- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Sequence Alignment Using Machine Learning for Accurate Template-based Protein Structure Prediction


Published: Vol 10, Iss 9, May 5, 2020 DOI: 10.21769/BioProtoc.3600 Views: 5682
Reviewed by: Prashanth N SuravajhalaJayaraman ValadiL N Chavali
Original research article
The authors used this protocol in:

Jan 2020
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Template-based modeling, the process of predicting the tertiary structure of a protein by using homologous protein structures, is useful when good templates can be available. Indeed, modern homology detection methods can find remote homologs with high sensitivity. However, the accuracy of template-based models generated from the homology-detection-based alignments is often lower than that from ideal alignments. In this study, we propose a new method that generates pairwise sequence alignments for more accurate template-based modeling. Our method trains a machine learning model using the structural alignment of known homologs. When calculating sequence alignments, instead of a fixed substitution matrix, this method dynamically predicts a substitution score from the trained model.
Background
Proteins are key molecules in biology, biochemistry and pharmaceutical sciences. To reveal the functions of proteins, it is essential to understand the relationships between proteins' structure and function. Protein structures can be determined by experimental; the protein structures are often registered to and accessible in the Protein Databank (PDB) (wwPDB consortium, 2018). However, despite improvements in experimental methods for determining protein structures, the speed at which amino acid sequences can be revealed has overtaken our ability to ascertain the corresponding proteins' structures (Muhammed et al. 2019). Therefore, protein structure prediction remains essential.
As one of various methods for protein structure prediction, template-based or homology modeling predicts structures based on templates and their sequence alignment to a target protein. Template structures are the structures of homologous proteins, often found by homology detection methods. Currently, template-based modeling methods are the most practical because the predicted models are often accurate if we can find good templates and protein sequence alignments. These accurate models by template-based modeling can be used for computer-aided drug design (CADD).
Indeed, recent homology search methods have been able to detect remote homologs (Boratyn et al., 2012; Zimmermann et al., 2018). Although, sometimes sufficiently accurate structure models cannot be obtained because the quality of the sequence alignment generated by homology detection program is poor. If a more accurate model is required, researchers must manually edit alignments to improve their quality before modeling. In structural alignment, the structural difference between a target protein structure and a template protein structure is minimized; thus, sequence alignments generated by structural alignment are almost ideal for template-based modeling. Often, the sequence alignments generated by the homology detection methods are dissimilar to those generated by structural alignment, especially for remote homologs. Thus far, a method’s ability to detect remote homologs has been prioritized because models cannot be generated without a template. However, to achieve higher-accuracy template-based modeling, the improvement of sequence alignment generation is a critical open problem. This problem has been mentioned in several studies (Kopp et al., 2007) in which researchers have tried to improve alignments manually based on their knowledge of biology; fully automated methods are still required.
Recently, machine learning methods have demonstrated power in various fields (Lyons et al., 2014; Cao et al., 2016; Wang, Peng, et al., 2016; Wei and Zou, 2016; Manavalan and Lee, 2017; Wang, Sun, et al., 2017). Machine learning also seems effective in tackling the problem of alignment generation for homology modeling. However, this topic has not been studied because it is challenging to treat alignment generation as a classification or regression problem.
For the problem, we proposed a new sequence alignment generation protocol based on a machine learning that learns the structural alignments of known homologs (Makigaki and Ishida, 2019). We use a dynamic programming algorithm during aligning sequences to dynamically predict a substitution score from the k-Nearest Neighbor (k-NN) model instead of a fixed substitution matrix or profile comparison. Machine learning is used in this substitution score prediction process.
The proposed method is valuable for researchers who use template-based modeling with remote homologs whose sequence identity is not high. In this paper, we show the overview of our method as a procedure, and more detailed usage of our tool and some examples are available in the source code repository (https://github.com/shuichiro-makigaki/exmachina).
Equipment
- Computer
> 128 GiB RAM and > 150 GiB free storage are recommended - Linux (> 3.10) or SUSE Linux Enterprise Server 12
Software
- PSI-BLAST (> v2.9)
To generate PSSM of an amino acid sequence
Download URL: https://blast.ncbi.nlm.nih.gov/Blast.cgi?CMD=Web&PAGE_TYPE=BlastDocs&DOC_TYPE=Download (Last access date: 2020-02-22)
Installation document URL: https://www.ncbi.nlm.nih.gov/books/NBK279690/ - TM-align (> v20190822) (Zhang and Skolnick, 2005)
To generate structural alignment of homologs
Download and installation document URL: https://zhanglab.ccmb.med.umich.edu/TM-align/ (Last access date: 2020-02-22) - Implementation: Source code and installation document are available in the source code repository.
Download URL: https://github.com/shuichiro-makigaki/exmachina/archive/master.zip
Installation Procedure: https://github.com/shuichiro-makigaki/exmachina#how-to-use
(Last access date: 2020-02-22)
Python 3.6: Required python packages are listed in the repository. - FLANN (Muja and Lowe, 2009): k-Nearest Neighbor implementation. The installation procedure also contains the FLANN installation document.
