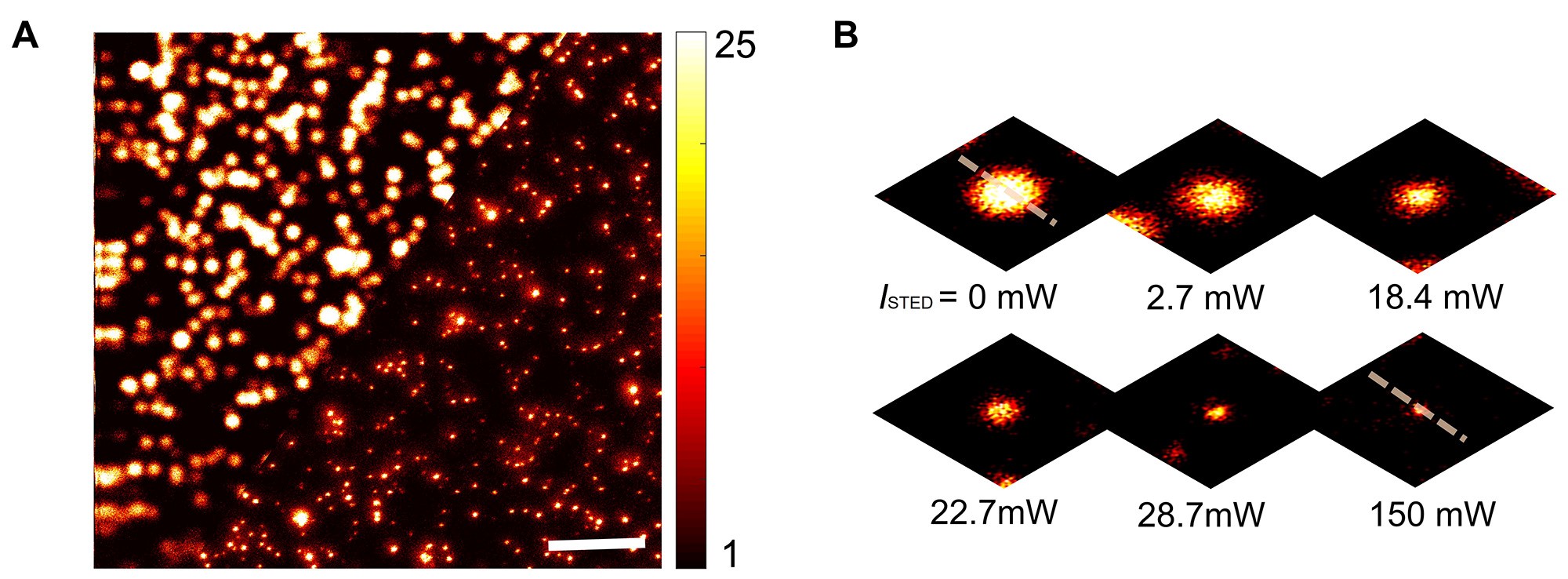
Figure 5. Dark-red bead images. A. Confocal (upper left) and STED (lower right) image of fluorescent beads on a glass coverslip, showing the enormous resolution enhancement due to STED imaging. Scale bar = 3 µm. B. Image of an individual bead taken at different powers of the depletion beam.
- Evaluation of the STED resolution using dark-red bead images (Day 3: ~1 h)
- For data evaluation, select ten beads for each STED intensity. For each bead, extract a line (with single-pixel width) crossing the bead through its center and fit with a Gaussian function.
Note: The fit yields a raw full width at half maximum (FWHMr), which includes the size of the dark-red beads (d0 = 40 nm). The real STED resolution (FWHM) can then be estimated as (FWHMr2 - d02)1/2. Based on the statistics of ten beads, the symbols and error bars in Figure 6A show, respectively, the means and standard deviations of the FWHMs at each STED intensity. The theoretical curve, which is fitted with the model FWHM = FWHM0/(1 + γ·ISTED2)1/2, describes the image resolution as a function of STED power. Here, FWHM0 is the resolution of the confocal microscope (with depletion beam off), ISTED is the intensity of the depletion beam, and γ is a coefficient that depends on the slope of the STED doughnut and the saturation intensity of the depletion beam (Harke et al., 2008). - Compare cross-sectional intensity distributions obtained by confocal (zero depletion power) and STED imaging (150 mW depletion power) of a 40 nm bead (Figure 6B). [Example Figure 6A: FWHMs of Gaussian fits (solid lines in Figure 6A) yield FWHMr = 339 ± 11 nm for confocal mode and 50 ± 3 nm for STED mode. Removing the size of the dark-red beads, we obtain an estimated resolution of our STED microscope as 32 ± 5 nm.]
