- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Construction of Antisense RNA-mediated Gene Knock-down Strains in the Cyanobacterium Anabaena sp. PCC 7120
Published: Vol 10, Iss 4, Feb 20, 2020 DOI: 10.21769/BioProtoc.3528 Views: 6075
Reviewed by: Dennis J Nürnberg Cheng Cai ZhangAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Feb 2017
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols


Abstract
Anabaena sp. PCC 7120 (hereafter Anabaena) is a model cyanobacterium to study nitrogen fixation, cellular differentiation and several other key biological functions that are analogous in plants. As with any other organism, many genes in Anabaena encode an essential life function and hence cannot be deleted, causing a bottleneck in the elucidation of its genomic function. Antisense RNA (asRNA) mediated approach renders the study of essential genes possible by suppressing (but not completely eliminating) expression of the target gene, thus allowing them to function to some extent. Recently, we have successfully implemented this approach using the strong endogenous promoter of the psbA1 gene (D1 subunit of Photosystem II) introduced into a high-copy replicative plasmid (pAM1956) to suppress the transcript level of the target gene alr0277 (encoding a sigma factor, SigJ/Alr0277) in Anabaena. This protocol represents an efficient and easy procedure to further explore the functional genomics, expanding the scope of basic and applied research in these ecologically important cyanobacteria.
Keywords: AntisenseRNA (asRNA)Background
Cyanobacteria, a diverse phylum of aerobic bacteria capable of photosynthesis, require light (solar energy), carbon dioxide and trace elements for growth. They are genetically amenable due to the availability of facile molecular biology techniques and efficient conjugation systems for the transfer of foreign genes into them (Wolk et al., 1984; Elhai et al., 1997). Anabaena is a filamentous cyanobacterium, which is capable of cellular differentiation, wherein specialised cells (termed heterocysts) carry out nitrogen fixation. Classical strategies such as gene knockout to disrupt the function of a “gene of interest” have been widely employed in cyanobacteria in order to understand their function. Following transformation, segregation of the polyploid genome is required to obtain homozygous mutants. In the case of many essential genes (for example, GroEL, LexA), however, the mutants are not viable or cannot segregate completely as the target protein is essential for its survival (Rajaram and Apte, 2008; Kumar et al., 2018). In such cases, an option is to knockdown the genes of interest by targeting them using asRNA (Blanco et al., 2011; Lin et al., 2013) or dCas9-based CRISPR approaches (Tian et al., 2017), and then study the down-regulated/knockdown phenotype.
The dCas9-based approach has its own limitations, for example, in some organisms, expression of the dCas9 protein may be toxic to cells wherein it is expressed (Lee et al., 2016; Zhang and Voigt, 2018). The reason for the toxicity of Cas9 in cyanobacteria remains unclear, nonetheless recently the employment of a novel RNA directed dsDNA nuclease Cpf1 from Francisella novicida showed far less toxicity in cyanobacteria (Ungerer and Pakrasi, 2016; Niu et al., 2018). Although CRISPR-based gene editing approaches are being rapidly optimized, they need further development to be efficiently used in future cyanobacterial gene knockdown procedures. In such cases, it would be more fruitful to use another methodology such as asRNA to create