- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Growth Recovery Assay and FACS-based Population Sorting Following Territorial Exclusion in Proteus mirabilis
Published: Vol 10, Iss 5, Mar 5, 2020 DOI: 10.21769/BioProtoc.3543 Views: 5283
Reviewed by: Kristin L. ShinglerYoko EguchiChao Jiang
Original research article
The authors used this protocol in:
Jul 2019

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Many bacteria take part in self recognition and kin discrimination behavior using contact-dependent effectors. Understanding the effects these effectors cause is important to explain bacterial community formation and population dynamics. Typically, kin discrimination effectors are toxins that kill target cells; their effect is therefore obvious and easily measurable. However, many self-recognition effectors, such as the Proteus mirabilis Ids system, are non-lethal and do not cause obvious physiological changes in target cells. Previously, experimental techniques to probe cells experiencing non-lethal kin recognition have been limited. Here we describe a technique to reliably isolate cells deemed self and non-self through Ids self-recognition for downstream phenotypic analysis. Liquid cultures of fluorescently labeled self-recognition mutants are mixed together and inoculated on swarm-permissive agar. Mixed swarms are harvested, and each strain is isolated through fluorescence-activated cell sorting (FACS). The growth rate of each strain is measured on a plate reader. This protocol is adaptable for other bacterial species. We describe briefly how sorted particles can be used for other analyses such as RNA-Seq library preparation.
Keywords: Contact-dependent effectorsBackground
Many organisms have evolved adaptations to community living. One common mechanism present throughout nature is kin discrimination: preferential treatment of close kin and hindrance of non-kin (Smith, 1964). Well studied examples of bacterial kin discrimination include contact-dependent inhibition (CDI) (Aoki et al., 2009; Garcia et al., 2016) and toxin transfer through type IV and type VI secretion systems (Brunet et al., 2013; Souza et al., 2015). Typically, kin discrimination in bacteria is mediated by exchange of toxic effector proteins, such as nucleases or peptidoglycan degrading enzymes (Alteri and Mobley, 2016). Each strain expresses characteristic effector-immunity pairs, and cells must either express the cognate immunity protein or die. Proteus mirabilis, however, makes use of a non-lethal self recognition system, the Ids system, to perform kin discrimination during swarming (Gibbs et al., 2008). P. mirabilis displays a sophisticated swarming phenotype on solid surfaces. After surface contact, a subset of cells differentiate into elongated “swarmer” cells that collectively travel outwards. Periods of swarming are regularly followed by non-expansion “consolidation” periods where swarmer cells differentiate into shorter stationary cells. Successive rounds of swarming and consolidation lead to a characteristic bullseye pattern on surfaces (Rauprich et al., 1996). While swarming, different P. mirabilis isolates express different variants of identity factors IdsD and IdsE. IdsD is transferred between cells in a contact-dependent fashion. If cells do not express a cognate IdsE they do not die but are instead forced out of cooperative swarm behavior in a phenomenon termed territorial exclusion. This behavior is due to forced induction into a persister-like state mediated by the alarmone (p)ppGpp (Tipping and Gibbs, 2019).
In lethal kin discrimination systems, progression can be measured through several methods: dilution assays, microscopy to visualize killing (e.g., Basler et al., 2013), or live-dead staining (Virta et al., 1998). Quantifying the efficacy of a non-lethal system is more difficult. The relative swarm extent in situ can be measured through microscopy or replica plating (Wenren et al., 2013), but these methods provide limited information about the swarm state, and none at all about the states of individual cells that have taken part in non-lethal kin discrimination. Isolation of cells from the greater swarm mass is therefore critical to enable their physiological state to be examined in more detail.
We have developed the first method for separating two populations of cells from a mixed P. mirabilis swarm using fluorescence-activated cell sorting (FACS), so that the state of cells that have taken part in self recognition behavior can be measured. This efficient and reliable protocol allows downstream examination of cells using several different methods. Here, we describe how to measure growth rate of cells that have experienced territorial exclusion. We have also used this method to generate samples for RNA-Seq library preparation, and describe our methods for doing so briefly. Furthermore, sorted cells can potentially be used to generate samples for other metabolic or biochemical assays. The protocol as described is optimized for separating P. mirabilis cells and could be adapted to isolate cells from swarms of other species such as Escherichia coli, Vibrio cholerae, Pseudomonas aeruginosa, and Providencia stuartii.
Materials and Reagents
Note: All materials and reagents are stable at room temperature. P. mirabilis strains are typically transported as stab cultures that are viable at room temperature.
