- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Super-resolution Microscopy-based Bimolecular Fluorescence Complementation to Study Protein Complex Assembly and Co-localization

(*contributed equally to this work) Published: Vol 10, Iss 4, Feb 20, 2020 DOI: 10.21769/BioProtoc.3524 Views: 6560
Reviewed by: David PaulSijie WeiYosuke Senju
Original research article
The authors used this protocol in:

May 2019
Advertisement




Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Numerous experimental approaches exist to study interactions between two subunits of a large macromolecular complex. However, most methods do not provide spatial and temporal information about binding, which are critical for dissecting the mechanism of assembly of nanosized complexes in vivo. While recent advances in super-resolution microscopy techniques have provided insights into biological structures beyond the diffraction limit, most require extensive expertise and/or special sample preparation, and it is a challenge to extend beyond binary, two color experiments. Using HyVolution, a super-resolution technique that combines confocal microscopy at sub-airy unit pinhole sizes with computational deconvolution, we achieved 140 nm resolution in both live and fixed samples with three colors, including two fluorescent proteins (mTurquoise2 and GFP) with significant spectral overlap that were distinguished by means of shifting the excitation wavelength away from common wavelengths. By combining HyVolution super-resolution fluorescence microscopy with bimolecular fluorescence complementation (SRM-BiFC), we describe a new assay capable of visualizing protein-protein interactions in vivo at sub-diffraction resolution. This method was used to improve our understanding of the ordered assembly of the Saccharomyces cerevisiae spindle pole body (SPB), a ~1 giga-Dalton heteromeric protein complex formed from 18 structural components present in multiple copies. We propose that SRM-BiFC is a powerful tool for examination of direct interactions between protein complex subunits at sub-diffraction resolution in live cells.
Keywords: Protein complex assemblyBackground
Protein complexes execute fundamental functions involved in various biological processes. To have these molecular machines assembled in a timely manner in a precise cellular location is critical for cellular activities, including protein synthesis, chromosome segregation, nuclear transport, signaling, motility and other diverse functions. Typically, the formation of many multimeric complexes is thought to occur in a stepwise manner via recruitment and integration of subunits or subcomplexes, which are bridged to existing molecules within the larger assembly by a physical protein-protein contact. Understanding the assembly of large macromolecular complexes is historically the job of biochemists and crystallographers who employ an arsenal of techniques: purification of complexes to determine the identity of subunits, co-purification of proteins, yeast two-hybrid screening, in vitro binding assays, chemical cross-linking studies and structural analysis to understand protein-protein interactions, and finally, reconstitution of the complex to determine the hierarchy of assembly.
While this traditional approach has been successful in many cases, a major challenge is incorporating knowledge from studies on a handful of proteins to the larger and more relevant problem of macromolecular complex assembly within the cell. In cells, the timing of protein production, protein localization and post-translational modifications affect distribution, local concentrations and interactors that in turn influence complex assembly. A number of imaging-based techniques have been developed to study protein-protein interactions within cells, including fluorescent resonance energy transfer (FRET), which examines energy transfer between pairwise donor-acceptor fluorophores linked to proteins of interest; fluorescence cross-correlation spectroscopy (FCCS), which measures the diffusion pattern of two fluorescent molecules through the focal volume; and bimolecular fluorescence complementational (BiFC), which is based on ability of proteins in close proximity to self-complement and create a functional fluorescent protein from two non-fluorescent protein fragments (Bacia et al., 2006; Slaughter et al., 2007; Kerppola, 2008; Miller et al., 2015; Unruh et al., 2018). These techniques can be used in live cells to study endogenously expressed protein-protein interactions in a pairwise manner provided the molecules of interest can be fused to fluorescent proteins (FP). In yeast, genes can be easily tagged at the N- or C-termini using PCR-based methods (Gardner and Jaspersen, 2014). With the development of CRISPR/Cas9 methodologies, genome editing is becoming increasingly feasible such that these approaches can be applied in a wide array of systems. The use of endogenous proteins, rather than transgenes, greatly minimizes false positives caused by high levels of protein expression. In comparison to in vitro assays, FRET, FCCS and BiFC have the advantage of indicating where within a cell the protein-protein interaction occurs (for example, see Muller et al., 2005; Cabantous and Waldo, 2006; Kerppola, 2006; Slaughter and Li, 2010; Sung et al., 2013; Chen et al., 2014; Smoyer et al., 2016; Hennen et al., 2018). However, these methods are limited in their spatial resolution due to the diffraction limit (~200 nm for most FPs), and combining FRET or FCCS with super-resolution methods in cells is extremely challenging.
