- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

RNA Sequencing of Single Myofibers from Mus musculus
(*contributed equally to this work) Published: Vol 10, Iss 4, Feb 20, 2020 DOI: 10.21769/BioProtoc.3525 Views: 7286
Reviewed by: Gal HaimovichRuth A. FranklinAnonymous reviewer(s)
Original research article
The authors used this protocol in:
Dec 2019

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Whole transcriptome analysis is a key method in biology that allows researchers to determine the effect a condition has on gene regulation. One difficulty in RNA sequencing of muscle is that traditional methods are performed on the whole muscle, but this captures non-myogenic cells that are part of the muscle. In order to analyze only the transcriptome of myofibers we combine single myofiber isolation with SMART-Seq to provide high resolution genome wide expression of a single myofiber.
Keywords: Single myofiberBackground
Differences in gene expression are key indicators used in studies to see the effect of disease and ageing on cell and tissue types. Switching Mechanism at 5′ end of RNA Templates Sequencing (SMART-Seq) is a powerful technique that enables high resolution sequencing of mRNA from a limited number of cells (Picelli et al., 2014).
Traditionally, RNA sequencing of muscle has been performed on the whole muscle (Pette et al., 1999). However, muscle is a very heterogeneous tissue that is composed of a variety of cell types, such as adipocytes, endothelial cells, mesenchymal cells, and fibroblasts (Arnold et al., 2007; Christov et al., 2007; Joe et al., 2010; Giordani et al., 2019). Adding to this problem is the variety of different types of fibers, with two main groups known as the fast and slow twitch fibers (Brooke and Kaiser, 1970; Pette and Staron, 1997). In order to clearly determine the changes in transcriptome that arise solely from the muscle fiber without the confounding presence of the other cell types, the fiber must be isolated from these cell types.
Here, we combine single myofiber isolation with SMART-Seq to perform a high-resolution sequencing of mRNA at a quality comparable to traditional whole muscle sequencing, while removing the presence of the other non-myogenic cell types. This method will allow researchers to analyze changes in the transcriptome in myofibers under different conditions, such as disease and ageing, and to distinguish the transcriptome of slow and fast fiber types. We successfully implemented this method to analyze the differences in the whole transcriptome of single myofibers isolated from young and old mice.
Materials and Reagents
- Materials
- Pipette tips
- 6-well plate (TC-tested) (Sarstedt, catalog number: 83.3920)
- Glass Pasteur pipettes (VWR, catalog number: 14672-200)
- 1.5 ml Microtubes (Sarstedt, catalog number: 72.690.300)
- 0.2 ml Microtubes (Sarstedt, catalog number: 72.737)
- Magnetic rack (Thermo Fisher, DynaMag-2 Magnet, catalog number: 12321D)
- 0.22 micron filter (UltiDent, catalog number: 229747)
- Large bore glass pipette
- Small bore glass pipette
- Animal
C57BL/6 mouse (Procedure has been successfully performed on mice aged from 1 month to 24 months of age) - Reagents
For digestion of the extensor digitorum longus- PBS (Wisent, catalog number: 311-425-CL)
- Collagenase from Clostridium histolyticum (Sigma, catalog number: C0130-1G)
- DMEM (Invitrogen, catalog number: 11995073)