- Disposable inoculation loop (Fisher, catalog number: 22-363-602 )
- Petri dishes (VWR, catalog number: 25384-168 )
- 50 ml conical tubes (Fisher, catalog number: 14-432-22 )
- 1.5 ml microcentrifuge tubes (Fisher, catalog number: 05-408-129 )
- 1-10 µl pipette tips (Fisher, catalog number: 07-000-854 )
- 20-100 µl pipette tips (Fisher, catalog number: 07-000-867 )
- 200-1,000 µl pipette tips (Fisher, catalog number: 07-000-350 )
- Disposable spreaders (Fisher, catalog number: 14-665-230 )
- Spectrophotometry cuvettes (VWR, catalog number: 97000-586 )
- 0.22 µm vacuum filter kit (Fisher, catalog number: 09-761-1 )
- FACS collection tubes (Fisher, catalog number: 50-187-1139 )
- 96-well flat-bottomed plates (Sigma-Aldrich, catalog number: M2936-100EA )
- Proteus mirabilis strain KAG107 (BB2000 constitutively expressing DsRed) (Gibbs et al., 2008) (available on request)
- Proteus mirabilis strain KAG966 (BB2000 ∆ids constitutively expressing GFPmut2) (Gibbs et al., 2008) (available on request)
- Tryptone (Fisher, catalog number: DF0123-17-3 )
- Yeast extract (Fisher, catalog number: DF0127-17-9 )
- Sodium chloride (NaCl) (Sigma-Aldrich, catalog number: S9888-500G )
- Oxoid CM55 agar (Fisher, catalog number: OXCM0055B )
- Glycerol (Sigma-Aldrich, catalog number: G7893-500ML )
- Bacto-agar (Fisher, catalog number: DF0140-01-0 )
- Magnesium sulfate (MgSO4) (Sigma-Aldrich, catalog number: M7506-500G )
- Nicotinic acid (Sigma-Aldrich, catalog number: N4126-100G )
- Sodium hydrogen phosphate dihydrate (Na2HPO4·2H2O) (Sigma-Aldrich, catalog number: 71645-1KG )
- Potassium hydrogen phosphate (K2HPO4) (Sigma-Aldrich, catalog number: 1051041000 )
- Sodium hydroxide (NaOH) (Sigma-Aldrich, catalog number: 795429-500G )
- Hydrochloric acid (HCl) (Sigma-Aldrich, catalog number: 320331-500ML )
- RNA-Protect solution (Qiagen, catalog number: 76526 )
- LB Broth (1 L) (see Recipes)
- CM55 swarm agar (1 L) (see Recipes)
- LSW- Agar (1 L) (see Recipes)
- Media (1 L)
- LSW supplemental mixture (50 ml)
- Phosphate-buffered saline (PBS) (see Recipes)
Equipment
- Vortexer (Fisher, catalog number: 14-955-163 )
- Hot plate (VWR, catalog number: 97042-690 )
- Stir rods (VWR, catalog number: 58947-114 )
- Glass culture tubes (VWR, catalog number: 47729-580 )
- 250 ml Duran bottles (VWR, catalog number: 89051-796 )
- 2 L Erlenmeyer flasks (VWR, catalog number: 89090-858 )
- Milli-Q water purification system (Millipore)
- pH meter (VWR, catalog number: 89231-662 )
- Genesys 10S UV-Vis spectrophotometer (Thermo Fisher, catalog number: 840-208100 )
- BD FACSAria II FACS system (BD Biosciences)
- Tecan Infinite 200 Pro plate reader (Tecan Group Ltd.)
Software
- BD FACSDiva 7.0 (BD Biosciences)
- Microsoft Excel 16.0 (Microsoft, Inc.)
- Tecan iControl 3.4.2 (Tecan Group Ltd.)
Procedure
This protocol describes how to set up mixed co-swarming assays between two different strains of Proteus mirabilis and how to recover cells of each strain from the mixed swarm using fluorescence-activated cell sorting (FACS). The cells were recovered to measure changes in growth rate caused by phenotypic changes induced by the Ids signaling system during swarm consolidation. We use two P. mirabilis strains, KAG107 and KAG966, which have red and green fluorescent markers respectively. The protocol can be adapted to recover cells at other swarm phases or for other downstream applications. An example of how this protocol can be used to generate cells for RNA-Seq library preparation is described at the end of this procedure.
Setup of apparatus, particularly the FACSAria cell sorter and Tecan plate reader, should be done in advance prior to preparation of experimental samples.
- Inoculation and growth of bacterial cultures
Day 1- Streak out stab cultures of strains of interest onto LSW- non-swarm agar plates using an inoculation loop and sterile technique. Incubate at 37 °C overnight.
- Pick a single colony with a sterile pipette tip and use to inoculate 5 ml LB media in a glass culture tube using sterile technique. Incubate at 37 °C overnight with shaking.