Structured illumination microscopy (SIM), stimulated emission depletion (STED), stochastic optical reconstruction microscopy (STORM) and photoactivated localization microscopy (PALM) are examples of super-resolution microscopy (SRM) approaches that are pushing cell biology in new directions once thought impossible by beating the diffraction limit (Huang et al., 2009; Schermelleh et al., 2010). The ~10-100 nm resolution achievable by these methods has enhanced our overall understanding of cell biology, particularly at the level of organelles, membranes and other cellular structures. The application of SRM to protein-protein interactions within nanosized protein complexes has been slower to develop, in part because it is difficult to study endogenous proteins with many of these methods, particularly if multiple fluorophores/FPs are needed to monitor interactions and to provide orientation of where within the complex or the assembly cycle the interaction is occurring. While a plethora of FPs have been developed, linear unmixing to image multiple fluorophores has not been integrated into SRM yet, limiting the use of many combinations of FPs. To overcome this obstacle, we combined a version of BiFC based on split-GFP to assay protein-protein interactions (Figure 1).To provide spatial and temporal information as to where and when binding within our complex of interest occurs at sub-diffraction resolution, we developed a three-color imaging strategy using HyVolution with 140 nm lateral resolution (Chen et al., 2019). Importantly, our protocol allows for imaging of endogenously expressed proteins in live cells fused to mTurquoise2 and GFP, along with mCherry, without spectral cross-talk. While the method was developed in Saccharomyces cerevisiae, it is suitable for a wide variety of systems, and it greatly expands our ability to inspect the step-wise assembly pathway of heteromeric macromolecular machines by elucidating protein-protein interactions in vivo at the nanoscale level.
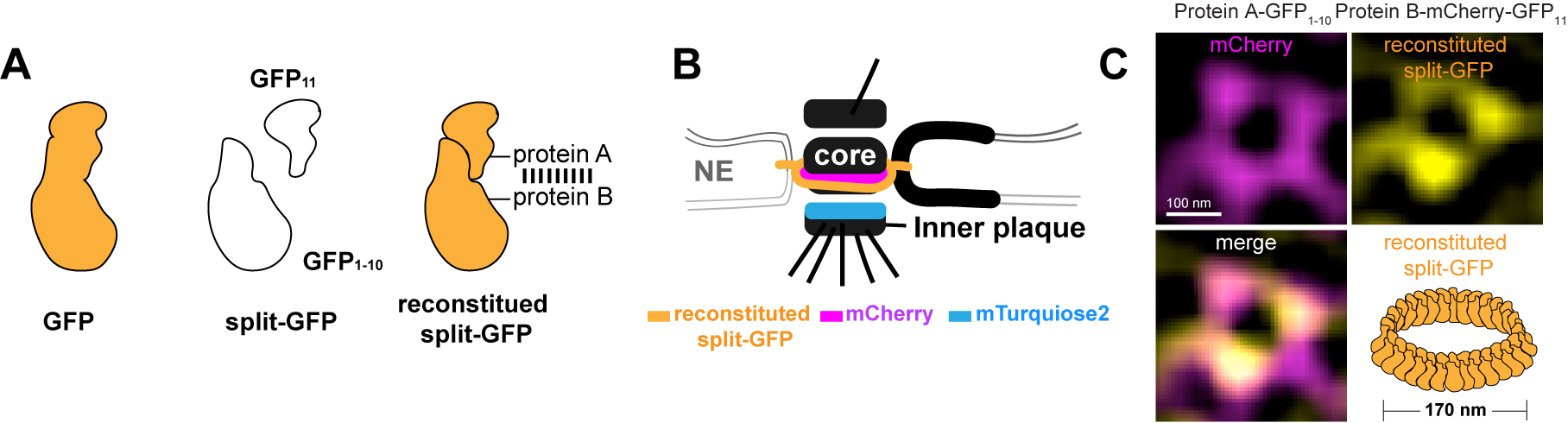
Figure 1. Structured illumination microscopy (SIM)-based split-GFP system to assay protein-protein interaction within nanosized protein complexes. A. Schematic of the bimolecular fluorescence complementation split-GFP system. Two parts of split-GFP can reconstruct and lead to GFP fluorescence when they associate with each other by interaction between their fusion proteins. B. An illustration of the yeast centrosome viewed from the side to show how it is embedded and anchored in the nuclear envelope (NE) by a series of integral membrane proteins, visualized by reconstituted split-GFP and mCherry tagging. The soluble inner-core plaque linker protein was fused to mTurquoise2. C. We showed that our spindle pole body (SPB) protein (fused to GFP1-10) is a component of the membrane complex. In a top down view of the cylinder-shaped SPB, we observed a 170 ring of reconstituted GFP when combined with a known membrane component (fused to mCherry-GFP11) using SIM-BiFC (Chen et al., 2019).