knockdown strains. The asRNA, which is the complement of its respective mRNA, binds specifically to the mRNA (i.e., forms a dsRNA), thereby reducing its ability to be translated by the ribosomal machinery. Due to decreased translation, a reduction in expression of the desired protein (knockdown) is achieved. Recently, we have used asRNA-mediated approach to downregulate a sigma factor SigJ (Alr0277) by using native psbA1 promoter for expression of asRNA and achieved a 3-fold reduction in the sigJ transcript (Srivastava et al., 2017). Similarly, this approach was also employed to downregulate in vivo expression of the Alr3183 protein (an atypical 2 cysteine-containing thiol peroxidase) in Anabaena (Tailor and Ballal, 2017). Here we present the detailed protocol to knockdown genes in Anabaena for applications in basic as well as applied research.
Materials and Reagents
- Vessel and consumable materials
- Pipette tips
- Petri dishes, 100 mm (Thermo Fisher Scientific, catalog number: FB0875713)
- 1.5 ml centrifuge tubes (Thermo Fisher Scientific, catalog number: 02-682-550)
- 15 ml conical centrifuge tubes (Thermo Fisher Scientific, catalog number: 05-527-90)
- 50 ml conical centrifuge tubes (Thermo Fisher Scientific, catalog number: 12-565-270)
- Supor® PES membrane disc filters (Pall corporation, catalog number: 60110)
- Biological materials
- pAM1956, a replicative vector in Anabaena (Yoon and Golden, 1998) (Addgene, plasmid number: 40251)
Note: The map of pAM1956 is available on Addgene; https://www.addgene.org/40251/. - pAM1956-PpsbA1 (control plasmid, available upon request)
- pRL443 (Elhai et al., 1997) (Addgene, plasmid number: 70261)
- pRL623 (Elhai et al., 1997) (Addgene, plasmid number: 58494)
- Anabaena PCC 7120 (can be obtained from Pasteur Culture Collection (PCC), Institut Pasteur, Paris, France)
- E. coli DH5α: F- recA41 endA1 gyrA96 thi-1 hsdR17 (rk- mk-) supE44 relAλ ΔlacU169 (Gibco-BRL)
- E. coli HB101: F- mcrB mrr hsdS20 (rB- mB-) recA13 leuB6 ara-14 proA2 lacY1 galK2 xyl-5 mtl-1 rpsL20 (SmR) glnV44 λ- (Promega, catalog number: L2015)
- E. coli HB101-R2: Donor strain carrying pRL623 (encoding methylases) and pRL443 (conjugal plasmid) (Elhai et al., 1997; Banerjee et al., 2012)
- pAM1956, a replicative vector in Anabaena (Yoon and Golden, 1998) (Addgene, plasmid number: 40251)
- Chemicals for preparing media
- MgSO4·7H2O (Sigma-Aldrich, catalog number: 63138)
- CaCl2·2H2O (Sigma-Aldrich, catalog number: C1016)
- Citric Acid (Sigma-Aldrich, catalog number: 251275)
- Ferric ammonium citrate (Sigma-Aldrich, catalog number: RES20400-A7)
- Na2-EDTA (Sigma-Aldrich, catalog number: E5134)
- Na2CO3 (Sigma- Aldrich, catalog number: 1613757)
- H3BO3 (Sigma-Aldrich, catalog number: B6768)
- Mn2Cl2·4H2O (Sigma-Aldrich, catalog number: 1375127)
- ZnSO4·7H2O (Sigma-Aldrich, catalog number: 1.08881)
- Na2MoO4·2H2O (Sigma-Aldrich, catalog number: M1003)
- CuSO4·5H2O (Sigma-Aldrich, catalog number: C8027)
- CoCl2·6H2O (Sigma-Aldrich, catalog number: 1.02539)
- NaNO3 (Sigma-Aldrich, catalog number: S5506)
- NaHCO3 (Sigma-Aldrich, catalog number: S5761)
- K2HPO4 (Sigma-Aldrich, catalog number: 1551128)
- NaCl (Sigma-Aldrich, catalog number: 63138)
- Tryptone (Casein Hydrolysate) (Sigma-Aldrich, catalog number: 22090)
- Yeast extract (Sigma-Aldrich, catalog number: Y1625)
- Agar (for E. coli) (Sigma-Aldrich, catalog number: A1296)
- Bacto Agar (for Anabaena) (BD Diagonistics, catalog number: 90000-767)
- Co(NO3)2·6H2O (Sigma-Aldrich, catalog number: 239267)
- Sodium thiosulfate pentahydrate (Sigma-Aldrich, catalog number: 217247)