- Set up CM55 swarm agar plates for the next day by melting premade agar in a microwave and pouring 25 ml aliquots into sterile Petri dishes. Leave to cure under a Bunsen burner or in a laminar-flow cabinet with lids partially covering the plates for 2 h. Plates can be kept at room temperature after curing for 24 h. Consistency in pouring and curing is important for the reproducibility of swarm behavior. Changing the amount of agar per plate, or curing time, can result in unpredictable swarm behavior after inoculation with P. mirabilis.
- Measure OD600 of overnight cultures using a spectrophotometer (typically, overnight cultures will reach OD600 3.0-3.5), then make 1 ml of OD600 1.0 normalized culture by diluting the relevant volume of overnight culture with fresh LB media in a sterile 1.5 ml Eppendorf tube. Cultures can optionally be normalized to OD600 0.1 rather than OD600 1.0; this serves to extend the swarming time in Step A5 by around six hours without affecting the experimental outcome. Altering the OD in this way can be useful when organizing experimental timings.
- For experiments using more than one strain (e.g., co-swarming assays), add portions of the normalized cultures (to a total volume of ~200 µl) at the desired proportions to a fresh Eppendorf tube. Vortex to mix.
- Inoculate CM55 swarm agar plates by pipetting 1 µl of mixed normalized cell culture onto the center of the plate. Incubate at 37 °C until completion of the first swarm ring, including consolidation, and emergence of the second swarm ring (around 8 h). It is important to begin the next step in the protocol at this time period to remove experimental errors caused by lack of swarm culture synchronization. To this end, plates should be checked by eye every 30 min after the emergence of the first swarm ring. Plates are most likely ready for harvesting as soon as the second swarm ring has begun to emerge from the consolidation area corresponding to the completed first swarm ring. Example images can be seen in Figure 1. Plates are ready once they have reached a state similar to Figure 1B.

Figure 1. Images of progressing swarm rings of a single P. mirabilis swarm culture. A. A swarm culture on a CM55 agar plate that has completed a single swarm ring and has begun consolidating. B. The same swarm culture after emergence of the second swarm ring, visible as a faint band outside the first swarm ring. C. The same swarm culture following completion of the second swarm ring. Arrows show position of outer edge of swarm ring. - Confirm emergence of the second swarm ring using light microscopy. Elongated swarmer cells should form visible rafts emerging from a densely-packed consolidation area, around 1 cm from the center of the plate. 10x or 40x magnification of a phase contrast, epifluorescence, brightfield or DIC image is sufficient to distinguish rafts of motile, elongated swarming cells from shorter, stationary consolidating cells by direct imaging of a plate. Figure 2 shows example phase contrast and Green Fluorescent Protein (GFP) epifluorescence images of swarming cells and consolidating cells.