Materials and Reagents
- 0.2 µm filtration unit-150 ml (Thermo Fisher Scientific, catalog number: 150-0020)
- 13 x 100 mm glass culture tube (Fisher, catalog number:14-961-27)
- 15 cm plain wood applicators (Fisher, catalog number: 23-400-102)
- 1.7 ml microcentrifuge tube (VWR, catalog number: 87003-294)
- 25 x 75 mm glass microscope slides (Fisher, catalog number: 12-544-4)
- 22 x 30 mm Number 1.5 coverslips (VWR, catalog number: 48393-150)
- 25-slide humidified chamber (Daigger Scientific, catalog number: EF15989A)
- Yeast strains (see Note 1)
- SLJ10800 (MATa/α SPC42-mTurquiose2-NATMX/Spc42-YFP-HIS3MX bar1/bar1 ADE2/ADE2 trp1-1/TRP1 lys2Δ/LYS2 leu2-3,112/leu2-3,112 his3-11,15/his3-11,15 ura3-1/ura3-1)
- SLJ6209 (MATa SPC42-mCherry-URA3MX bar1 ADE2 trp1-1 lys2Δ leu2-3,112 his3-11,15 ura3-1)
- SLJ7904 (MATa/α SPC110-GFP-KANMX/Spc110 bar1/bar1 ADE2/ade2 trp1-1/TRP1 lys2Δ/LYS2 leu2-3,112/leu2-3,112 his3-11,15/his3-11,15 ura3-1/ura3-1)
- SLJ12889 (MATa Spc110-mTurquiose2-HIS3MX bar1 ADE2 TRP1 lys2Δ leu2-3,112 his3-11,15 ura3-1)
- Bacto-yeast extract
- Bacto-peptone
- Glucose
- Bacto-agar
- Yeast-nitrogen base with ammonium sulfate without amino acids
- CSM powder (Sunrise Science, catalog number: 1001-100)
- 16% paraformaldehyde (Ted Pella, catalog number: 18505)
- Sucrose
- Sodium chloride (NaCl)
- Potassium chloride (KCl)
- Disodium phosphate
- Monopotassium phosphate
- ProLong Diamond mounting medium (Thermo Fisher, catalog number: P36961)
- DeltaVision Immersion Oil Kit; contains bottles of 18 oils with refractive indices from 1.500 to 1.534 (GE Healthcare, catalog number: 29163068)
- Ethanol (Amresco, catalog number: 64-17-5)
- YPD plate (see Recipes)
- SC complete broth (see Recipes)
Note: We typically do not culture cells in YPD liquid media since it produces high background levels of fluorescence. - 4% paraformaldehyde solution (see Recipes)
- PBS (see Recipes)
Equipment
- 2 L flask
- Rotating tube mixer (Fisher Scientific, catalog number: 346)
- 23 °C incubator (VWR, catalog number: 7774)
- Culture roller (New Brunswick TC-7, catalog number: M1053-4004)
- Desktop centrifuge (Eppendorf 5424, catalog number: 022620401)
- Pipettes (Gilson P10, catalog number: F144802)
- BioPhotometer (Eppendorf, model: 6131, catalog number: 62111-460)
- GE Healthcare OMX structured illumination microscope (GE Healthcare DeltaVision OMX Blaze V4) with three scientific complementary metal-oxide semiconductor (sCMOS) cameras and 6 laser lines, or a similar microscope (e.g., Nikon N-SIM, Zeiss Elyria)
- Olympus PLANAPO N 60x, 1.42 NA oil immersion objective
- DeltaVision OMX channel/image registration slide (GE Healthcare, catalog number: 53-852891-000)
- Leica Inverted Confocal SP8 equipped with:
- White Light Laser (470-670 nm range)
- An Argon Laser
- 3 Leica HyD Detectors
- Leica Application Suite software with Hyvolution Module
- Leica 100x, 1.4-NA HC PL APO OIL CS2 Objective
Software
- OMX acquisition computer and software v3.70 (GE Healthcare, http://incelldownload.gehealthcare.com/bin/download_data/OMX/V3.70/DeltaVisionOMX.htm)
- OMX image-processing station with softWoRx v6.52 (GE Healthcare, http://incelldownload.gehealthcare.com/bin/download_data/SoftWoRx/6.5.2/SoftWoRx.htm) software installed
- ImageJ (Java software for image-processing analysis; freely available at http://rsbweb.nih.gov/ij/ or http://Fiji.sc) (Schindelin et al., 2012)
- ImageJ plugins (created in the microscopy center of The Stowers Institute for Medical Research; freely available at http://research.stowers.org/imagejplugins/index.html)
- Leica Application Suite X (LAS X) software for image acquisition (version 3.1.5.16308) (Leica Microsystems)
- Huygens Professional version 17.10.0p5.64b, Scientific Volume Imaging BV, Hilversum, The Netherlands
Procedure
- Sample preparation
- Yeast growth and culture
- Pick up a single colony from the YPD plate and inoculate into 2 ml SC complete broth. Culture at 23 °C overnight in a rotating cell culture tube mixer at 50 rpm.
Note: Keep SC complete broth in closed cabinet or wrapped in foil as light exposure over long periods will result in the formation of a precipitate that increases autofluorescence. - The next day, measure the overnight culture OD600 with BioPhotometer, so that the culture can be diluted back an OD600 0.2-0.3 in 2 ml SC complete broth. Continue growing at 23 °C for 3-5 h until the cells reach an OD600 of ~0.8. If studying temperature-sensitive alleles, after 2 h of growth at 23 °C, shift to 34-36 °C for another 4 h.
- Pick up a single colony from the YPD plate and inoculate into 2 ml SC complete broth. Culture at 23 °C overnight in a rotating cell culture tube mixer at 50 rpm.
- Yeast sample preparation
- Harvest cultures when the OD600 reaches 0.8 or after the temperature shift. Pellet 1-2 ml of cells in a microcentrifuge tube by spinning for 1 min at 13,400 x g in a microcentrifuge at room temperature.
- Aspirate media and resuspend cells in 1 ml 4% paraformaldehyde solution. Place onto rotating tube mixer at 23 °C for 15 min.
- Spin for 1 min at 13,400 x g in an microcentrifuge at room temperature.
- Aspirate media and resuspend cells in 1 ml PBS.
- Spin for 1 min at 13,400 x g in an microcentrifuge at room temperature and remove PBS. Repeat two more times. After the last wash carefully remove all the remaining liquid.