- Horse Serum (Wisent, catalog number: 065-250)
- Trypsin (Gibco, catalog number: 15090-046)
- Isoflurane (Fresenius Kabi, catalog number: CP0406V2)
- CO2 (Praxair, catalog number: GP-700500)
- Digestion buffer (see Recipes)
- Coating media (see Recipes)
- RNase free water
- RNase inhibitors (from SMART-Seq HT kit, Takara BIO USA, catalog number: 634456)
- SMART-Seq Reaction Buffer (see Recipes)
For cDNA library preparation- SMART-Seq HT kit (Takara BIO USA, catalog number: 634456)
For library purification- Ampure XP beads (Beckman Coulter Life Sciences, catalog number: A63882)
- Ethanol (Commercial Alcohols, catalog number: P016EAAN)
- RNase free water
- Nextera XT DNA Library Preparation kit (Illumina, catalog number: FC-131-1024)
- Nextera XT Index kit (Illumina, catalog number: FC-131-1001)
- Quant-IT Picogreen dsDNA Assay kit (Invitrogen, catalog number: P7589)
For purification and size selection- Ampure XP beads (Beckman Coulter Life Sciences, catalog number: A63882)
- Ethanol (Commercial Alcohols, catalog number: P016EAAN)
- RNase free water
- Quant-IT Picogreen dsDNA Assay kit (Invitrogen, catalog number: P7589)
For quality control- Agarose (Bioshop, catalog number: AGA001.500)
- Tris (Hydroxymethyl) Amino Methane (Multicell, catalog number: 600-125-LG)
- Glacial acetic acid (Fisherbrand, catalog number: 351272-212)
- Na2EDTA·2H2O (Bioshop, catalog number: EDT 001)
- GelGreen Nucleic Acid Gel Stain (Biotium, catalog number: 41005)
- SYBR Green (Applied Biosystems, catalog number: A25742)
- Primers (Integrated DNA Technologies, standard desalting, 25nmole) (Table 1)
- 100 bp DNA ladder (GenedireX, catalog number: DM001-R500)
- TAE Buffer (see Recipes)
Table 1. Primer sequences used for quality control of libraries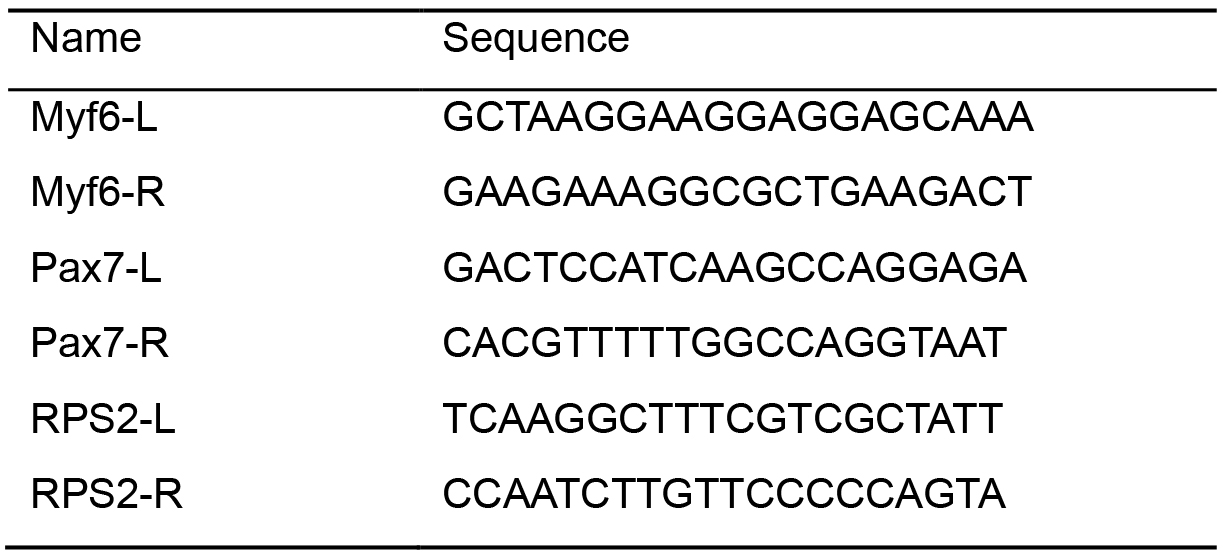
Equipment
- Dissection tools
- Fine point high precision forceps (Fisher Scientific, catalog number: 22-327379)
- Sharp-Pointed dissecting scissors (Fisher Scientific, catalog number: 08-935)
- Pipettes (P20, P200)
- Diamond pen (VWR, catalog number: 201-0392)
- Microscope (Fisher Scientific, inverted microscope, equipped with transmitted light and a 4x objective)
- Centrifuge (Thermo Scientific, Sorval Legend Micro 21R Microcentrifuge, catalog number: 75002447)
- Thermocycler (Bio-Rad, C1000 Touch Thermal Cycler, catalog number: 1851148)
- Mini-centrifuge (Mandel, Benchmark MyFuge Minicentrifuge, catalog number: BEN-C1008-R)
- Gel imager (LI-COR, Odyssey FC Imaging System)
- qPCR machine (Applied Biosystems, QuantStudio7 Flex)
- Bioanalyzer (Agilent, BioAnalyzer 2100)
- Cell incubator (Thermo Scientific, Forma Series II Water-Jacketed CO2 Incubator, catalog number:3110)
- Vortex mixer (Mandel)
- Power supply (Bio-Rad, PowerPac, catalog number: 1645050)
- Microplate reader (BioTek, synergy4)
Procedure
Procedure Overview:
- Dissection and Digestion of Extensor Digitorum Longus (EDL) from Mus musculus (1.5-2 h)
- Myofiber RNA Extraction (30 min)
- cDNA Library Preparation (3 h)
- Purification of cDNA (30 min)
- Preparing Sequencing ready libraries (2.5 h)
- Purification and Size Selection (30 min)
- Quality Control (2-3 h)
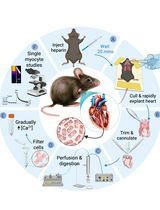
- Dissection and digestion of Extensor Digitorum Longus (EDL) from Mus musculus
- Prepare a digestion buffer of 1,000 U/ml of collagenase in unsupplemented DMEM and filter the buffer with a 0.22 micron filter. 800 µl of digestion buffer is required per EDL muscle.