- Structural Classification of Proteins (SCOP) database
The SCOP database classifies proteins by class, folds, superfamily (SF), family and domain based on manually curated function/structure classifications and contains redundant sequences. Thus, we used the SCOP40 database instead, which contains only domains whose sequence identity is < 40% to avoid overfitting and reduce execution time.
Download URL: https://scop.berkeley.edu/astral/pdbstyle/ver=1.75 (Last access date: 2020-02-22) - UniRef (The UniProt Consortium, 2016) database
For Position Specific Scoring Matrix (PSSM) generation, we used three-iteration PSI-BLAST (Altschul et al., 1997) with the UniRef90 database.
Download URL: https://www.uniprot.org/downloads#unireflink (Last access date: 2020-02-22)
Procedure
The primary purpose of the training phase is to generate k-NN model that will be used for substitution score prediction in the prediction and alignment generation phase. The prediction phase consists of score prediction and alignment generation. Figure 1 shows the overview of the method. More detailed step-by-step commands and the example are available at source code repository (https://github.com/shuichiro-makigaki/exmachina).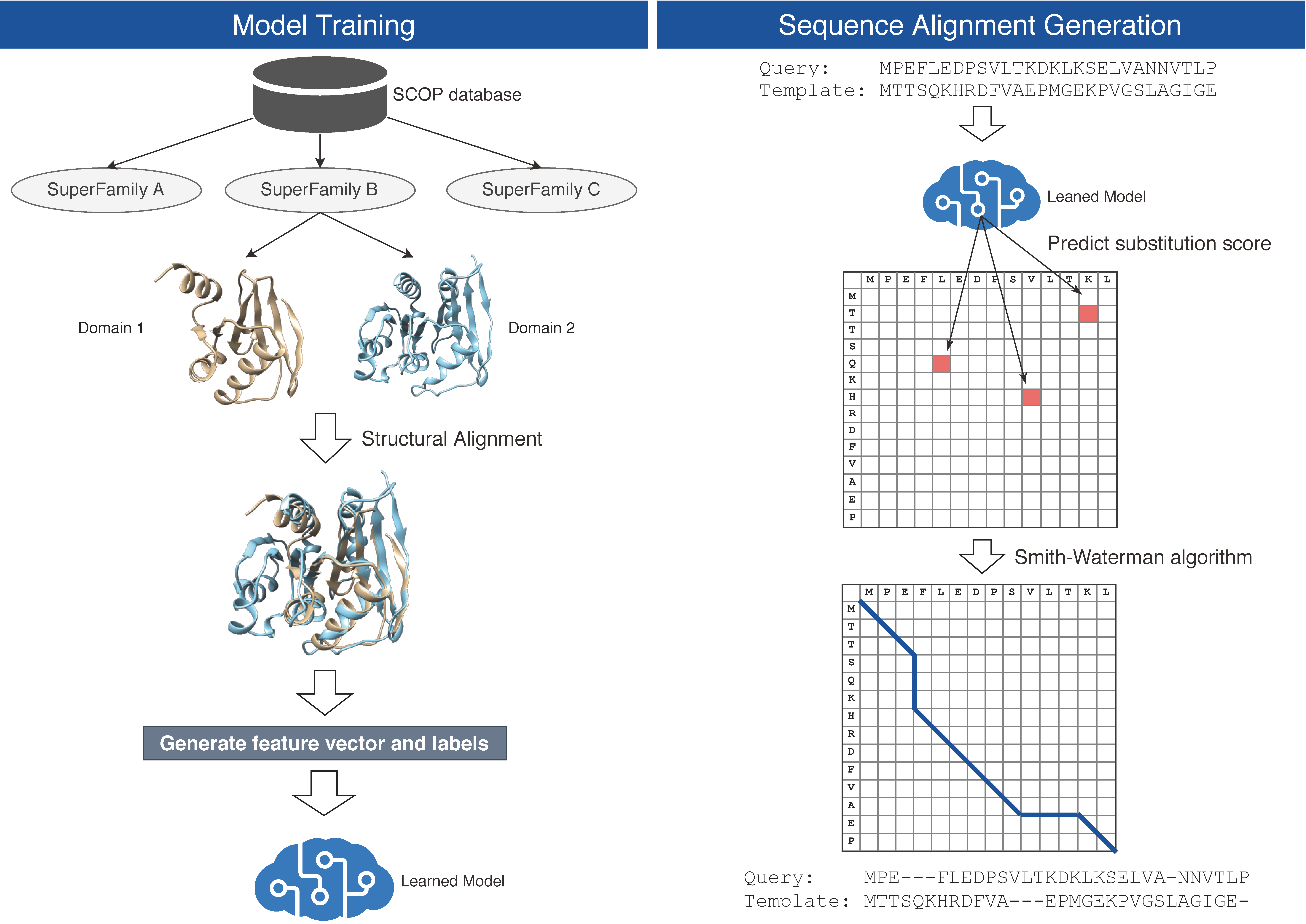
Figure 1. Overview of the proposed method
- Model training
- Download SCOP40 database.