- Add appropriate volume of mounting medium ProLong Diamond to attain desired cell intensity in the specimen, resuspend cell pellet by vortexing briefly at low speed to avoid bubble generation.
- Pipet 5 µl onto a cleaned 22 mm x 30 mm coverslip. Use the pipet tip to spread cells over surface.
Note: Both coverslips and glass slides should be cleaned using 70% ethanol and allowed to dry on a lint-free surface such as lens paper. - Overlay with a clean glass slide, pressing extremely hard to ensure that the cells form a single monolayer between the two glass surfaces.
Note: When preparing the specimen, it is important that cells are less than 10 microns from the coverslip surface for SIM imaging. - Place in a sealed humidified chamber with the coverslip facing down.
- Incubate in the dark for at least 18-24 h at room temperature. The slides can be stored at 4 °C for up to 3 days, after which time the signal decreases and the background increases. If you anticipate your specimen will result in a dim signal, we recommend that it be imaged immediately for optimal results.
- Yeast growth and culture
- Acquire multiple color SIM images with GE Healthcare OMX
We acquire multiple color 3D-SIM images using a GE Healthcare DeltaVision OMX Blaze V3 fitted with an Olympus PlanApo N 100x 1.42 NA oil objective. We first begin by aligning the cameras, then we proceed to our samples. This ensures day to day reproducibility. This protocol is specifically for the DeltaVision OMX Blaze; if using another instrument, follow your manufacturer’s recommended instructions.- Slide alignment
- Place the GE registration slide in the slide holder. Move the stage to locate the center of slider and find an area with the 20 x 20 grid of 100 nm holes with 5 µm spacing.
- Choose “conventional” as the light path, set up the illumination intensity (DIC in the software) to 50% or 100%. Enable multiple cameras (for example, mCherry, GFP, DAP) and adjust the exposure time of individual channel.
- Monitor the intensity histogram and adjust exposure time for individual channels so the mean intensity across all channels are similar.
- Set up the image size to 1024 x 1024 (the maxiumum size on OMX system) and acquire a 3 µm z-stack image with z-spacing of 125 nm.
- We use the central part of acquired image to perfom alignment due to uneven illumination on the corners. We crop the image (set up “Output Options”: X/Y/Width/Height as 112/112/800/800 or similar values) using the software softWoRx, the intergated software on OMX system for imaging processing. Load the cropped image to softWoRx and process it under “Process” and then “Create OMX Image Alignment”.
- Ensure the “Show aligned target image” check box is selected (Demmerle et al., 2017). Display all channels and closely examine the image, especially at the corners. Make sure the holes in each channel co-localize. The manufacturer’s instruction shows a good example of aligned image on page 4 (http://incelldownload.gehealthcare.com/bin/download_data/SoftWoRx/7.0.0/DVOMXSR_ImageAlignment_04-720165-000CC.pdf).
- mTurquiose2 and YFP alignment evaluation
Notes:- To display single images, we typically scale the images four times in ImageJ (Image>Scale) with bilinear interpolation. For fitting and analysis, we leave the images at their original scale.
- We typically prefer to image mTurquoise2 and YFP together as they have similar folding kinetics in yeast. A similar experiment could be easily extended to three color with mCherry or other common red fluorescent proteins, e.g., mKate2 and TagRFP657. Both mKate2 and TagRFP657 have high excitation and emission wavelengths and therefore have no cross-talk with GFP and mTurquoise2 with our current settings. In the current study, the slow folding kinetics of mCherry relative to GFP makes experimental data more difficult to interpret, a problem that does not exist for mTurquoise2 and YFP.
- Switch the system to “SI” Module. Next, acquire a 2 color image 1.5 µm z-stack (spacing = 0.125 µm) with diploid yeast strain SLJ10800. In the strain, Spc42 is tagged at the endogenous loci with mTurquiose2 and YFP separately. Even if one does not work on spindle pole bodies, because they form small, bright puncta of a defined size and distribution, this yeast strain useful to evaluate color alignment with SIM.
- Set up 2 channel SIM acquisition with the correct excitation laser (514 nm and 445 nm) and emission filter, and image YFP first and followed by mTurquiose2.
- We use 50-100 ms exposure time for both channels if possible. Longer exposure times may cause artifacts due to sample and/or mechanical drift; shorter exposure times require higher laser power and may cause photobleaching.
- We use 10% or 1% laser power (maximum laser output 100 mW) to excite both channels. Monitor the camera histogram and adjust the exposure time and laser power accordingly to achieve a signal to noise ratio of 5 or more.
- We use immersion oil with a refractive index (RI) of 1.514 or 1.516 for mTurquiose2 and YFP; we use immersion oil with RI of 1.516-1.518 for mCherry and GFP in other experiments. The optimal immersion oil plays critical role for successful reconstruction and it largely depends on the distance between the signal and coverslip. Due to small size, the distance between region of interest (SPB) in yeast cells and coverslip is very constant and less than a few microns. For other samples, the distance may vary significantly. Please refer to two recent protocol papers to adjust the optimal immersion oil (Demmerle et al., 2017; Wang et al., 2018).
- Align the successfully reconstructed images by running the softWoRx alignment protocol, utilizing the alignment files created in Step B2.