- BG11 medium (see Recipes)
- BG11 agar plate (see Recipes)
- 1 M TES (see Recipes)
- Molecular biology reagents
- Ampicillin sodium salt (Sigma-Aldrich, catalog number: A9393)
- Chloramphenicol (Sigma-Aldrich, catalog number: C0378)
- Kanamycin sulfate (Sigma-Aldrich, catalog number: 60615)
- Neomycin trisulfate salt (Sigma-Aldrich, catalog number: N1876)
- Wizard® SV Gel and PCR Clean-Up System (Promega, catalog number: A9281)
- QIAGEN Plasmid Mini Kit (100) (Qiagen, catalog number: A9281)
- Agarose (Sigma-Aldrich, catalog number: A9539)
- Cfr9I (XmaI) (10 U/μl) (Thermo Fisher Scientific, catalog number: ER0171)
- Reverse Transcriptase (Roche Diagnostics GmbH, catalog number: 38823100)
- AmbionTM DNase I (RNase-free) (Thermo Fisher Scientific, catalog number: AM2222)
- Gibson assembly master mix (NEB, catalog number: E2611L)
- Phenol (Sigma-Aldrich, catalog number: P1037)
- Glycerol (Sigma-Aldrich, catalog number: G5516)
- 8-hydroxyquinoline (Sigma-Aldrich, catalog number: 252565)
- Na2-EDTA (Sigma-Aldrich, catalog number: E5134)
- Sodium acetate (Sigma-Aldrich, catalog number: S2889)
- Guanidine thiocyanate (Sigma-Aldrich, catalog number: G9277)
- Guanidine hydrochloride (Sigma-Aldrich, catalog number: G3272)
- Triton X-100 (Sigma-Aldrich, catalog number: X100)
- PGTX solution (see Recipes; compositions listed points 12-19)
Equipment
- Pipetts
- Tweezers, 120 mm (Ideal-Tek, catalog number: 36A-SA)
- Erlenmeyer flask (Thermo Fisher Scientific, catalog number: FB-500-150)
- NanoDrop (Thermo Fisher Scientfic, catalog number: ND-1000)
- Gel documentation machine (Bio-Rad, Gel Doc 2000 Imaging System)
- Fluorescence microscope, excitation filter BP470/40 nm, emission filter BP 525/50, Leica DFC360FX black and white camera (Leica Camera Inc., catalog number: DM5500B)
- Light microscope, 100/1.3 oil objective, Leica DFC420C camera (Leica Camera Inc., catalog number: DM2500)
- PCR thermocycler (Thermo Fisher Scientific, Veriti® Thermal Cycler)
- UV-Vis spectrophotometer (Analytik Jena AG, Germany, SPECORD® 205)
- Water bath sonicator (Bandelin Souvrex NK51)
- Centrifuge (Eppendorf 5415R Refrigerated Centrifuge)
- Incubator (New BrunswickTM Innova® 43)
- Vacuum filter system (Thermo Fisher Scientific, NalgeneTM 300-4100)
Software
- ImageJ (NIH, USA, https://imagej.nih.gov/ij/)
- Geneious (Biomatters Ltd., https://www.geneious.com)
Procedure
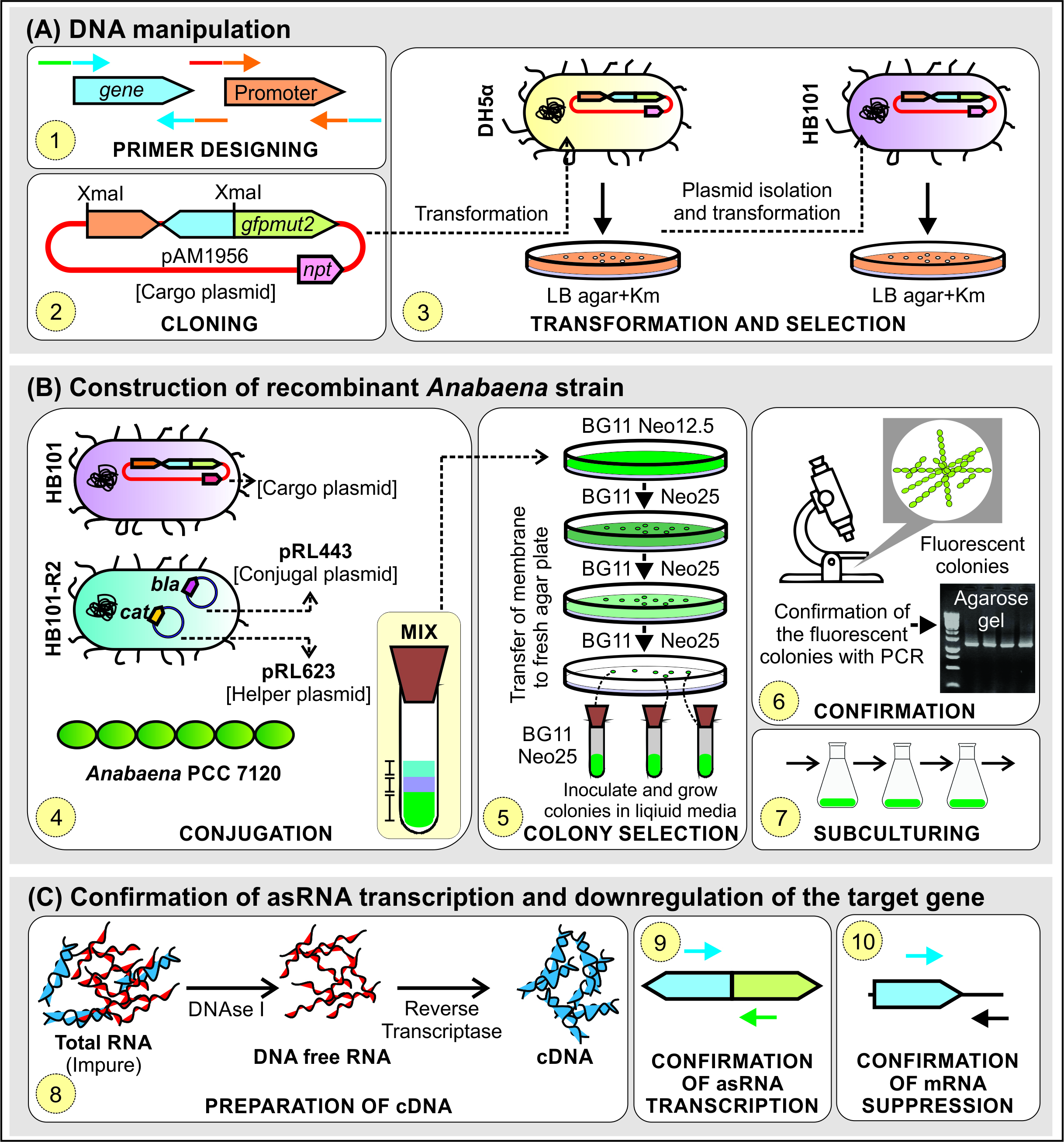
Figure 1. Schematic representation of steps for construction of asRNA mediated gene knockdown mutant in Anabaena. Abbreviations: Km, Kanamycin; Neo12.5, Neomycin (12.5 µg/ml); Neo25, Neomycin (25 µg/ml); npt, neomycin phosphotransferase; bla, beta-lactamase; cat, chloramphenicol acetyltransferase.
Note: The handling of cyanobacterial cultures should be done exclusively under sterile conditions.
- DNA manipulation
- Design primers for (a) promoter e.g., PpsbA1 (alr4866; encoding Photosystem II protein D1) and (b) target gene in reverse orientation (hereafter asGENE) and amplify the DNA fragments by PCR. All the steps in this section are illustrated in Figure 1A. The primers can be designed either manually or using programs such as Geneious (https://www.geneious.com). Primers used for construction and confirmation of knock-down mutant of alr0277 gene in Anabaena (Srivastava et al., 2017) are listed in Table 1.
Note: In this protocol, use of the Gibson assembly, as described by the manufacturer (Gibson et al., 2009), was employed. Primer designing in the Gibson assembly method does not require the addition of restriction sites in primers.