- Generate structural alignments of every domain pair in the same SF by TM-align.
- Select only pairs that the TM-score is ≥ 0.5.
- Generate a PSSM of the domain by three-iteration PSI-BLAST with the UniRef90 database.
- Generate training data and labels.
As a hyper-parameter, window size is 5. - Reduce training dataset to 1/10 by random sampling.
Because the original training dataset became too large to process within a reasonable computation time. - Save the training dataset and the labels as FLANN-acceptable data format.
- Score prediction and sequence alignment generation
- Prepare two homologous amino acid sequences
As a current limitation, our implementation expects that the inputs are sub-domains. When the protein consists of multiple domains, it should be split into domains. Usually, the domain regions can be predicted by homology detection. - Generate PSSMs of each sequence by three-iteration PSI-BLAST with the UniRef90 database.
- Predict all substitution scores of each residue pairs.
- Query vector format is the same as the training phase, and the k-NN's classification scores are used for the substitution score directly.
- As hyper-parameters, the window size is 5, and the number of the neighbor is 1,000.
- Save predicted substitution score matrix.
- Generate local sequence alignment by Smith-Waterman algorithm implemented in Biopython (https://biopython.org/).
During the dynamic-programming, the predicted substitution scores are used for score calculation.
- Prepare two homologous amino acid sequences
Acknowledgments
This work was supported by JSPS KAKENHI [18K11524] and (Makigaki and Ishida, 2019).
Competing interests
The authors declare no competing interests.
References
- Altschul, S. F., Madden, T. L., Schäffer, A. A., Zhang, J., Zhang, Z., Miller, W. and Lipman, D. J. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25(17): 3389-3402.
- Boratyn, G. M., Schaffer, A. A., Agarwala, R., Altschul, S. F., Lipman, D. J. and Madden, T. L. (2012). Domain enhanced lookup time accelerated BLAST. Biol Direct 7: 12.
- Cao, R., Bhattacharya, D., Hou, J. and Cheng, J. (2016). DeepQA: improving the estimation of single protein model quality with deep belief networks. BMC Bioinformatics 17(1): 495.
- Kopp, J., Bordoli, L., Battey, J. N., Kiefer, F. and Schwede, T. (2007). Assessment of CASP7 predictions for template-based modeling targets. Proteins 69 Suppl 8: 38-56.
- Lyons, J., Dehzangi, A., Heffernan, R., Sharma, A., Paliwal, K., Sattar, A., Zhou, Y. and Yang, Y. (2014). Predicting backbone Calpha angles and dihedrals from protein sequences by stacked sparse auto-encoder deep neural network. J Comput Chem 35(28): 2040-2046.
- Makigaki, S. and Ishida, T. (2019). Sequence alignment using machine learning for accurate template-based protein structure prediction. Bioinformatics.
- Manavalan, B. and Lee, J. (2017). SVMQA: support-vector-machine-based protein single-model quality assessment. Bioinformatics 33(16): 2496-2503.
- Muhammed, M. T. and Aki-Yalcin, E. (2019). Homology modeling in drug discovery: Overview, current applications, and future perspectives. Chem Biol Drug Des 93(1): 12-20.
- Muja, M. and Lowe, D. (2009). Fast approximate nearest neighbors with automatic algorithm configuration. In Muja M. and Lowe D. (Eds). VISAPP International Conference on Computer Vision Theory and Applications. Lisboa, Portugal, February 5-8, 2009 - Volume 1.
- The UniProt Consortium (2016). UniProt: the universal protein knowledgebase. Nucleic Acids Research 45(D1): D158-D169.
- Wang, S., Peng, J., Ma, J. and Xu, J. (2016). Protein secondary structure prediction using deep convolutional neural fields. Sci Rep 6: 18962.
- Wang, S., Sun, S., Li, Z., Zhang, R. and Xu, J. (2017). Accurate de novo prediction of protein contact map by ultra-deep learning model. PLoS Comput Biol 13(1): e1005324.
- Wei, L. and Zou, Q. (2016). Recent progress in machine learning-based methods for protein fold recognition. Int J Mol Sci 17(12).
- wwPDB consortium. (2018). Protein Data Bank: the single global archive for 3D macromolecular structure data. Nucleic Acids Res 47(D1): D520-D528.
- Zhang, Y. and Skolnick, J. (2005). TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res 33(7): 2302-2309.
- Zimmermann, L., Stephens, A., Nam, S. Z., Rau, D., Kubler, J., Lozajic, M., Gabler, F., Soding, J., Lupas, A. N. and Alva, V. (2018). A completely reimplemented MPI bioinformatics toolkit with a new HHpred server at its core. J Mol Biol 430(15): 2237-2243.
Article Information
Copyright
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Makigaki, S. and Ishida, T. (2020). Sequence Alignment Using Machine Learning for Accurate Template-based Protein Structure Prediction. Bio-protocol 10(9): e3600. DOI: 10.21769/BioProtoc.3600.
Category
Systems Biology > Transcriptomics > RNA-seq
Biochemistry > Protein > Structure
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.