- Aliquot 800 µl of the digestion buffer to a 1.5 ml tube and place in an incubator, with the caps closed, at 37 °C and 5% CO2 for 30 min before dissection to warm up the buffer and infuse it with CO2.
Notes:- The Myofibers are very sensitive to oxygen, all buffers and media used on them should be first placed in the incubator for at least 30 min. When working outside the incubator do not do so for longer than 5 min at a time if possible. If more time is needed to work on the fibers, allow them to recover in the incubator for 10 min before reusing them.
- Sacrifice the mouse by anesthetizing it with 5% isoflurane, in an induction chamber, and chemically inducing death with CO2, followed by cervical dislocation.
- Remove the skin on the hindlimb of the mouse with a pair of scissors by cutting the skin around the ankle and making an incision along the ventral side of the leg.
- Using a pair of sharp forceps, remove the epimysium covering the Tibialis Anterior (TA) muscle.
- Cut the TA tendon at the ankle, being careful to only cut the top tendon as the bottom tendon belongs to the Extensor Digitorum Longus (EDL) (Figure 1A).
- Using forceps, gently peel off the TA up the leg until you hit the knee, exposing the EDL. Cut out the TA as close to the knee as possible (Figure 1B).
- The biceps femoris is covering the proximal EDL tendon at the patella and must be removed to expose the tendon (Figure 1A).
- Slide a pair of sharp forceps underneath the EDL and gently run it along its length to ensure that both tendons are exposed and accessible (Figure 1B).
- Cut the distal EDL tendon at the ankle. Lift the EDL upright by the tendon with a pair of forceps and, without pulling too hard, cut the proximal tendon as close to the knee as possible.
Notes:- It is very important to cut the EDL at the tendons, cutting into the muscle will result in most of the fibers being lost.
- It should take about 5 minutes to dissect an EDL.
- A video demonstrating the EDL isolation procedure has been made available by Pasut et al. (2013).
- Place the dissected EDL into the digestion buffer and incubate for an hour in the incubator, with the cap closed, while periodically inverting the tube to mix the solution. Trypsin can be added to the digestion buffer at a final concentration of 0.25% at this moment if the removal of fiber-associated satellite cells is needed.
- During the digestion process, coat a 6-well plate with enough coating media (10% horse serum (HS) in unsupplemented DMEM) to cover the bottom of the wells (approximately 3-4 ml), for at least 30 min.
Note: Myofibers are very sticky and will become stuck to any surface that is not coated with horse serum. - Replace the coating media with 2 ml of unsupplemented DMEM and place the plate in the incubator for at least 30 min.
- Coat a large bore glass pipette with HS and rinse with coating media. Use the large bore pipette to transfer the EDL from the digestion buffer to the 6-well plate, being careful to transfer as little of the digestion buffer as possible with it.
Notes:- At this point, the EDL muscle is still intact and will be at the bottom of the tube. It helps to invert the tube to dislodge the EDL from the bottom and then use the large bore pipette to pick up the EDL. The EDL will sink to the tip of the pipette and can then be transferred to the coated 6-well plate in a single drop of digestion buffer.
- A large bore glass pipette can be made by cutting a Pasteur pipette with a diamond pen where the area begins to widen, and then flame polishing the edges with a Bunsen burner (Figure 1C).
- Disassociate the fibers by gently pipetting the EDL up and down with the large bore pipette. Transfer the EDL to subsequent wells as fibers are isolated to avoid damaging them (Figures 1D-1E).