Table 1. Primers used for construction and confirmation of knock-down mutant of alr0277 gene in Anabaena (Srivastava et al., 2017)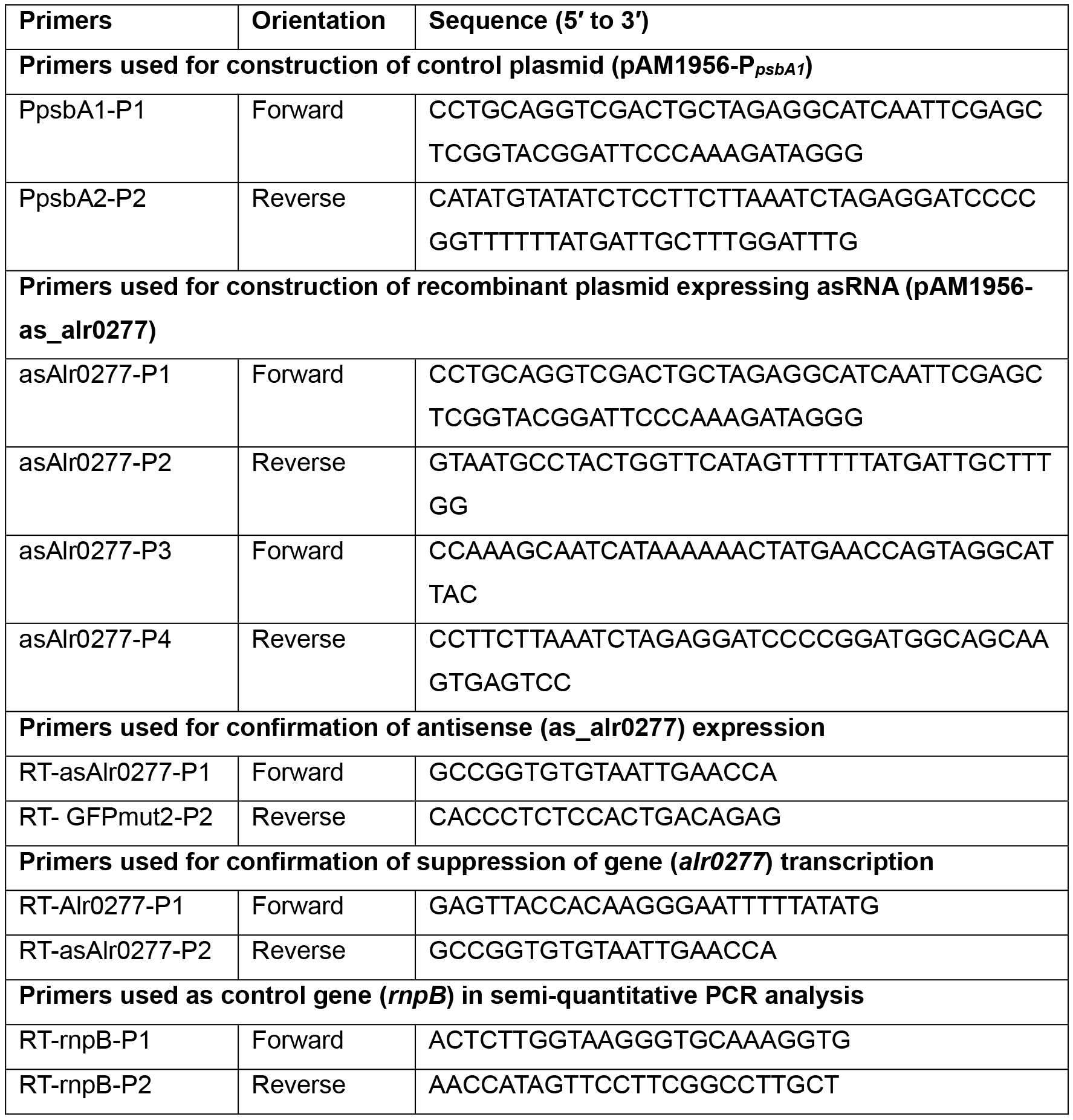
- Linearize the promoterless-gfpmut2 containing replicative shuttle vector pAM1956 (Yoon and Golden, 1998) using the XmaI restriction enzyme.
- Check all three fragments on 1.5% (w/v) agarose gel for the desired size and recover them by gel-purification. Measure the concentration and purity of the purified fragments on a NanoDrop instrument.
- Combine promoter, asGENE and linearized plasmid (pAM1956-XmaI), in the Gibson assembly reaction (contains the enzymes: T5 Exonuclease, Phusion DNA Polymerase and Taq DNA Ligase) and follow the manufacturer's instructions.
- Transform the ligation mix into competent E. coli HB101, spread on a LB agar plate (1.5% agar) supplemented with kanamycin (50 μg/ml) and wait for the colonies to grow overnight at 37 °C. The competent E. coli HB101 cells can be prepared by treatment with CaCl2 (Sambrook et al., 2006).
- Once the colonies have appeared, they can be confirmed by colony-PCR by using the forward primer of PpsbA1 and the reverse primer of asGENE.
- Design primers for (a) promoter e.g., PpsbA1 (alr4866; encoding Photosystem II protein D1) and (b) target gene in reverse orientation (hereafter asGENE) and amplify the DNA fragments by PCR. All the steps in this section are illustrated in Figure 1A. The primers can be designed either manually or using programs such as Geneious (https://www.geneious.com). Primers used for construction and confirmation of knock-down mutant of alr0277 gene in Anabaena (Srivastava et al., 2017) are listed in Table 1.
- Construction of recombinant Anabaena strain
- Grow Anabaena in 25 ml BG11 medium (Rippka et al., 1979) (Recipe 1) under continuous light illumination (40 μmol photons/m2/s) at 30 °C and shaking at 150 rpm for 4-5 days until it is in the exponential phase (OD730 ~0.5). All the steps in this section are illustrated in Figure 1B.
- Collect the cells by centrifugation at 4,000 x g for 10 min at room temperature and resuspend the cell pellet in 25 ml BG11 media in a 150 ml Erlenmeyer flask.
- Break filaments by sonicating the cultures for 60-120 s, using a water bath sonicator to an average 3-5 cell length and confirm it by bright field microscopy.
- Grow the broken filaments for one day under continuous light illumination (40 μmol photons/m2/s) at 30 °C and shaking at 150 rpm.
- On the same day, inoculate the colonies of E. coli HB101 bearing the cargo plasmid (KmR), which has been constructed in the step 5 (section A), and E. coli HB101-R2 harboring pRL443 (ApR) and pRL623 (CmR) in 2 ml LB broth with 50 μg/ml kanamycin and 100 μg/ml ampicillin + 25 μg/ml chloramphenicol respectively for 12-16 h at 37 °C at 200 rpm.
- Next day, sub-inoculate 100 μl of the overnight-grown cultures of the above-mentioned E. coli strains into 5 ml of fresh LB liquid media supplemented with appropriate antibiotics and allow them to grow until they reach an exponential phase (OD600 ~0.5).
- Harvest the 5 ml culture by centrifugation at 4,000 x g in a table centrifuge for 10 min at 25 °C (room temperature).
- Wash the cell pellets three times with 1 ml LB to remove antibiotics completely, and then add 200 μl LB to re-suspend the pellets in both. Do not vortex the cells and if required, use a sterile pipette to gently re-suspend the E. coli cells.