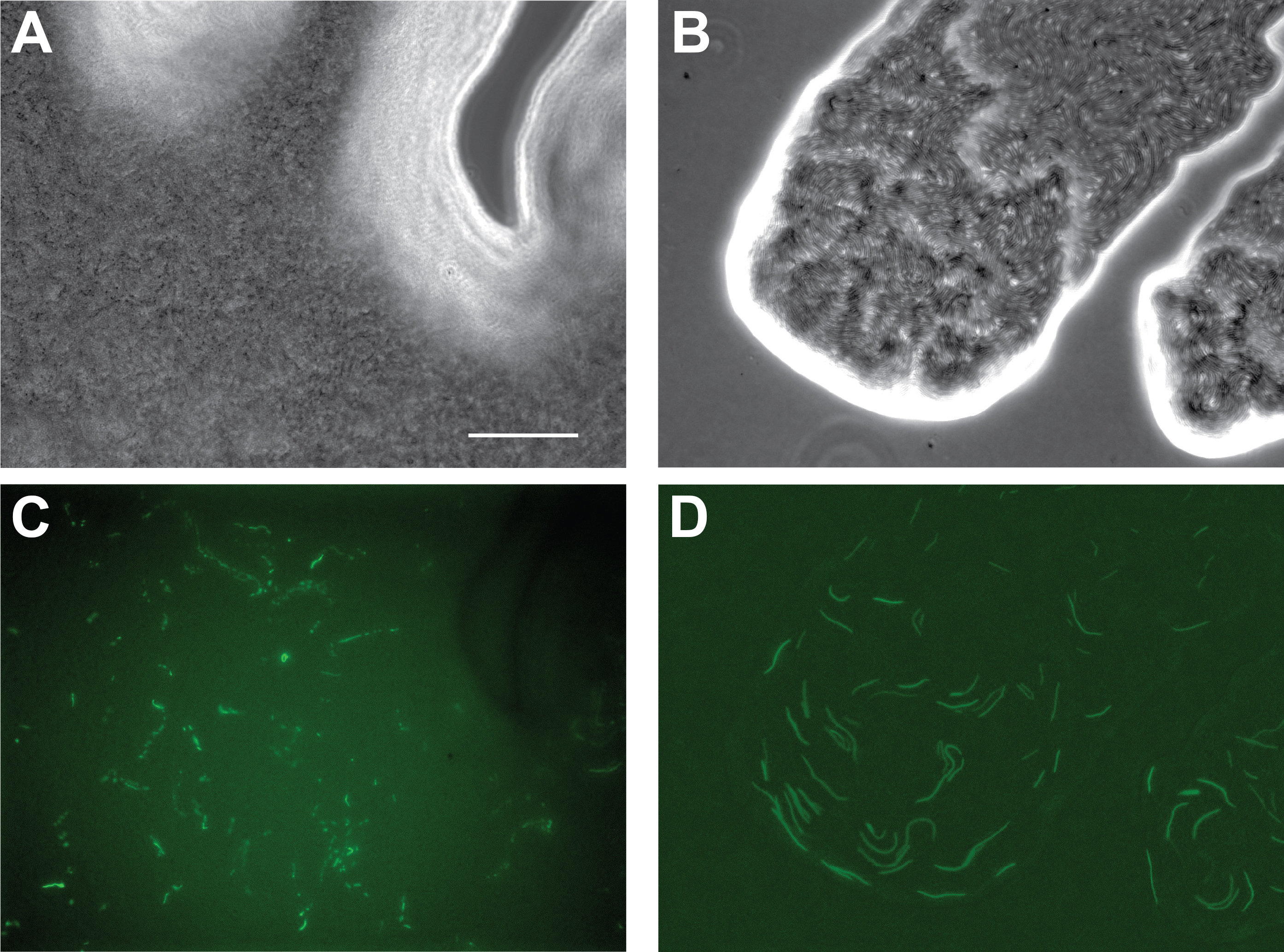
Figure 2. Phase contrast (A, B) and GFP epifluorescence (C, D) microscopy images of consolidating cells (C) and swarming cells (D). Images taken at 40x magnification. Fluorescent cells were added to a non-fluorescent population at a ratio of 1:10 to allow individual cells to be seen. Scale bar = 40 µm.
- Preparing cells for FACS
- Remove and dispose of the inoculum from the rest of the experimental sample by punching through the agar with a disposable pipette tip. It is necessary to discard this subset of cells as it does not take part in self recognition behavior. A demonstration can be seen in Video 1.
 Video 1. Removing inoculum from swarm plates using a pipette tip
Video 1. Removing inoculum from swarm plates using a pipette tip - Wash the remaining cells off the agar plate by pipetting 1-1.5 ml sterile PBS onto the plate and gently lifting cells off the surface with an inoculation plate spreader. Once all cells have been harvested, tilt the plate slightly to pool the PBS/cell mix at one side, then pipette into a 1.5 ml microcentrifuge tube chilled on ice.
- Dilute the cell suspension using ice-cold PBS in a 50 ml conical tube until mixture is barely turbid (approximately OD600 0.05) and decant 2 ml into a chilled FACS collection tube.
- Remove and dispose of the inoculum from the rest of the experimental sample by punching through the agar with a disposable pipette tip. It is necessary to discard this subset of cells as it does not take part in self recognition behavior. A demonstration can be seen in Video 1.
- FACSAria setup
Here we describe the settings for sorting fluorescently labeled bacteria on a BD FACSAria. We assume familiarity with operation of a FACSAria and precalibration of the C&ST, droplet delay, side stream angle and breakoff. Users should receive training in operating the FACSAria prior to performing this assay. We recommend setting up a practice run to determine sorting settings prior to generating experimental samples for downstream analysis.- Turn on FACSAria and controlling PC 30 min prior to experimental start. Load FACSDiva software.
- Install the high pressure 70 PSI nozzle.
- Set Sample Temperature in the FACSDiva Cytometer menu to 4 °C. If the sort collection chamber can be temperature controlled, set that to 4 °C also.
- Insert labeled FACS collection tubes into the sort collection chamber.
- Install the sample into the FACSAria sample injection chamber and load.
- Arrange the FACSDiva workspace so that forward scatter (FSC) and side scatter (SSC) can be charted against one another.
- Set up sorting gates to retain particles with forward scatter (FSC) and side scatter (SSC) of approximately 1,000-50,000 (Figure 3).