Note: Typically over 300 intact fibers will be isolated from a single EDL.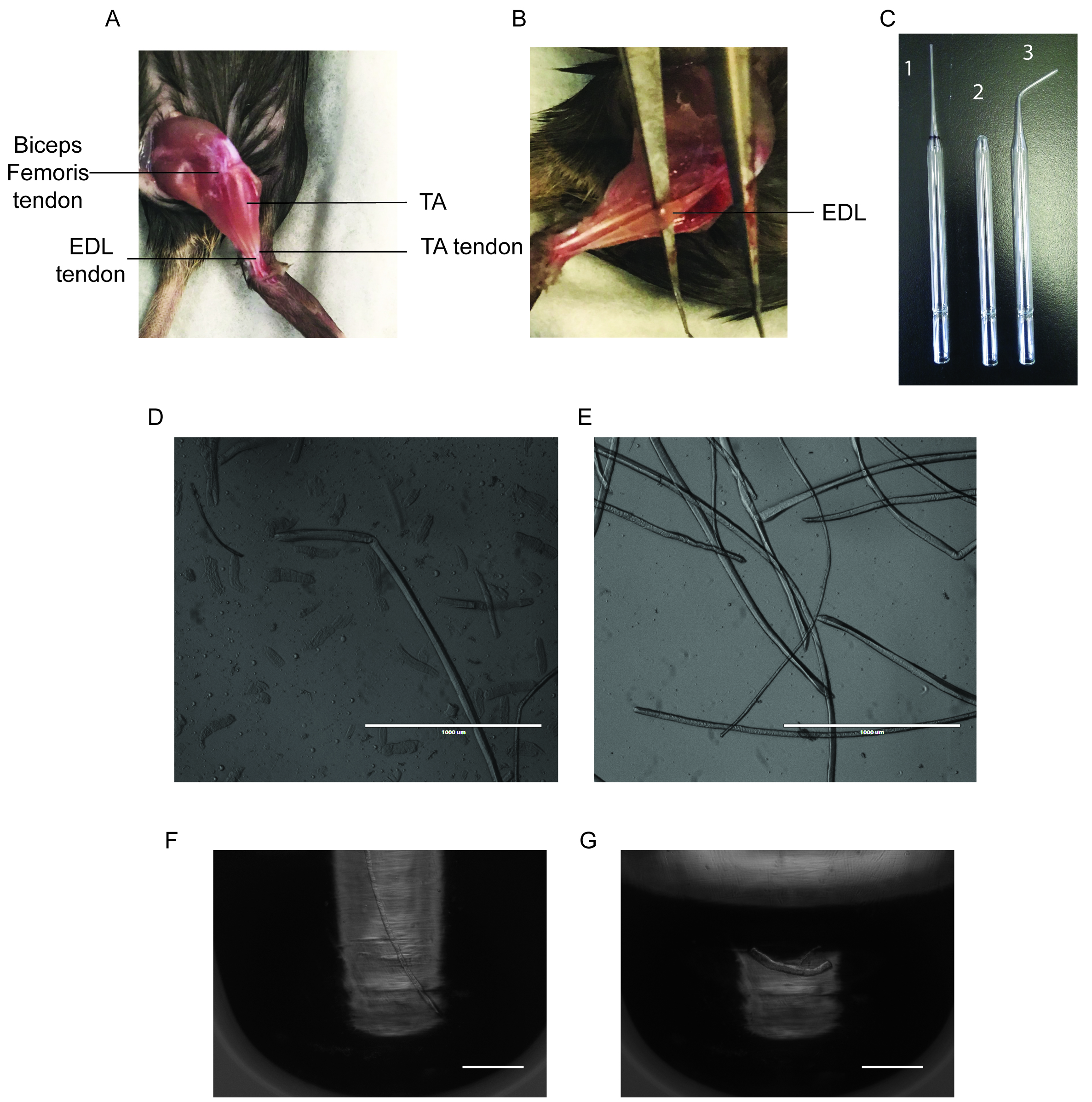
Figure 1. Representative Results of Fiber Isolation. A. Photo of the hindlimb of a 5-week old mouse, highlighting the location of the TA muscle and the tendons of the biceps femoris, TA and EDL muscles. B. Photo of the hindlimb of a 5-week old mouse, with the TA removed exposing the EDL muscle. C. Photo of glass pipettes used for the disassociation of the EDL myofibers. 1. Uncut glass pipette, the black band indicates where to cut with a diamond pen. 2. Large bore glass pipette. 3. Small bore glass pipette. D. Microscope image of an unsuccessful myofiber isolation. E. Microscope image of a successful myofiber isolation. F. Microscope image of a live fiber in a microtube. G. Microscope image of a single myofiber in a microtube after RNA extraction. Scale bars = 1 mm (in D and E). Scale bars = 200 µm (F and G)
- RNA Extraction from a Single Myofiber
- Coat a 6-well plate with coating media and then replace it with 2 ml of PBS. Place the plate in the incubator for at least 30 min.