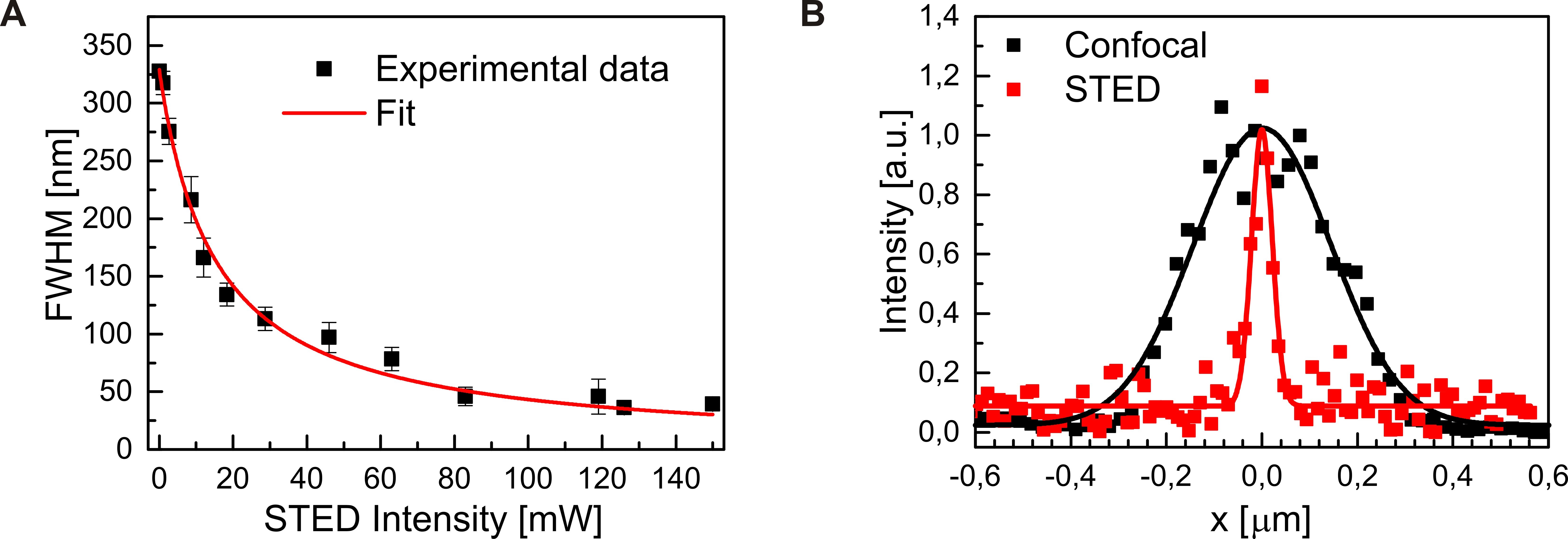
Figure 6. Determination of the STED resolution. A. Solid squares: Quantification of the widths of cross-sections through individual beads as a function of the power of the depletion laser (see Figure 5B). Data were corrected for the intrinsic size of the beads and are shown as means ± standard deviations from ten bead images. Red line: fit with a square-root function according to STED theory. B. Cross sections through a single bead imaged at 0 and 150 mW depletion laser power (see Figure 5) showing the enormous resolution enhancement offered by STED microscopy. Gaussian fits are shown as solid lines.
- For data evaluation, select ten beads for each STED intensity. For each bead, extract a line (with single-pixel width) crossing the bead through its center and fit with a Gaussian function.
- Imaging GFP-SiRA5 mRNA in live E. coli with STED microscopy (Day 3: ~1-2 h)
- Attach the temperature-controlled sample chamber to the microscope, prewarm to 37 °C and add the 8-well imaging slide used in Step E2.
- Set the power of the excitation and depletion beams (Example Figure 7: Excitation 5 μW, depletion 150 mW). Scan repeatedly a square region of 10 x 10 μm2, covering a group of E. coli cells, for five frames of 1,024 x 1,024 pixels (Pixel dwell time in Figure 7A: 30 μs, the main image of Figure 7A corresponds to the inset with t = 126 s).
- After taking the STED images, switch off the depletion laser and take a confocal image (Figure 7B).