- For the mating experiment, mix 100 μl HB101 bearing the cargo plasmid with 100 μl HB101-R2 (harboring pRL443 and pRL623) and incubate at room temperature for 30 min (experimental group). For negative control, mix 100 μl LB with 100 μl HB101-R2 and incubate at room temperature for 30 min (control group).
Note: We have used an E. coli strain (HB101) that contains both plasmids pRL443 and pRL623, which differs from the original method (Elhai and Wolk, 1988) where one strain contains conjugal plasmid (pRL443), and other strain contains the helper plasmid (pRL623) and the cargo plasmid. - The same day, harvest 15 ml Anabaena culture by centrifugation at 4,000 x g for 10 min at room temperature. Re-suspend the cell pellet in 1 ml BG11, transfer the cells into a 1.5 ml Eppendorf tube, and then centrifuge at 6,000 x g for 1 min at room temperature. Re-suspend the pellet in 400 μl of 2x BG11; then divide cells equally into two tubes.
- Mix 200 μl of Anabaena resuspension with 200 μl of the mated E. coli mixture (experimental group) or the control mixture (control group), respectively, and incubate at room temperature and low light (20 μmol photons/m2/s) overnight.
- Spread the conjugal mixtures onto the autoclaved Supor® PES Membrane Disc Filters (Supor-800, 0.8 μm, 47 mm) placed on BG11 agar plate (1.5% agar, Recipe 2) containing 12.5 μg/ml neomycin and incubate at 30°C, with continuous low light illumination (20 μmol photons/m2/s).
- Transfer the membrane to a fresh BG11 agar plate (25 μg/ml neomycin) every fourth day until neomycin-resistant colonies gradually appear after 15-20 days.
- Observe the Anabaena colonies under a fluorescent microscope and select the colonies emitting GFP fluorescence (100x/1.3 oil objective lens, an excitation filter BP 470/40 nm and emission filter BP 525/50 nm) for further experiments.
- Pick up the desired colony with a sterile inoculation loop or a toothpick and inoculate into a sterile glass tube (3-4 ml total volume) containing 250 μl of BG11 medium with 25 μg/ml neomycin. Keep this tube under continuous low light illumination (20 μmol photons m2/s). After 3-4 days, once the visual increase in growth is observed, add another 250-500 μl of BG11 medium (25 μg/ml neomycin). Once the OD730 of this culture reaches 0.3-0.4, transfer cells into 5 ml of BG11 medium (in a 25 ml sterile glass tube) containing neomycin as mentioned above. Now the tube can be placed under continuous light illumination of 40 μmol photons/m2/s. After sufficient growth is observed (OD730 ~0.6-1.0), cells can be transferred to a 150 ml sterile Erlenmeyer flask containing 25 ml BG11 medium (25 μg/ml neomycin). Now cells can be subcultured as required in the above-mentioned medium for routine maintenance or for performing experiments. All these steps can be performed at 24-26 °C.
- Confirmation of expression of asRNA and downregulation of target gene
- To confirm the expression of asRNA by a semi-quantitative PCR approach, design the forward primer internal to asGENE and reverse primer internal to gfpmut2 gene. To confirm the downregulation of the target gene, design the forward primer from the upstream sequence of the target gene that is present in the genome but absent in the antisense construct so that it can be selectively amplified from cDNA derived from GENE mRNA and not from its asRNA. All the steps in this section are illustrated in Figure 1C.
Note: A regular PCR was used for amplification after the reverse transcriptase reaction. However, qRT-PCR can also be employed to evaluate differences in the expression of mRNA between the wild-type and the knockdown strain, which is a more sensitive technique. Alternatively, if antibodies against the proteins that are knocked down are available, then Western blotting may also be employed to monitor the reduction in the protein content. - Filter 20 ml of exponential phase (OD730 ~0.5) Anabaena culture on the membrane filter (Supor® PES membrane disc filters-supor-800, 0.8 μm, 47 mm) using a vacuum pump assembly for harvesting the cells. Alternatively, cells may be also harvested by centrifugation (4,000 x g, 10 min) and reagents such as TRI Reagent (Sigma)/Trizol (Invitrogen) may be used to isolate total RNA using the manufacturer’s protocol.
- Insert the filter containing cells in the vial containing PGTX solution (see Recipes) using tweezers and vortex until the filter is broken into small flakes and mixed with phenol properly. Keep samples on ice.
- Freeze the samples in liquid nitrogen immediately, store at -80 °C and perform the RNA extraction exactly as described by Pinto et al. (2009).
- Detect the RNA on a denaturing formaldehyde-agarose gel (1.5-2.0% w/v agarose). After staining with ethidium bromide, intact total RNA on migration will show sharp, clear 23S and 16S rRNA bands. Measure the RNA concentration and purity using the NanoDrop instrument as described by the manufacturer.
- Treat the RNA (usually 1:10 dilution of original stock) with the DNase I enzyme and subsequently inactivate the DNase I by DNase inactivation reagent according to the manufacturer’s instruction.
- DNA contamination of RNA preparations is not necessarily detected by gel electrophoresis or similar methods. To test if a detectable amount of genomic DNA is absent, use the diluted RNA sample as a template for PCR using primer pairs for a constitutive gene rnpB. Measure the RNA concentration and purity using the NanoDrop instrument.
- Prepare cDNA using Reverse transcriptase with 1 μg of RNA as template and 0.5 μM of random primers, following the manufacturer's instructions.
- PCR amplify a small fragment of the housekeeping gene rnpB as a control and target gene using specific primers in different PCR reactions, increasing the number of cycles from 25 to 36.
- Detect the PCR products on an agarose gel and document the image.
- Measure the intensity of PCR bands using ImageJ software for comparison of the transcript level.