- Coat a small-bore glass pipette with HS and rinse with coating media. Use the small-bore pipette to transfer fibers to the PBS filled 6-well plate (Figure 1C).
Note: A small bore glass pipette can be made by using a Bunsen burner to bend the thin portion of a Pasteur pipette into a curve. Flame polish the edges of the opening (Figure 1C). - Using a small-bore pipette under a microscope, transfer a single fiber to a 200 µl PCR tube and remove the PBS with a P200 pipette(Figure 1F). Visually confirm that only a single fiber is in the PCR tube by observing the tube under a microscope.
- Prepare a 10x SMART reaction buffer (Recipe 3).
- Lyse the fiber with 10 µl of 1x reaction buffer. Pipette the fiber up and down for 3 min with a P20 pipette. Put the tube on ice for 5 min and vortex as needed (Figure 1G).
Note: The fiber will never fully breakdown, but it does so enough for the RNA to be extracted in sufficient quantity. - To remove the residual fiber pieces, spin down the sample and, while visualizing the fiber pieces under a microscope, transfer the supernatant to a new 200 µl PCR tube.
- Preparing cDNA library
- Bring the volume of the sample up to 11.5 µl with RNase free water.
- Using the DNA SMART-Seq HT kit perform the following steps:
a.Add 1 µl of 3′ SMART-Seq CDS primer II A to the sample.
b.Mix by pipetting or gently vortexing and spin down. - Incubate for 3 min at 72 °C, then place immediately on ice.
- Prepare template switching master mix. For one reaction use the following reagents: 0.7 µl nuclease free water, 8 µl one-step buffer, 1 µl SMART-Seq HT oligonucleotide, 0.5 µl RNase inhibitor, 0.3 µl SeqAmp DNA polymerase, 2 µl SMARTscribe reverse transcriptase, total volume 12.5 µl per reaction.
Note: Add the enzymes last to the master mix and use immediately. - Add 12.5 µl per reaction. Mix and spin down.
- Place samples in a thermal cycler using the following settings:
42 °C 90 min
95 °C 1 min
98 °C 10 s65 °C 30 s
 10-12 cycles
10-12 cycles 68 °C 3 min
72 °C 10 min
4 °C forever
Note: This is a stopping point. cDNA can be left at 4 °C overnight or at -20 °C until purification.
- cDNA Purification
- Add AMPure XP beads directly into the PCR tube at a 1:1 (v/v) ratio. Mix thoroughly and allow the DNA to bind to the beads at room temperature for 8 min (Figure 2A).
Note: Allow AMPure XP beads to warm to room temperature before pipetting. Vortex AMPure XP beads vigorously before use. - Place the tubes on a magnetic strip and wait for the solution to clear, approximately 5 min (Figure 2B).
- Using a P200 pipette, discard the supernatant while being careful to not remove the magnetic beads. Wash the beads twice with 200 µl of 80% ethanol for 20 s.
Note: On the last wash, make sure to remove all the ethanol and use an aspirator to remove any residual droplets on the side of the tube, if necessary. - Elute the DNA using 17 µl of elution buffer provided with the SMART-Seq HT kit. Add the buffer to the tube, mix thoroughly until the beads go into solution, and incubate at room temperature for 5 min.
Note: Be careful not to allow your beads to over dry. When dried the DNA becomes difficult to elute, resulting in low yield. If the beads are dry place them in elution buffer at 45 °C and vortex periodically until no clumps are visible and the beads do not settle at the bottom of the tube. - Place the tubes on a magnetic strip until the solution clears and keep the supernatant (Figure 2B).