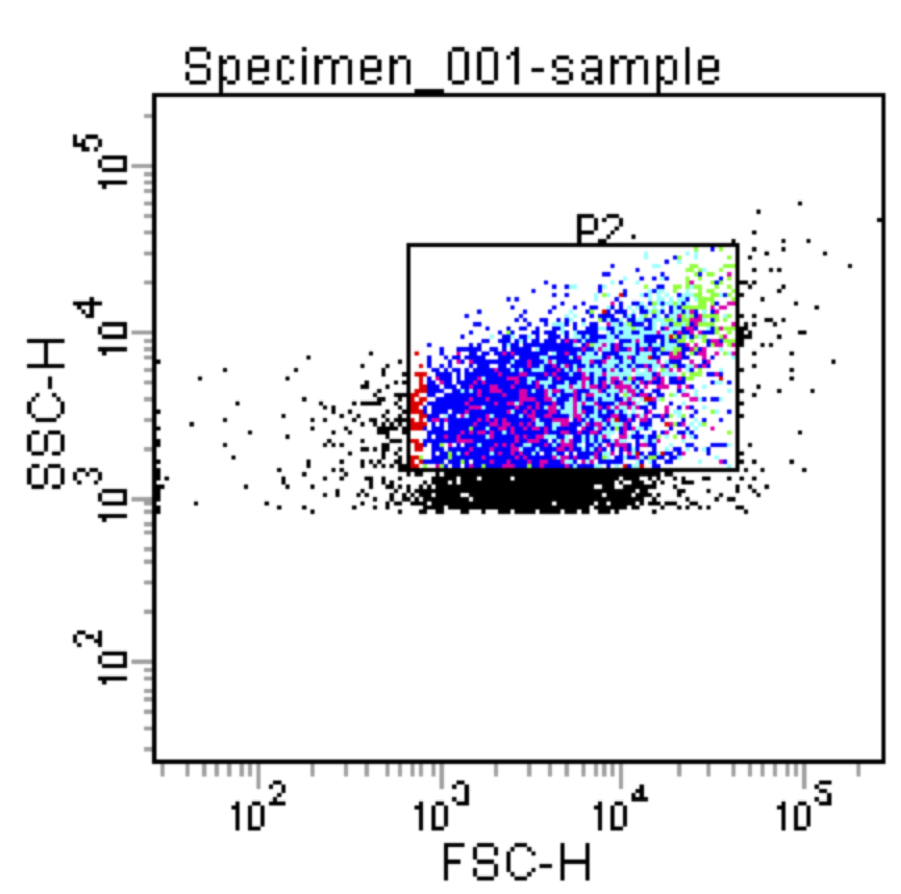
Figure 3. Chart of side-scatter (SSC) vs forward-scatter (FSC) of P. mirabilis particles run through a FACSAria. The area labeled P2 represents a sorting gate that has been set up to isolate single particles from doublets. - Set up a second chart measuring red fluorescence intensity against green intensity: each color corresponds to a strain of interest. Set up sorting gates to isolate particles with fluorescence intensity of approximately 103 or higher (Figure 4).
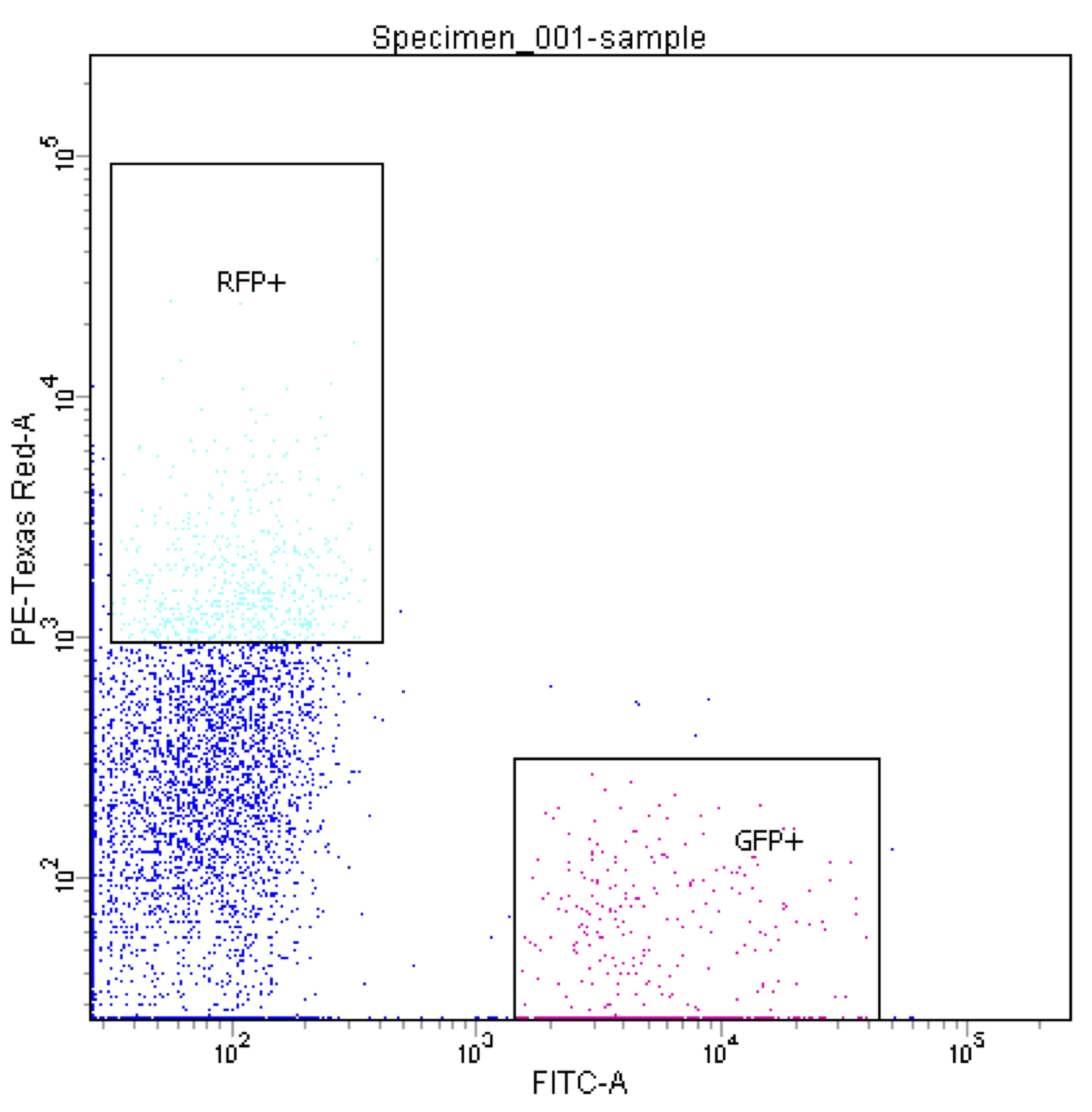
Figure 4. Chart of Red Fluorescent Protein (RFP) intensity (PE-Texas Red-A, Y-axis) versus GFP intensity (FITC-A, X-axis) for single P. mirabilis sorted particles (the P2 subset from Figure 2). RFP+ and GFP+ gates are set up to isolate red and green particles respectively. - In the Sort Layout window, select the relevant gates for the collection tubes, set Precision to “Purity” and Particle Count to 1,000,000.
- Press “Sort” in the Sort Layout window to begin sorting into the collection tubes. Check periodically that the tubes do not overfill. If they do, pause the sort from the Sort Layout window, turn off the deflection plates from the Acquisition Dashboard, swap out the collection tubes, and restart the sort from the Sort Layout window.
- Once the sorted particle count has been reached, unload the sample. To clean the FACSAria, load and run a tube containing 20% bleach solution through the cell sorter for 10 min, then load and run a tube containing Milli-Q water for 10 min.
- Measuring growth recovery following swarming