- To confirm the expression of asRNA by a semi-quantitative PCR approach, design the forward primer internal to asGENE and reverse primer internal to gfpmut2 gene. To confirm the downregulation of the target gene, design the forward primer from the upstream sequence of the target gene that is present in the genome but absent in the antisense construct so that it can be selectively amplified from cDNA derived from GENE mRNA and not from its asRNA. All the steps in this section are illustrated in Figure 1C.
Data analysis
The gene expression data can be analyzed using ImageJ software (NIH, USA, https://imagej.nih.gov/ij/) as following:
- Record the picture of gel with a gel documentation machine and "invert" image color so that the cDNA bands appear black. Open your final gel image file (tiff) in ImageJ software for analysis.
- All the bands in the image should be selected individually. Choose the “rectangular” selection tool to select the widest band as the first band. The height of the selected area should be almost double of this band.
- Mark the selected box as first by pressing CTRL + 1. As a result, number 1 will be displayed inside the selected box.
- Move the pointer (cursor) inside the first selected box, click and drag the box area to the next band in the image.
- Mark the selected box as second by pressing CTRL + 2. Number 2 will be displayed inside the selected box.
- Repeat steps 4 and 5 to select rest of the bands in the image.
- Once all the bands are selected press CTRL + 3 to display the histogram of each band.
- To measure the area below each peak, first separate the histogram peaks by choosing the “draw line” tool to draw a line across the lowest part of each of the histogram. Then choose the “Magic wand” tool and click inside the histogram. The selected area will be outlined in yellow color and a “Result” window will appear.
- The Result window will show the quantitative intensity (value) of the each band.
The size of the peak will be registered as a percentage of the total size of the entire highlighted peaks. The peak percentage of test gene should be divided by the peak percentage of the housekeeping gene (control gene) to demonstrate the relative percentage of expression of each experimental gene. The relative expression should be measured in triplicates and statistical analyses should be performed considering the mean, standard deviation, minimum and maximum of the values of each sample. For details of the ImageJ procedure please refer to the user manual, section 30.13 (Ferreira and Rasb, 2012).
Notes
For appropriate comparison with the knockdown strain that has been constructed as described above, it is recommended to construct a control strain by conjugating into the wild-type Anabaena, a plasmid that contains only the PpsbA1 promoter fused with promoter-less gfpmut2 gene in pAM1956 (i.e., vector control).
Recipes
- BG11 media (modified from Rippka et al., 1979)