Note: No deconvolution was carried out on either the STED or the confocal images. Different color maps can be used for the STED and confocal images to optimally display cell structures.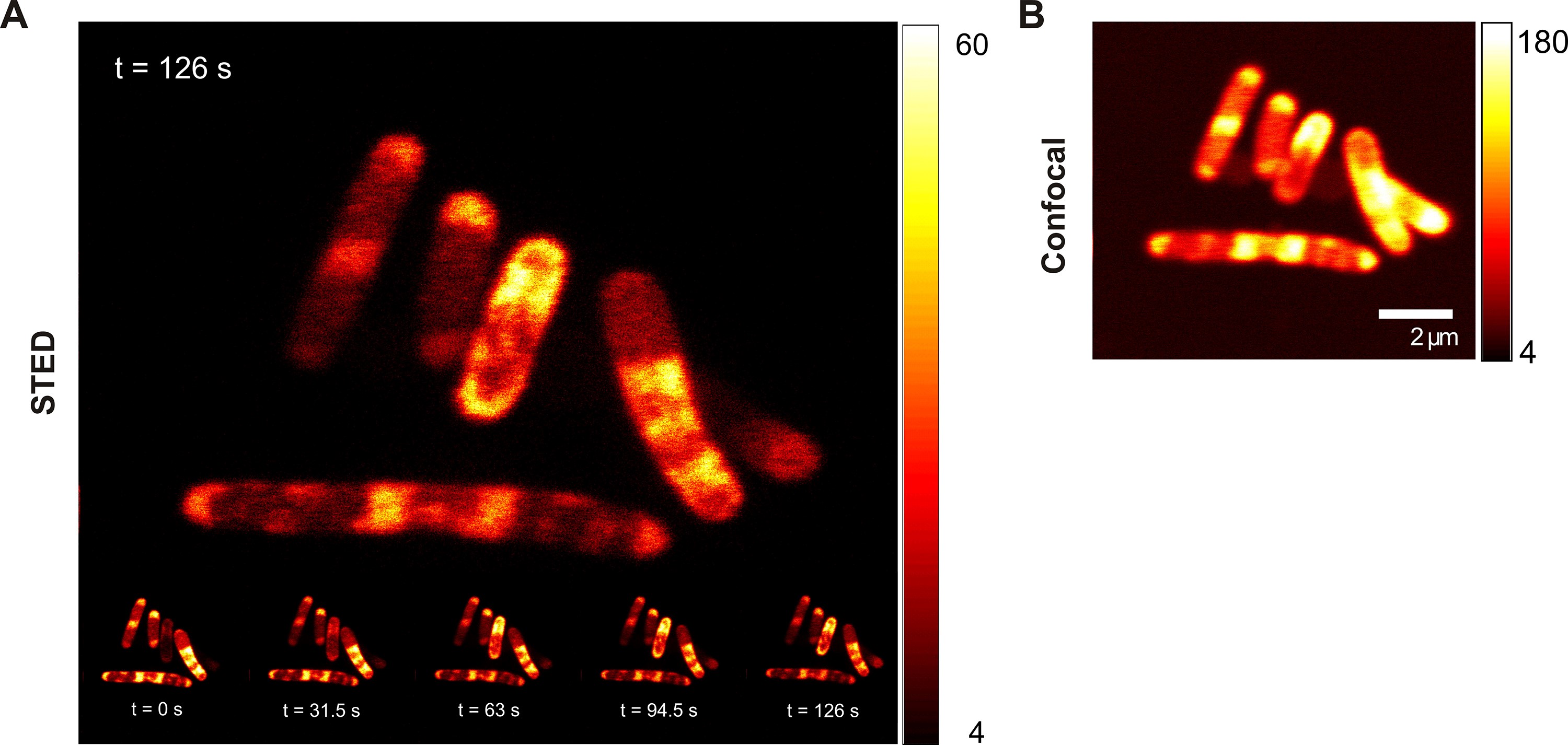
Figure 7. STED imaging of live E. coli bacteria expressing GFP-SiRA 5 mRNA. A. Time series of STED images taken in 31.5 s intervals for a total of 126 s; top: magnified image at 126 s; B. Regular confocal image taken directly after the STED time series.
Data analysis
- To determinate the STED resolution, statistics with multiple beads are necessary. In our experiment, ten individual beads for each STED intensity were selected to calculate the raw full width at half maximum (FWHMr) with the cross-section intensity distribution of the bead. The data points and error bars in Figure 6A present the means and standard deviations of the ten beads.