- Record the number of sorted particles for each condition. Typically, 100,000 to 1,000,000 particles will be sorted. One strain will have a higher sorted particle count than the other.
- Normalize cell mixtures to a standardized concentration (typically 100,000 cells/ml) by adding defined volumes of sorted cell mixture to a defined volume of sterile PBS. Keep normalized cell mixtures on ice.
- Immediately after sorting and normalization, set up growth recovery assays on flat-bottomed 96-well plates. For the Tecan plate reader described here, plates need not be covered or sealed. For each sorted cell mixture, add 50 µl sorted cell mixture to a well containing 250 µl LB broth. If sufficient volume of cell mixture is available, set up technical duplicates in adjacent wells. Run negative control wells containing 300 µl LB broth. Carefully note which mixtures are in which well.
- Turn on Tecan plate reader and the controlling PC. Insert the 96-well plate with samples into the plate reader. Ensure that the 595 nm absorbance filterset is in the filter slot.
- Measure OD595 of wells using the Tecan plate reader. Open the Tecan iControl software and run the following program:
Temperature 37 °C
Plate: Costar 96 Flat Transparent
Part of Plate: Select the wells containing experimental samples
Kinetic cycle: duration 23 h:50 min:0 s- Absorbance measurement 595 nm, 25 flashes
- Shaking 900 s, amplitude 1 mm, linear mode
- Shaking 898 s, amplitude 1 mm, linear mode
- Wait (Timer) 2 s
- The Tecan iControl software will automatically export to an open Excel spreadsheet. Save the resulting files for further analysis.