- Open the reconstructed and aligned (SIR_ALX) images in ImageJ. Do a Z-projection (Image>Stacks>Z Project) with Max Intensity to cover the entire SPB in the z-dimension. Use the line tool in ImageJ to draw a line 4 pixels wide across the 2 SPBs in the image (Figure 2A). Measure the average intensity along the line using “polyline kymograph jru v1”.
- Verify that SPB intensity measured along the line from the two channels has a similar pattern. The intensity peak for both SPBs should overlap well with an offset of less than 1 pixel (40 nm) (Figure 2B).
Note: Due to mechanical shift or sample drift during acquisition, slight misalignment always occurs with SIM. Single-particle averaging will further increase resolution and alignment precision (Burns et al., 2015). To minimize misalignment, make sure to closely follow the alignment calibration and be certain that yeast are properly immobilized on the coverslip.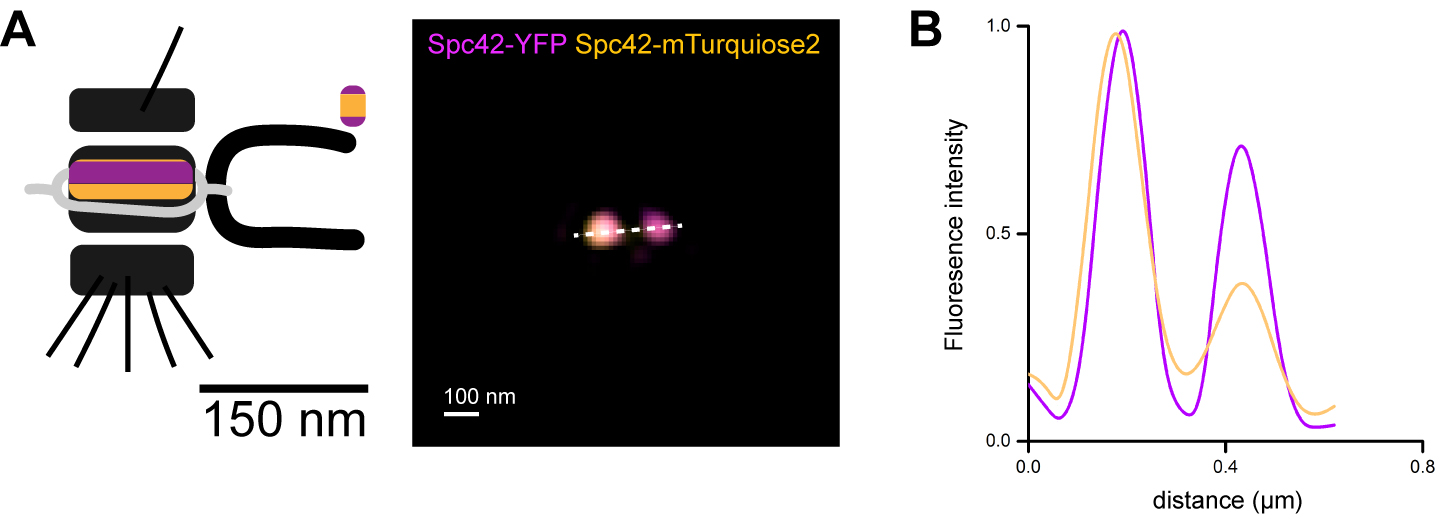
Figure 2. mTurquiose2 and YFP alignment evaluation at yeast SPBs. A. Visualization of Spc42-YFP and Spc42-mTurquiose2 signals at two SPB sub-structures ~150 nm apart in the strain SLJ10800, shown here in a maximum intensity projection along the z-axis. B. Line profiles to measure the intensity between the two spots in both YFP and mTurquiose2 channels shows that the color alignment error is less than 1 pixel (40 nm). - If the channels do not align, repeat the alignment protocol starting with the color alignment slide.
- To align mCherry with mTuruqoise2 and YFP, repeat the alignment procedure starting with the color alignment slide. Use a multi-color sample for alignment using Steps B2b-B2f.
- Slide alignment
- Acquire three-color HyVolution images with Leica SP8
We routinely image three-color samples by SIM and conventional confocal systems. GFP (or split-GFP) has a single absorption peak centered at about 488 nm, which is one of the most common laser lines in the lab and has been routinely used to excite GFP. However, the combination of GFP (or split-GFP) and mTurquiose2 is problematic due to significant cross-talk both in terms of emission and excitation profiles. Traditional imaging approaches on conventional systems that use 488 nm and 455 nm excitation for GFP and mTurquiose2, respectively, will not result in cross-talk free images. Corrective measures must be taken to subtract off cross-talk. Another alternative is to use spectral imaging (Zimmermann et al., 2003). However, dim samples particularly in the mTurquiose2 channel, combined with significant cross-talk from GFP, can lead to misinterpretation of positive signal. Below we describe another solution to this problem.
Spectrum setting for three colors- Run the Software LAS X in the TCS SP8 mode. Set up a sequential mode with 3 color images in the order of mCherry, reconstituted split-GFP/GFP and mTurquiose2. Choose “between frame” as the default setting for channel switching.