Note: This medium is used for growing Anabaena.- Prepare 200 ml of each stock solution, 100 ml of trace element stock solution and 50 ml of 1 M NaHCO3 (Table 2, Table 3 and Table 4 respectively) and sterilize them by autoclaving.
Note: Store citric acid and ferric citrate at room temperature, protected from light; whereas other solutions should be stored at 4 °C. Filter sterilize the trace elements and NaHCO3 stock. - To prepare 1 L BG11 media (1x), add 5 ml of each 200x st.ock solution and 1 ml of trace element stock solution (1,000x) in 964 ml Milli-Q water
Table 2. Stock solutions (200x) for BG11 media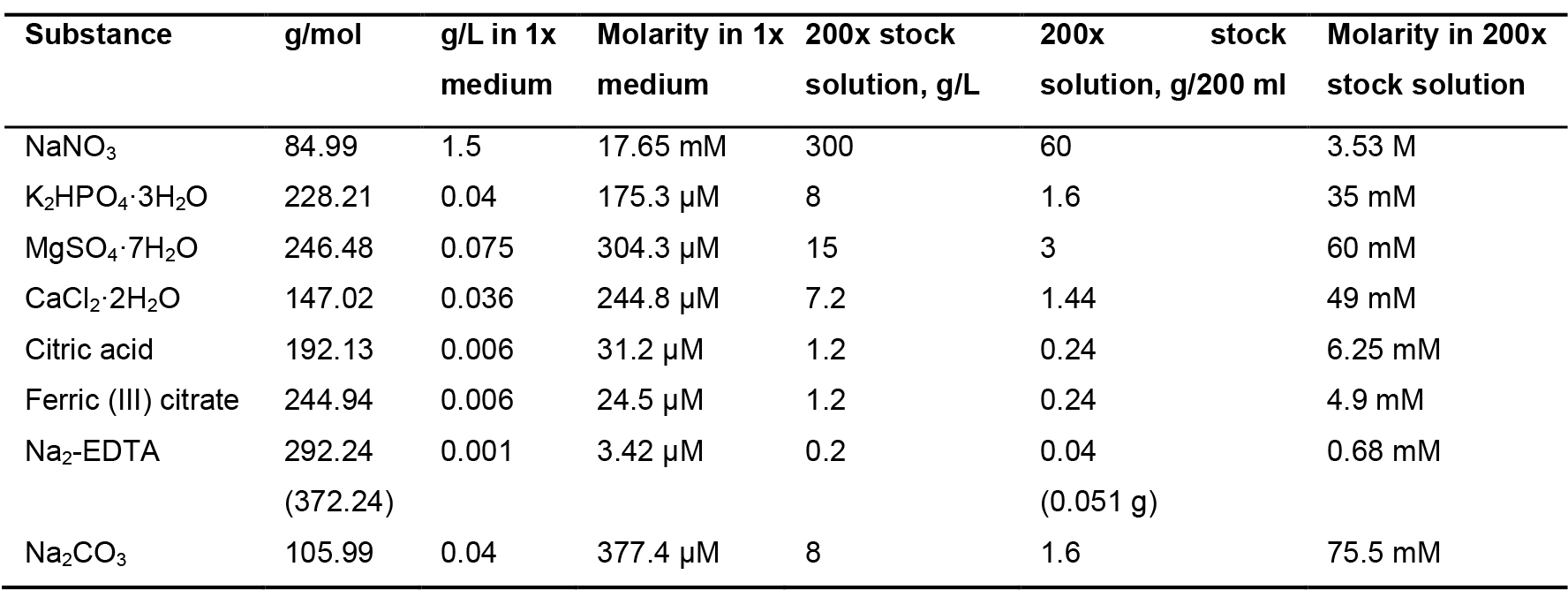
Note: The modified BG11 media includes adjustment in the level of following reagents:
Na2CO3: Modified: 0.04 g/L; Original: 0.02 g/L
Co(NO3)2·6H2O: Modified: 0.078 g/L; Original: 0.0494 g/L
Table 3. Trace elements stock solution (1,000x)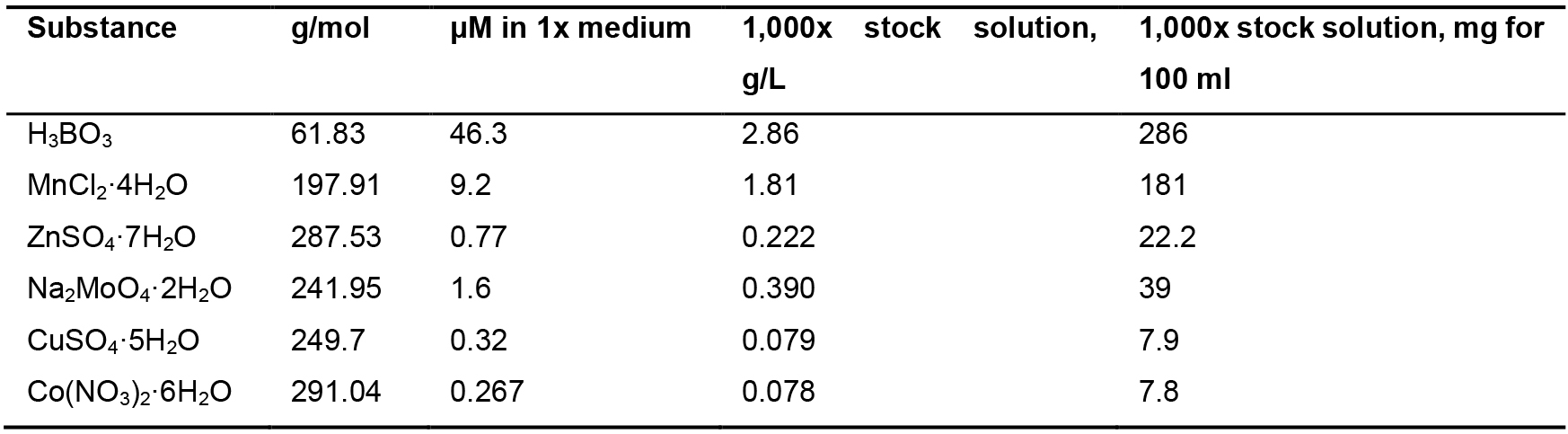
Table 4. 1 M NaHCO3 stock solution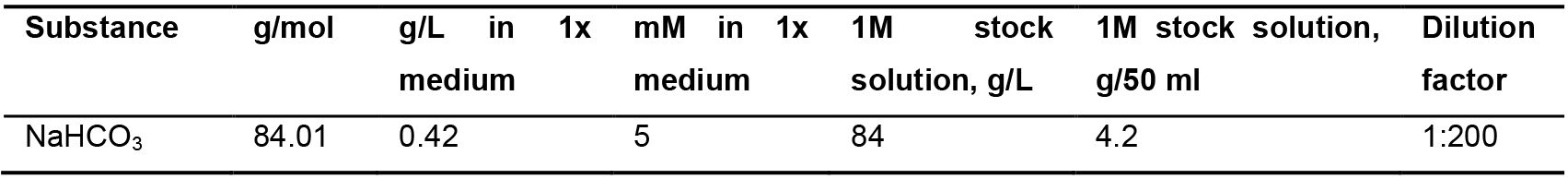
- Prepare 200 ml of each stock solution, 100 ml of trace element stock solution and 50 ml of 1 M NaHCO3 (Table 2, Table 3 and Table 4 respectively) and sterilize them by autoclaving.
- BG11 1.5% (w/v) Agar plates
- To prepare 500 ml of 2x concentrated BactoTM agar in 1,000 ml bottle, add 30 g BactoTM Agar in 500 ml of Milli-Q water
- Then prepare 2x BG11 medium in 500 ml bottle by adding 464 ml Milli-Q water and 5 ml of each 200x stock, 1 ml of trace element stock solution (1,000x)
- In addition, add 3 g/L Sodium thiosulfate pentahydrate and add 10 ml 1 M (1:100) TES, pH 7.6-8.2
- Autoclave both 2x BG11 media and 2x agar
- Combine both solutions when cooled to approx. 60 °C under the sterile hood
- Add NaHCO3 to a final concentration of 5 mM before pouring the plates
- Add antibiotics if desired
- TES buffer
- To prepare 1 M TES buffer, add 229.25 g of TES [N-{Tris(hydroxymethyl)methyl}-2-aminoethanesulfonic acid] in 750 ml of Milli-Q water
- Adjust to pH 8.2 using 10 N NaOH
- Fill to final volume of 1 L with dH2O
- Filter sterilize (recommended) or autoclave
- Store at 4 °C
- PGTX solution (100 ml)
Phenol 39.6 g Glycerol 6.9 ml 8-hydroxyquinoline 0.1 g Na2-EDTA 0.58 g Sodium acetate 0.8 g Guanidine thiocyanate 9.5 g Guanidine hydrochloride 4.6 g Triton X-100 2 ml - Weigh all components of the PGTX solution and add dH2O to 100 ml
- The solution should be stirred at 50-60 °C to create a homogenous solution
- This mixture forms a monophasic solution at room temperature
- The final pH of the solution should reach ~4.2 without any manual adjustment
Acknowledgments
The protocol is based on the publications “Conjugal transfer of DNA to cyanobacteria” (Elhai and Wolk, 1988) and “Down-regulation of the alternative sigma factor SigJ confers a photoprotective phenotype to Anabaena PCC 7120” (Srivastava et al., 2017). This work is supported by the Department of Science and Technology (DST), New Delhi (grant sanctioned to A.K.T., A.B. and A.S.). A.S. is also supported by German Academic Exchange Service (DAAD) and Ministry of Education, Youth and Sports of the Czech Republic (project LO1416).