Data analysis
The Tecan iControl software outputs timecourse files as .xlsx spreadsheet files, which can be analyzed and visualized using most statistics and graphing packages. Typically, growth recovery can be easily visualized by graphing the change in optical density over time for each strain of interest. An example graph, from Tipping and Gibbs (2019), can be seen in Figure 5; here, no difference was observed between strains.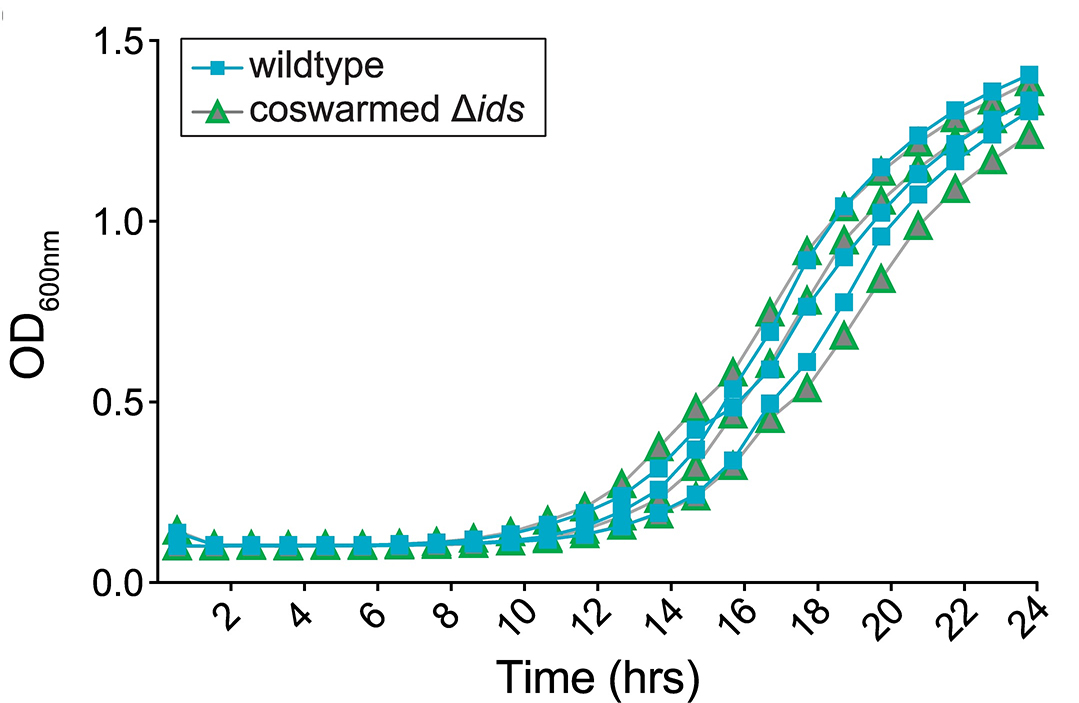
Figure 5. Territorial exclusion does not result in long-term growth defects. Optical density at 600 nm was measured over time for liquid cultures of KAG107 and the coswarmed strain KAG966. Liquid cultures were inoculated using cells isolated by FACS from co-swarm colonies where strain KAG966 was actively excluded from the swarm front. Reprinted with permission from Tipping and Gibbs (2019).
Statistical significance of differences between strains can be measured by calculating Kruskal-Wallis tests for each timepoint. We recommend running at least three biological repeats for each experiment.
Notes
It is important that FACS is begun immediately after harvesting into PBS to avoid variability between samples. Likewise, measurement of growth recovery should be done immediately after sufficient cells have been separated through FACS. Consider growing bacterial cultures and swarm plates in the vicinity of the FACS apparatus to avoid variability caused by transport time. Other FACS apparatus and plate readers can be used for this assay, but the protocol will vary; seek training in equipment prior to use.
Recipes
- LB Broth (1 L)
Tryptone 10 g
Yeast extract 5 g
Sodium chloride 5 g
Milli-Q H2O to 1 L
Mix ingredients in a 2.0 L Erlenmeyer flask, heat with stirring on a hot plate until all ingredients are dissolved, make 250 ml aliquots and autoclave. Recipe adapted from Belas et al. (1991). - CM55 swarm agar (1 L)
Oxoid CM0055 40 g
Milli-Q H2O to 1 L
Mix ingredients in a 2.0 L Erlenmeyer flask, heat with stirring on a hot plate until all ingredients are dissolved, make 250 ml aliquots and autoclave. Recipe adapted from Belas et al. (1991).
Note: CM55 agar is sensitive to repeated reheating: once melted, aliquots should be used to pour plates and the remainder thrown away. - LSW- Agar (1 L)
This media has two components: the main media, and a supplemental mixture that is added immediately before pouring plates. Add 12 ml of supplemental mixture per liter of media.
Media (1 L)
Tryptone 10 g
Yeast extract 5 g
Glycerol (ultrapure) 5 ml
Sodium chloride 0.4 g
Bacto-agar 20 g
Milli-Q H2O to 1 L
Mix ingredients in a 2.0 L Erlenmeyer flask, heat with stirring on a hot plate until all ingredients are dissolved, make 250 ml aliquots and autoclave. Recipe adapted from Belas et al. (1991).
LSW supplemental mixture (50 ml)
1 M MgSO4 solution 2 ml
Glycerol 4 ml
1 % (w/v) nicotinic acid solution 2 ml
Milli-Q water to 50 ml
Add ingredients to a 50 ml conical tube, mix through shaking or vortexing, then sterilize by passing through a 0.22 µm filter into a fresh, sterile conical tube. - Phosphate-buffered saline (PBS)
It is convenient to make this as a 10x concentrated stock for storage. Dilute to the working concentration with sterile Milli-Q water.