- We found that shifting the excitation of GFP from 488 nm to 496 nm dramatically reduced excitation of mTurquiose2. Excitation efficacy of reconstituted split-GFP/GFP is 89% at 496 nm, which is only 10% less than the value at 488 nm, while the shift to 496 nm excitation leads to absorption efficacy of mTurquiose2 that is less than 0.02% (Lambert, 2019). Thus, using 496 nm laser line to excite reconstituted split-GFP, along with careful selection of emission filters, effectively eliminates bleed through of mTurquiose2 to reconstituted split-GFP/GFP (Figure 3A).
- We set up the excitation of the 3 channels as follows: mCherry (561 nm, white light laser or argon laser), reconstituted split-GFP (496 nm, argon laser), mTurquiose2 (445 nm, white laser or argon laser). Due to weak signal of reconstituted split-GFP and low output of white laser, we use the higher power laser line (argon laser) to excite split-GFP.
- There is no Notch filter for 496 nm laser line in our SP8 system. Therefore, we used the internal polarizer to remove reflected light in the reconstituted split-GFP/GFP channel.
- In the “Fluorifier Disc Settings” panel, set up a value of “90” for the “Polarizer” to remove the reflected light from microscope.
- For collection of emission photons with the 3 HyD detectors, the emission filters are set up as follows: mCherry: 570-635 nm or a similar range, reconstituted split-GFP/GFP: 508-550 nm; mTurquiose2: 463-495 nm. The emission filter setting at mTurquoise2 channel will block ~94% emission photons from the reconstituted split-GFP/GFP channel (Figure 3B).
- Switch the image mode to HyVolution in the acquisition software LAS X and focus the sample using mCherry. We begin by using the default settings within HyVolution. In LAS X, increase microscope resolution by shrinking the size of pinhole. A pinhole close to 0.6 AU (airy unit) compensates between resolution and signal intensity (Borlinghaus and Kappel, 2016).
- The SPB contains ~1,000 copies of each component so it is a common source of cross-talk and bleed-through in multicolor imaging experiments (for example, Muller et al., 2005). Using the HyVolution mode, we verified 3 color imaging settings with the yeast strains tagged with mCherry (SLJ6209), GFP (SLJ7904) and mTurquoise2 (SLJ12889) only. For the yeast strain SLJ7904 containing GFP only, we did not observe emission in the mTurquoise2 channel (463-495 nm) when we excited at 445 nm (mTurquoise2 excitation) (Figure 3C, top panel, left). Also, no mTurquoise2 emission was observed when we excited mTurquoise2 only in the strain SLJ12889 at 496 nm in the GFP channel (508-550 nm) (Figure 3C, center panel, right). For visualization purposes, we apply a filter (gaussian blur, 1 pixel) and set the contrast from 0-30 for all images except Spc110-GFP excited with 488 nm, setting contrast from 1-67 due to the strong signal (Figure 3C). The GFP channel has higher background due to high reflection, there is no mCherry signal for both SLJ7904 and SLJ12889 (data not shown).
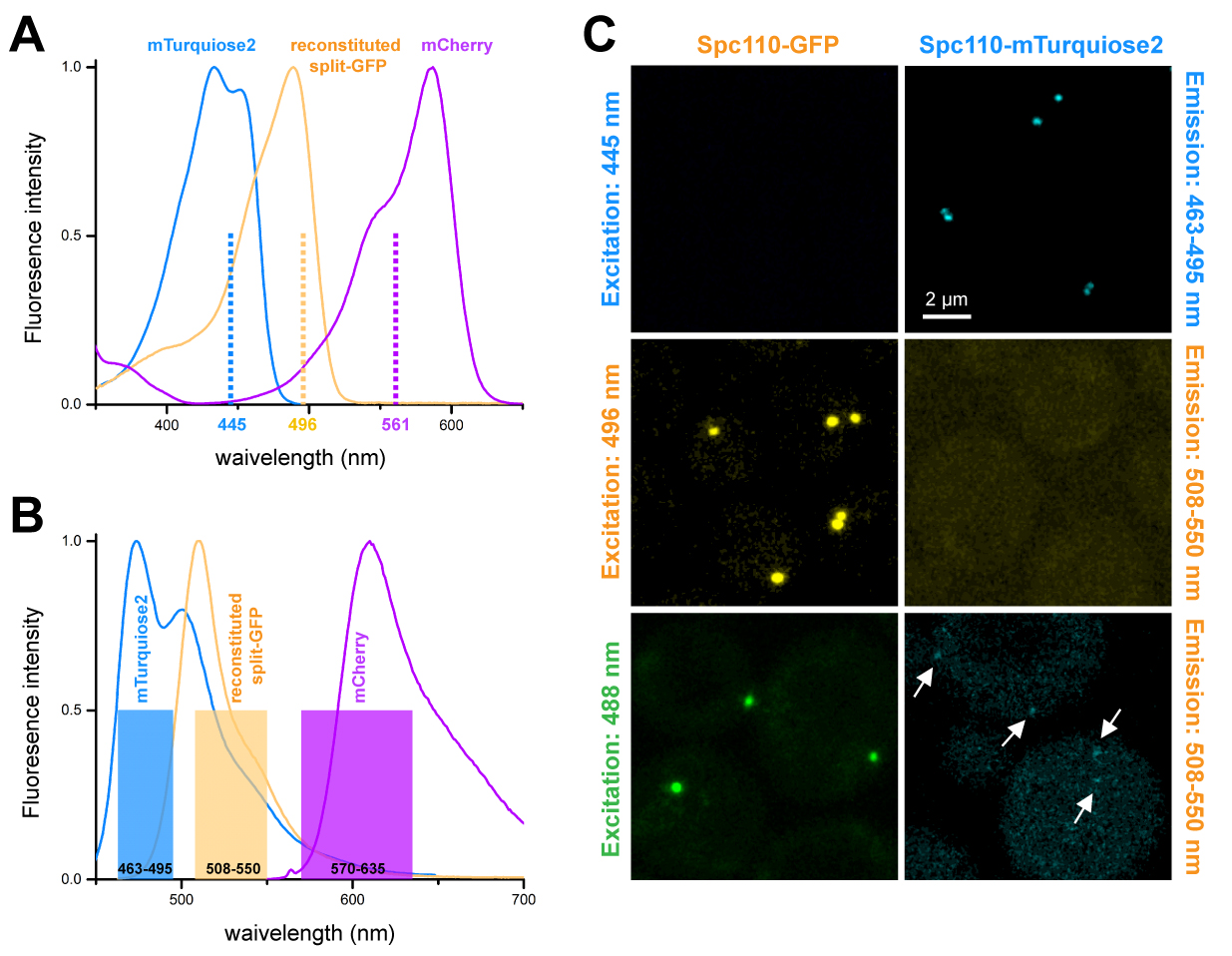
Figure 3. Three color HyVolution image to study multiple SPB components. A. absorption spectrum and laser illumination for 3 channels (mCherry, split-GFP/GFP and mTurquiose2). B. emission spectrum and emission filter setting for these 3 channels. C. In HyVolution mode, SLJ12889 (Spc110-mTurquiose2) and SLJ7904 (Spc110-GFP) were imaged with the indicated spectrum setting. mTurquiose2 shows signal crosstalk (arrows pointed) by using 488 nm excitation (bottom panel, right) with 508-550 nm emission filter, however, no bleed-through was observed at 496 nm excitation with the same emission filter (center panels, right).
Notes