Note: To avoid selection of a cluster of beads (rather than single DR beads), those were excluded (and replaced with new selections) when their cross-section intensity distribution has the fitted Gaussian amplitude 1.5 times higher than the averaged amplitude. - To reduce the influence of sample defocusing on STED resolution determination, for each STED intensity two images were taken (from a new sample area), and the in-focus position of the sample was checked each time, which provides the maximal brightness for the beads in the confocal mode. Generally, five beads from each image were selected for the statistics of the STED resolution. However, for the case that one image has systematically-larger FWHMs than the other image (due to defocusing), ten beads from the in-focus image solely will be selected for the FWHM analysis.
Recipes
- M9 imaging solution (50 ml)
- Preparation of the 5x M9 salt broth:
- Add 26.25 g of M9 broth powder [Na2HPO4 (15 g), KH2PO4 (7.5 g), NaCl (1.25 g), NH4Cl (2.5 g)] into a 1,000 ml PYREX® glass bottle
- Add 500 ml of MilliQ water, shake thoroughly and check the pH (7.4 at 37 °C)
- Autoclave with a standard liquid program
- Fill 10 ml of the 5x M9 into a 50 ml Falcon and supplement the solution with 250 μl of MgSO4 (1 M stock, 5 mM final concentration), 5 μl of CaCl2 (1 M stock, 100 μM final concentration) and 500 μl of Glucose (40% stock, final concentration 0.4%)
- Fill the Falcon to 50 ml with MilliQ water and filter-sterilize the solution before using it for the imaging experiment
- Preparation of the 5x M9 salt broth:
Acknowledgments
M.S. and A.J. were supported by the Deutsche Forschungsgemeinschaft (Grant # Ja794/11-1), and G.U.N. by the Helmholtz Association (Program Science and Technology of Nanosystems) and the Karlsruhe School of Optics and Photonics (KSOP). This protocol was adopted from our previously published work (Wirth et al., 2019).
Competing interests
The authors declare no competing financial interest.
References
- Alexander, S. C. and Devaraj, N. K. (2017). Developing a fluorescent toolbox to shed light on the mysteries of RNA. Biochemistry 56(39): 5185-5193.
- Arora, A., Sunbul, M. and Jäschke, A. (2015). Dual-colour imaging of RNAs using quencher- and fluorophore-binding aptamers. Nucleic Acids Res 43(21): e144.
- Autour, A., S, C. Y. J., A, D. C., Abdolahzadeh, A., Galli, A., Panchapakesan, S. S. S., Rueda, D., Ryckelynck, M. and Unrau, P. J. (2018). Fluorogenic RNA Mango aptamers for imaging small non-coding RNAs in mammalian cells. Nat Commun 9(1): 656.
- Bouhedda, F., Autour, A. and Ryckelynck, M. (2017). Light-up RNA aptamers and their cognate fluorogens: from their development to their applications. Int J Mol Sci 19(1).
- Braselmann, E., Wierzba, A. J., Polaski, J. T., Chromiński, M., Holmes, Z. E., Hung, S. T., Batan, D., Wheeler, J. R., Parker, R., Jimenez, R., Gryko, D., Batey, R. T. and Palmer, A. E. (2018). A multicolor riboswitch-based platform for imaging of RNA in live mammalian cells. Nat Chem Biol 14(10): 964-971.
- Dolgosheina, E. V., Jeng, S. C., Panchapakesan, S. S., Cojocaru, R., Chen, P. S., Wilson, P. D., Hawkins, N., Wiggins, P. A. and Unrau, P. J. (2014). RNA mango aptamer-fluorophore: a bright, high-affinity complex for RNA labeling and tracking. ACS Chem Biol 9(10): 2412-2420.
- Filonov, G. S., Moon, J. D., Svensen, N. and Jaffrey, S. R. (2014). Broccoli: rapid selection of an RNA mimic of green fluorescent protein by fluorescence-based selection and directed evolution. J Am Chem Soc 136(46): 16299-16308.
- Gao, P., Prunsche, B., Zhou, L., Nienhaus, K. and Nienhaus, G. U. (2017). Background suppression in fluorescence nanoscopy with stimulated emission double depletion. Nature Photonics 11: 163-169.
- Harke, B., Keller, J., Ullal, C. K., Westphal, V., Schonle, A. and Hell, S. W. (2008). Resolution scaling in STED microscopy. Opt Express 16(6): 4154-4162.