Note: This is a stopping point. cDNA can be kept at 4 °C or -20 °C.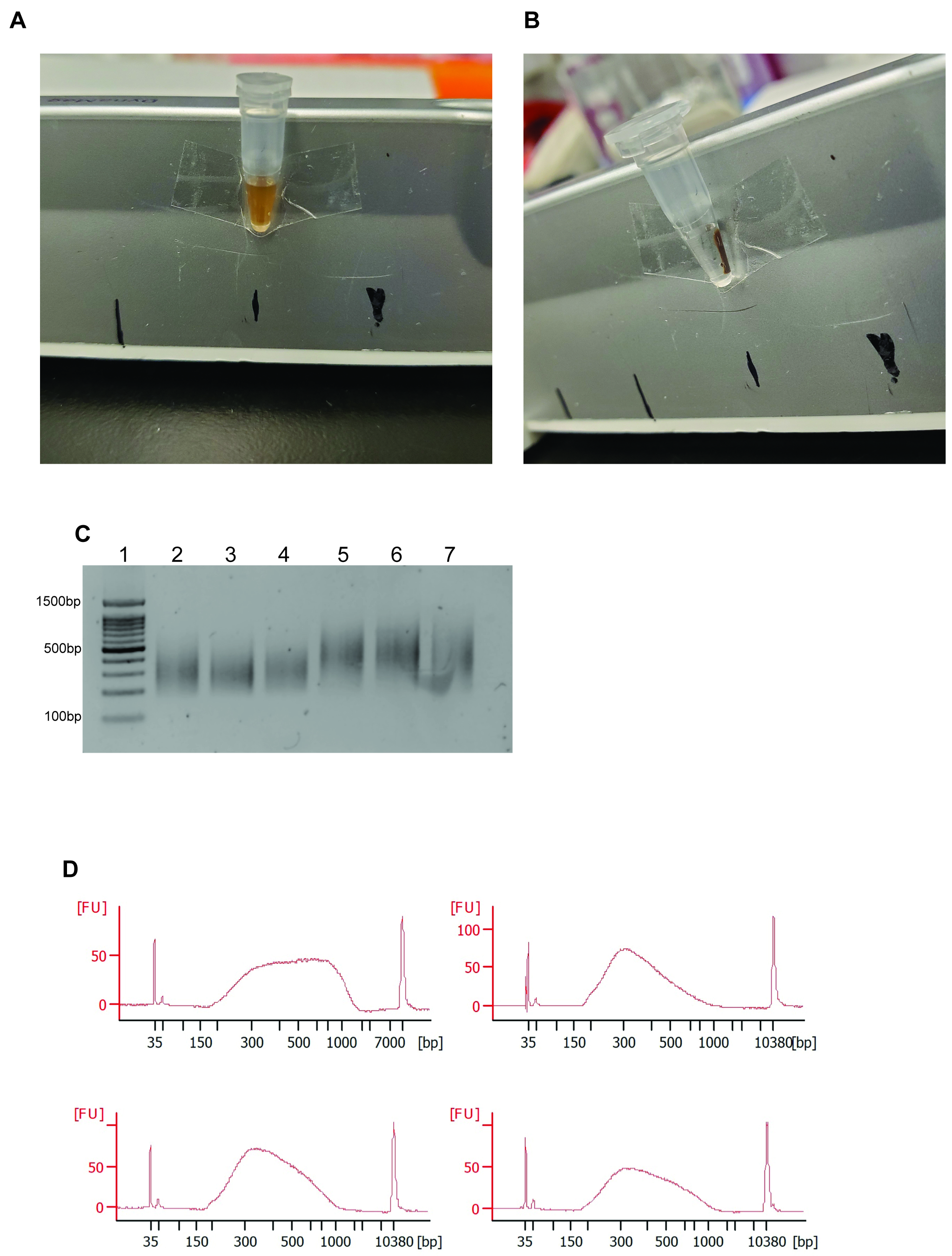
Figure 2. Representative Results in Preparing Sequencing Ready Libraries. A. Photo of a 0.2 ml microtube with AMPure beads in solution. B. Photo of a 0.2 ml microtube with AMPure beads separated out of solution with a magnetic rack. C. Agarose gel of appropriately-sized sequencing ready libraries. A 100 bp DNA ladder is shown in lane 1. D. Typical bioanalyzer profiles of RNA-Seq libraries made from single fibers.
- Add AMPure XP beads directly into the PCR tube at a 1:1 (v/v) ratio. Mix thoroughly and allow the DNA to bind to the beads at room temperature for 8 min (Figure 2A).