Competing interests
The authors have no conflicts of interest to declare.
References
- Blanco, N. E., Ceccoli, R. D., Segretin, M. E., Poli, H. O., Voss, I., Melzer, M., Bravo-Almonacid, F. F., Scheibe, R., Hajirezaei, M. R. and Carrillo, N. (2011). Cyanobacterial flavodoxin complements ferredoxin deficiency in knocked-down transgenic tobacco plants. Plant J 65(6): 922-935.
- Elhai, J. and Wolk, C. P. (1988). Conjugal transfer of DNA to cyanobacteria. Methods Enzymol 167: 747-754.
- Elhai, J., Vepritskiy, A., Muro-Pastor, A. M., Flores, E. and Wolk, C. P. (1997). Reduction of conjugal transfer efficiency by three restriction activities of Anabaena sp. strain PCC 7120. J Bacteriol 179(6): 1998-2005.
- Ferreira, T. and Rasb, W. (2012). ImageJ user guide: IJ 1.46 r. (Most recent accessed date: 9/15/2019)
- Gibson, D. G., Young, L., Chuang, R. Y., Venter, J. C., Hutchison, C. A., 3rd and Smith, H. O. (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6(5): 343-345.
- Kumar, A., Kirti, A. and Rajaram, H. (2018). Regulation of multiple abiotic stress tolerance by LexA in the cyanobacterium Anabaena sp. strain PCC7120. Biochim Biophys Acta Gene Regul Mech 1861(9): 864-877.
- Lee, Y. J., Hoynes-O'Connor, A., Leong, M. C. and Moon, T. S. (2016). Programmable control of bacterial gene expression with the combined CRISPR and antisense RNA system. Nucleic Acids Res 44(5): 2462-2473.
- Lin, C. H., Tsai, Z. T. and Wang, D. (2013). Role of antisense RNAs in evolution of yeast regulatory complexity. Genomics 102(5-6): 484-490.
- Niu, T. C., Lin, G. M., Xie, L. R., Wang, Z. Q., Xing, W. Y., Zhang, J. Y. and Zhang, C. C. (2018). Expanding the potential of CRISPR-Cpf1-based genome editing technology in the cyanobacterium Anabaena PCC 7120. ACS Synth Biol 8(1): 170-180.
- Pinto, F. L., Thapper, A., Sontheim, W. and Lindblad, P. (2009). Analysis of current and alternative phenol based RNA extraction methodologies for cyanobacteria. BMC Mol Biol 10: 79.
- Rajaram, H. and Apte, S. K. (2008). Nitrogen status and heat-stress-dependent differential expression of the cpn60 chaperonin gene influences thermotolerance in the cyanobacterium Anabaena. Microbiology 154(Pt 1): 317-325.
- Rippka, R., Deruelles, J., Waterbury, J. B., Herdman, M. and Stainer, R. Y. (1979). Generic assignments, strain histories and properties of pure cultures of cyanobacteria. J Gen Microbiol 111(1): 1-61.
- Sambrook, J. and Russell, D. W. (2006). Preparation and transformation of competent E. coli using calcium chloride. Cold Spring Harb Protoc (1): pdb-prot3932.
- Srivastava, A., Brilisauer, K., Rai, A. K., Ballal, A., Forchhammer, K. and Tripathi, A. K. (2017). Down-regulation of the alternative sigma factor SigJ confers a photoprotective phenotype to Anabaena PCC 7120. Plant Cell Physiol 58(2): 287-297.
- Tailor, V. and Ballal, A. (2017). Novel molecular insights into the function and the antioxidative stress response of a Peroxiredoxin Q protein from cyanobacteria. Free Radic Biol Med 106: 278-287.
- Tian, P., Wang, J., Shen, X., Rey, J. F., Yuan, Q. and Yan, Y. (2017). Fundamental CRISPR-Cas9 tools and current applications in microbial systems. Synth Syst Biotechnol 2(3): 219-225.
- Ungerer, J. and Pakrasi, H. B. (2016). Cpf1 is a versatile tool for CRISPR genome editing across diverse species of cyanobacteria. Sci Rep 6: 39681.
- Wolk, C. P., Vonshak, A., Kehoe, P. and Elhai, J. (1984). Construction of shuttle vectors capable of conjugative transfer from Escherichia coli to nitrogen-fixing filamentous cyanobacteria. Proc Natl Acad Sci U S A 81(5): 1561-1565.
- Yoon, H. S. and Golden, J. W. (1998). Heterocyst pattern formation controlled by a diffusible peptide. Science 282(5390): 935-938.
- Zhang, S. and Voigt, C. A. (2018). Engineered dCas9 with reduced toxicity in bacteria: implications for genetic circuit design. Nucleic Acids Res 46(20): 11115-11125.
Article Information
Copyright
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Srivastava, A., Ballal, A., Forchhammer, K. and Tripathi, A. K. (2020). Construction of Antisense RNA-mediated Gene Knock-down Strains in the Cyanobacterium Anabaena sp. PCC 7120. Bio-protocol 10(4): e3528. DOI: 10.21769/BioProtoc.3528.
Category
Plant Science > Plant molecular biology > DNA > Mutagenesis
Biochemistry > DNA > Single-molecule Activity
Molecular Biology > DNA > Mutagenesis
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.