- Holeman, L. A., Robinson, S. L., Szostak, J. W. and Wilson, C. (1998). Isolation and characterization of fluorophore-binding RNA aptamers. Fold Des 3(6): 423-431.
- Lukinavičius, G., Umezawa, K., Olivier, N., Honigmann, A., Yang, G., Plass, T., Mueller, V., Reymond, L., Corrêa, I. R., Jr., Luo, Z. G., Schultz, C., Lemke, E. A., Heppenstall, P., Eggeling, C., Manley, S. and Johnsson, K. (2013). A near-infrared fluorophore for live-cell super-resolution microscopy of cellular proteins. Nat Chem 5(2): 132-139.
- Murata, A., Sato, S., Kawazoe, Y. and Uesugi, M. (2011). Small-molecule fluorescent probes for specific RNA targets. Chem Commun (Camb) 47(16): 4712-4714.
- Neubacher, S. and Hennig, S. (2019). RNA structure and cellular applications of fluorescent light-up aptamers. Angew Chem Int Ed Engl 58(5): 1266-1279.
- Nienhaus, K. and Nienhaus, G. U. (2016). Where do we stand with super-resolution optical microscopy? J Mol Biol 428(2 Pt A): 308-322.
- Ouellet, J. (2016). RNA fluorescence with light-up aptamers. Front Chem 4: 29.
- Paige, J. S., Wu, K. Y. and Jaffrey, S. R. (2011). RNA mimics of green fluorescent protein. Science 333(6042): 642-646.
- Reshes, G., Vanounou, S., Fishov, I. and Feingold, M. (2008). Cell shape dynamics in Escherichia coli. Biophys J 94(1): 251-264.
- Song, W., Filonov, G. S., Kim, H., Hirsch, M., Li, X., Moon, J. D. and Jaffrey, S. R. (2017). Imaging RNA polymerase III transcription using a photostable RNA-fluorophore complex. Nat Chem Biol 13(11): 1187-1194.
- Sunbul, M. and Jäschke, A. (2013). Contact-mediated quenching for RNA imaging in bacteria with a fluorophore-binding aptamer. Angew Chem Int Ed Engl 52(50): 13401-13404.
- Sunbul, M. and Jäschke, A. (2018). SRB-2: a promiscuous rainbow aptamer for live-cell RNA imaging. Nucleic Acids Res 46(18): e110.
- Sunbul, M., Arora, A. and Jäschke, A. (2018). Visualizing RNA in live bacterial cells using fluorophore- and quencher-binding aptamers. Methods Mol Biol 1649: 289-304.
- Tan, X., Constantin, T. P., Sloane, K. L., Waggoner, A. S., Bruchez, M. P. and Armitage, B. A. (2017). Fluoromodules consisting of a promiscuous RNA aptamer and red or blue fluorogenic cyanine dyes: selection, characterization, and bioimaging. J Am Chem Soc 139(26): 9001-9009.
- Tyagi, S. (2009). Imaging intracellular RNA distribution and dynamics in living cells. Nat Methods 6(5): 331-338.
- Wang, L., Frei, M. S., Salim, A. and Johnsson, K. (2019). Small-molecule fluorescent probes for live-cell super-resolution microscopy. J Am Chem Soc 141(7): 2770-2781.
- Wirth, R., Gao, P., Nienhaus, G. U., Sunbul, M. and Jäschke, A. (2019). SiRA: a silicon rhodamine-binding aptamer for live-cell super-resolution RNA imaging. J Am Chem Soc 141(18): 7562-7571.
- Xia, Y., Zhang, R., Wang, Z., Tian, J. and Chen, X. (2017). Recent advances in high-performance fluorescent and bioluminescent RNA imaging probes. Chem Soc Rev 46(10): 2824-2843.
- Yerramilli, V. S. and Kim, K. H. (2018). Labeling RNAs in live cells using malachite green aptamer scaffolds as fluorescent probes. ACS Synth Biol 7(3): 758-766.
Article Information
Copyright
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Wirth, R., Gao, P., Nienhaus, G. U., Sunbul, M. and Jäschke, A. (2020). Confocal and Super-resolution Imaging of RNA in Live Bacteria Using a Fluorogenic Silicon Rhodamine-binding Aptamer. Bio-protocol 10(9): e3603. DOI: 10.21769/BioProtoc.3603.
Category
Biochemistry > RNA > RNA structure
Molecular Biology > RNA > RNA labeling
Molecular Biology > RNA > RNA detection
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.