- Preparing Sequencing ready libraries
- Quantify the samples using Quant-IT PicoGreen dsDNA assay kit.
Note: Typically we obtain a total cDNA yield between 20 and 100 ng. - Prepare 250 pg of single fiber cDNA in 1.25 µl of water in a microtube.
Note: It is important to accurately quantify the cDNA concentration. The amount of starting material can alter the library fragment size. Too much DNA results in larger fragments while too little gives small fragments that will be lost during size selection. - Using the Nextera XT DNA Library Preparation kit, add to the mix 2.5 µl of TD buffer, and 1.25 µl of ATM, mix and spin down.
- Incubate at 55 °C for 5 min.
- Remove from heat and add 1.25 µl of NT, mix and spin down.
Note: Add NT buffer directly after the tagmentation reaction (Step E4) to deactivate the Tn5 transposase, since the enzyme is still active at this point. - Incubate the sample for 5 min at RT.
- To amplify your libraries, add 1.25 µl of an i7 adaptor and 1.25 µl of an i5 adaptor and 3.75 µl of NPM. Vortex and spin down.
Note: When incorporating sequencing adapters, care must be taken to make sure the choice of i5 and i7 adaptors is color-balanced for the type of sequencer used (two vs four channel). Briefly, the goal is to have signal in both color channels (red and green) at each nucleotide position for the pool of adapters. For example, on a NextSeq 500, which is a two-channel system, T appears as red, C appears as green, A appears in both color channels and G is colorless. In this case, at a single nucleotide position, a G must be pooled with at least an A, or a mix of T and C. In the case of a four-channel system, the same rules apply, but A and C both appear as red, and T and G both appear as green. For further information of sequencing pooling on both two- and four-channel systems, visit:
https://support.illumina.com/content/dam/illumina-support/documents/documentation/chemistry_documentation/experiment-design/illumina-adapter-sequences-1000000002694-11.pdf
https://support.illumina.com/content/dam/illumina-support/documents/documentation/chemistry_documentation/experiment-design/index-adapters-pooling-guide-1000000041074-05.pdf - Run the PCR program using the following steps:
72 °C for 3 min
95 °C for 30 s
95 °C for 10 s55 °C for 30 s
12 cycles 72 °C for 30 s
72 °C for 5 min
10 °C forever
Note: This is a stopping point.
- Quantify the samples using Quant-IT PicoGreen dsDNA assay kit.
- Purification and size selection
- Add AMPure XP beads directly into the PCR tube at a 0.85x (v/v) ratio to the volume of sample. Mix thoroughly and incubate at room temperature for 8 min.
Note: This ratio will remove fragments smaller than 200 bp. - Place the tubes on a magnetic strip and wait for the solution to clear, approximately 5 min.
- Discard the supernatant. Wash the beads twice with 200 µl of 80% ethanol for 20 s.
Note: On the last wash, make sure to remove all the ethanol and use an aspirator to remove droplets on the side of the tube, if necessary. - Elute the DNA fragments by adding 20 µl of resuspension buffer that is provided with the Nextera XT kit and mixing thoroughly until the beads go into solution. Incubate at room temperature for 5 min.
- Place the tubes on the magnetic strip until the solution clears and keep the supernatant.
- Quantify your samples using Quant-IT PicoGreen dsDNA assay kit.
Note: Typically we have a final concentration between 4-7 ng/µl. - Store at -20 °C until ready for sequencing.
- Add AMPure XP beads directly into the PCR tube at a 0.85x (v/v) ratio to the volume of sample. Mix thoroughly and incubate at room temperature for 8 min.
- Quality control
- Run a qPCR of marker genes after the preparation of the cDNA libraries to assess the quality of the RNA.
Note: Here we used Myf6 as a positive control and Pax7 as a negative control. However, any genes that are expressed in myofibers can be used as a positive control and any non-expressed genes can be used for a negative control. - To verify tagmentation and library size, run 20 ng of sample on a 1.25% agarose gel with dsGreen (Figure 2C).
Note: Run only 2 µl of DNA ladder that has been diluted 1:2 before use to prevent washing out the signal from the library. - Run the sequencing ready libraries on a bioanalyzer to assess the size of the libraries and the concentration (Figure 2D).
- Run a qPCR of marker genes after the preparation of the cDNA libraries to assess the quality of the RNA.
Data analysis
Follow standard RNA-Seq analysis pipeline.