- This protocol was developed for S. cerevisiae but could be applied to other systems. We use yeast that do not contain mutations in the adenine biosynthesis pathway to limit the amount of autofluorescence (Weisman et al., 1987). This dramatically improves signal-to-noise, which is critical for dim samples (Unruh et al., 2018). Genes are tagged using PCR-based approaches as previously described (Gardner and Jaspersen, 2014). We generally grow yeast at 23 °C rather than 30 °C to slow down cell growth, which results in more fluorescent protein due to improved fluorophore maturation.
- When using BiFC to study protein-protein interactions, it is important to consider the topology of target proteins, especially integral membrane proteins. The two halves of split-GFP must be in the same compartment, such as the nucleus or cytoplasm, to reconstitute a functional GFP. For this reason, tagging at the N- or C-terminus may be preferred. It is also imperative to perform appropriate controls, such as a vector only and a mutant/different subunit to confirm specificity in the interaction (see Chen et al., 2019).
- One part of split-GFP, GFP11, is only 25 amino acids long. We have been unable to find a commercially available antibody that recognizes GFP11 so we typically use mCherry as part of our GFP11 fusion. mCherry can be added to the opposite terminus, or in between the target and GFP11. It is important that both GFP1-10 and GFP11 are at the extreme N- or C-terminus of the final fusion protein so they are able to interact.
- In our study, the N-terminally tagged mCherry-GFP11 fusion protein is expressed under the CDC42 promoter. Expression from this promoter is slightly higher than the endogenous expression, but it is sufficiently low that two control proteins do not interact (Smoyer et al., 2016; Chen et al., 2019). To facilitate optimal expression and stability in yeast, we use yeast codon optimized versions of all fluorescent proteins, including mCherry-GFP11 and GFP1-10. These PCR tagging plasmids and reporters have been deposited at AddGene (https://www.addgene.org/search/advanced/?q=jaspersen).
Recipes
- YPD plate (1 L)
10 g Bacto-yeast extract
20 g Bacto-peptone
20 g Glucose
20 g Bacto-agar
950 ml ddH2O
Dissolve the first three ingredients in water in a 2 L flask, then add the Bacto-agar along with a large stir bar. Sterilize for 30 min. After autoclaving, place on a stir plate until the liquid has cooled to ~50-60 °C. Pour roughly 25 ml of liquid into 15 cm Petri plates using sterile technique. Let the media fully cool and the plates dry for 1-2 days. - SC complete broth (1 L)
6.7 g Yeast-nitrogen base with ammonium sulfate without amino acids
20 g Glucose
2.0 g CSM powder
Mix together then autoclave to sterilize - 4% Paraformaldehyde solution (80 ml)
20 ml 16% paraformaldehyde (EM grade, methanol free)
11.7 g sucrose and 60 ml ddH2O
Pass through a 0.2 µm filter
Store at 4 °C in a container shielded from light. Use this solution within 1-2 months
Note: Both coverslips and glass slides should be cleaned using 70% ethanol and allowed to dry on a lint-free surface such as lens paper. - PBS (1 L)
8 g sodium chloride
0.2 g potassium chloride
1.42 g disodium phosphate
0.24 g monopotassium phosphate
1,000 ml ddH2O
Adjust the pH to 7.4
Acknowledgments
We are grateful to members of the Microscopy Center and the Jaspersen lab for advice during the course of this project. Research reported in this publication was supported by the Stowers Institute for Medical Research and the National Institutes of Health, National Institute of General Medical Sciences under award number R01GM121443 (to SLJ). The authors declare no competing financial interests.