10x PBS stock
Sodium chloride 80 g
Potassium chloride 2 g
Sodium hydrogen phosphate dihydrate (Na2HPO4·2H2O) 14.4 g
Potassium dihydrogen phosphate (KH2PO4) 2.4 g
Milli-Q H2O to 1 L- Mix ingredients in a 2.0 L Erlenmeyer flask, make 250 ml aliquots in Duran bottles and autoclave.
- Dilute to working concentration immediately before use. The 10x stock should be pH 6.8, rising to pH 7.4 in the working 1x stock.
- Measure with a pH meter or litmus paper and adjust with concentrated hydrochloric acid (HCl) or 10 M sodium hydroxide solution (NaOH) if necessary. Recipe adapted from Sambrook et al. (1989).
Acknowledgments
We thank the Harvard University Bauer Core Facility, particularly Claire Reardon and Zachary Nizioleck, for assistance with FACS, and Jacob Austerman for swarm plate images. This work was funded by the David and Lucile Packard Foundation, the George W. Merck Fund and Harvard University.
Competing interests
The authors have no competing interests.
References
- Alteri, C. J. and Mobley, H. L. T. (2016). The Versatile Type VI Secretion System. Microbiology spectrum 4(2): 10.1128/microbiolspec.VMBF-0026-2015.
- Aoki, S. K., Webb, J. S., Braaten, B. A. and Low, D. A. (2009). Contact-dependent growth inhibition causes reversible metabolic downregulation in Escherichia coli. Journal of Bacteriology 191(6): 1777-1786.
- Basler, M., Ho, Brian T. and Mekalanos, John J. (2013). Tit-for-Tat: Type VI secretion system counterattack during bacterial cell-cell interactions. Cell 152(4): 884-894.
- Belas, R., Erskine, D. and Flaherty, D. (1991). Transposon mutagenesis in Proteus mirabilis. Journal of Bacteriology 173(19): 6289-6293.
- Brunet, Yannick R., Espinosa, L., Harchouni, S., Mignot, T. and Cascales, E. (2013). Imaging Type VI secretion-mediated bacterial killing. Cell Reports 3(1): 36-41.
- Garcia, E. C., Perault, A. I., Marlatt, S. A. and Cotter, P. A. (2016). Interbacterial signaling via Burkholderia contact-dependent growth inhibition system proteins. Proc Natl Acad Sci U S A 113(29): 8296-8301.
- Gibbs, K. A., Urbanowski, M. L. and Greenberg, E. P. (2008). Genetic determinants of self identity and social recognition in bacteria. Science 321(5886): 256-259.
- Sambrook, J., Fritsch, E. F. and Maniatis, T. (1989). Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Smith, J. M. (1964). Group selection and kin selection. Nature 201(4924): 1145-1147.
- Rauprich, O., Matsushita, M., Weijer, C. J., Siegert, F., Esipov, S. E. and Shapiro, J. A. (1996). Periodic phenomena in Proteus mirabilis swarm colony development. J Bacteriol 178(22): 6525-6538.
- Souza, D. P., Oka, G. U., Alvarez-Martinez, C. E., Bisson-Filho, A. W., Dunger, G., Hobeika, L., Cavalcante, N. S., Alegria, M. C., Barbosa, L. R. S., Salinas, R. K., Guzzo, C. R. and Farah, C. S. (2015). Bacterial killing via a type IV secretion system. Nature Communications 6(1): 6453.
- Tipping, M. J. and Gibbs, K. A. (2019). Peer pressure from a Proteus mirabilis self-recognition system controls participation in cooperative swarm motility. PLOS Pathogens 15(7): e1007885
- Virta, M., Lineri, S., Kankaanpää, P., Karp, M., Peltonen, K., Nuutila, J. and Lilius, E. M. (1998). Determination of Complement-Mediated Killing of Bacteria by Viability Staining and Bioluminescence. Appl Environ Microbiol 64(2): 515-519.
- Wenren, L. M., Sullivan, N. L., Cardarelli, L., Septer, A. N. and Gibbs, K. A. (2013). Two independent pathways for self-recognition in Proteus mirabilis are linked by type VI-dependent export. MBio 4(4). DOI: 10.1128/mBio.00374-13.
Article Information
Copyright
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Tipping, M. J. and Gibbs, K. A. (2020). Growth Recovery Assay and FACS-based Population Sorting Following Territorial Exclusion in Proteus mirabilis. Bio-protocol 10(5): e3543. DOI: 10.21769/BioProtoc.3543.
Category
Microbiology > Microbial signaling > Interspecies communication
Cell Biology > Cell-based analysis > Flow cytometry
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.