Recipes
- Digestion buffer
- Dissolve collagenase in DMEM at a concentration of 1,000 U/ml
- Filter the solution with a syringe and 0.22 µm filter
- Add trypsin to a final concentration of 0.25%
- Use fresh and do not store
- Coating media
- Add horse serum (HS) to DMEM at a final concentration of 10%
- Coating media can be stored at 4 °C and reused multiple times
- SMART-Seq reaction buffer
- Prepare 10x reaction buffer by adding 1 µl of RNase inhibitor to 19 µl 10x lysis buffer which is provided with the SMART-Seq kit
- Prepare 1x buffer by adding 1 µl of 10x reaction buffer to 9 µl of RNase free water
- TAE buffer
- Prepare 50x stock solution by dissolving 242 g of tris base, 57.1 ml of glacial acetic acid, and 41.2 g of Na2EDTA·2H2O in 1 L of dH2O
- Dilute 50x stock solution to 1x working solution
Acknowledgments
Funding for this project was provided by grants from the Canadian Institute of Health Research (CIHR) [PJT-15087] and a Natural Sciences and Engineering Council (NSERC) discovery grant to VDS.
This protocol benefited from Pasut et al., 2013 for isolation of single myofibers and Picelli et al. (2014) for SMART-Seq technology.
Competing interests
The authors have no conflict of interest to declare.
Ethics
Animal experimentation: all procedures used on animals were approved by the McGill University Animal Care Committee (UACC) (protocol #2014-7512, valid 07/01/2017 to 07/01/2020).
References
- Arnold, L., Henry, A., Poron, F., Baba-Amer, Y., van Rooijen, N., Plonquet, A., Gherardi, R. K. and Chazaud, B. (2007). Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J Exp Med 204(5): 1057-1069.
- Brooke, M. H. and Kaiser, K. K. (1970). Three "myosin adenosine triphosphatase" systems: the nature of their pH lability and sulfhydryl dependence. J Histochem Cytochem 18(9): 670-672.
- Christov, C., Chretien, F., Abou-Khalil, R., Bassez, G., Vallet, G., Authier, F. J., Bassaglia, Y., Shinin, V., Tajbakhsh, S., Chazaud, B. and Gherardi, R. K. (2007). Muscle satellite cells and endothelial cells: close neighbors and privileged partners. Mol Biol Cell 18(4): 1397-1409.
- Giordani, L., He, G. J., Negroni, E., Sakai, H., Law, J. Y. C., Siu, M. M., Wan, R., Corneau, A., Tajbakhsh, S., Cheung, T. H. and Le Grand, F. (2019). High-dimensional single-cell cartography reveals novel skeletal muscle-resident cell populations. Mol Cell 74(3): 609-621 e606.
- Joe, A. W., Yi, L., Natarajan, A., Le Grand, F., So, L., Wang, J., Rudnicki, M. A. and Rossi, F. M. (2010). Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat Cell Biol 12(2): 153-163.
- Pasut, A., Jones, A. E. and Rudnicki, M. A. (2013). Isolation and culture of individual myofibers and their satellite cells from adult skeletal muscle. J Vis Exp (73): e50074.
- Pette, D., Peuker, H. and Staron, R. S. (1999). The impact of biochemical methods for single muscle fibre analysis. Acta Physiol Scand 166(4): 261-277.
- Pette, D. and Staron, R. S. (1997). Mammalian skeletal muscle fiber type transitions. Int Rev Cytol 170: 143-223.
- Picelli, S., Faridani, O. R., Bjorklund, A. K., Winberg, G., Sagasser, S. and Sandberg, R. (2014). Full-length RNA-seq from single cells using Smart-seq2. Nat Protoc 9(1): 171-181.
Article Information
Copyright
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Blackburn, D. M., Lazure, F. and Soleimani, V. D. (2020). RNA Sequencing of Single Myofibers from Mus musculus. Bio-protocol 10(4): e3525. DOI: 10.21769/BioProtoc.3525.
- Blackburn, D. M., Lazure, F., Corchado, A. H., Perkins, T. J., Najafabadi, H. S. and Soleimani, V. D. (2019). High-resolution genome-wide expression analysis of single myofibers using SMART-Seq. J Biol Chem 294(52): 20097-20108.
Category
Developmental Biology > Cell growth and fate > Myofiber
Molecular Biology > RNA > RNA sequencing
Cell Biology > Cell isolation and culture > Cell isolation
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.