Competing interests
The authors declare no conflicts of interest or competing interests.
References
- Bacia, K., Kim, S. A. and Schwille, P. (2006). Fluorescence cross-correlation spectroscopy in living cells. Nat Methods 3(2): 83-89.
- Borlinghaus, R. T. and Kappel, C. (2016). HyVolution—the smart path to confocal super-resolution. Nature Methods 13: 276.
- Burns, S., Avena, J. S., Unruh, J. R., Yu, Z., Smith, S. E., Slaughter, B. D., Winey, M. and Jaspersen, S. L. (2015). Structured illumination with particle averaging reveals novel roles for yeast centrosome components during duplication. Elife 4: e08586.
- Cabantous, S. and Waldo, G. S. (2006). In vivo and in vitro protein solubility assays using split GFP. Nat Methods 3(10): 845-854.
- Chen, J., Gardner, J. M., Yu, Z., Smith, S. E., McKinney, S., Slaughter, B. D., Unruh, J. R. and Jaspersen, S. L. (2019). Yeast centrosome components form a noncanonical LINC complex at the nuclear envelope insertion site. J Cell Biol 218(5): 1478-1490.
- Chen, J., Smoyer, C. J., Slaughter, B. D., Unruh, J. R. and Jaspersen, S. L. (2014). The SUN protein Mps3 controls Ndc1 distribution and function on the nuclear membrane. J Cell Biol 204(4): 523-539.
- Demmerle, J., Innocent, C., North, A. J., Ball, G., Muller, M., Miron, E., Matsuda, A., Dobbie, I. M., Markaki, Y. and Schermelleh, L. (2017). Strategic and practical guidelines for successful structured illumination microscopy. Nat Protoc 12(5): 988-1010.
- Gardner, J. M. and Jaspersen, S. L. (2014). Manipulating the yeast genome: deletion, mutation, and tagging by PCR. Methods Mol Biol 1205: 45-78.
- Hennen, J., Saunders, C. A., Mueller, J. D. and Luxton, G. W. G. (2018). Fluorescence fluctuation spectroscopy reveals differential SUN protein oligomerization in living cells. Mol Biol Cell 29(9): 1003-1011.
- Huang, B., Bates, M. and Zhuang, X. (2009). Super-resolution fluorescence microscopy. Annu Rev Biochem 78: 993-1016.
- Kerppola, T. K. (2006). Design and implementation of bimolecular fluorescence complementation (BiFC) assays for the visualization of protein interactions in living cells. Nat Protoc 1(3): 1278-1286.
- Kerppola, T. K. (2008). Bimolecular fluorescence complementation (BiFC) analysis as a probe of protein interactions in living cells. Annu Rev Biophys 37: 465-487.
- Lambert, T. J. (2019). FPbase: a community-editable fluorescent protein database. Nat Methods 16(4): 277-278.
- Miller, K. E., Kim, Y., Huh, W. K. and Park, H. O. (2015). Bimolecular fluorescence complementation (BiFC) analysis: advances and recent applications for genome-wide interaction studies. J Mol Biol 427(11): 2039-2055.
- Muller, E. G., Snydsman, B. E., Novik, I., Hailey, D. W., Gestaut, D. R., Niemann, C. A., O'Toole, E. T., Giddings, T. H., Jr., Sundin, B. A. and Davis, T. N. (2005). The organization of the core proteins of the yeast spindle pole body. Mol Biol Cell 16(7): 3341-3352.
- Schermelleh, L., Heintzmann, R. and Leonhardt, H. (2010). A guide to super-resolution fluorescence microscopy. J Cell Biol 190(2): 165-175.
- Schindelin, J., Arganda-Carreras, I., Frise, E., Kaynig, V., Longair, M., Pietzsch, T., Preibisch, S., Rueden, C., Saalfeld, S., Schmid, B., Tinevez, J. Y., White, D. J., Hartenstein, V., Eliceiri, K., Tomancak, P. and Cardona, A. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9(7): 676-682.
- Slaughter, B. D. and Li, R. (2010). Toward quantitative "in vivo biochemistry" with fluorescence fluctuation spectroscopy. Mol Biol Cell 21(24): 4306-4311.
- Slaughter, B. D., Schwartz, J. W. and Li, R. (2007). Mapping dynamic protein interactions in MAP kinase signaling using live-cell fluorescence fluctuation spectroscopy and imaging. Proc Natl Acad Sci U S A 104(51): 20320-20325.
- Smoyer, C. J., Katta, S. S., Gardner, J. M., Stoltz, L., McCroskey, S., Bradford, W. D., McClain, M., Smith, S. E., Slaughter, B. D., Unruh, J. R. and Jaspersen, S. L. (2016). Analysis of membrane proteins localizing to the inner nuclear envelope in living cells. J Cell Biol 215(4): 575-590.
- Sung, M. K., Lim, G., Yi, D. G., Chang, Y. J., Yang, E. B., Lee, K. and Huh, W. K. (2013). Genome-wide bimolecular fluorescence complementation analysis of SUMO interactome in yeast. Genome Res 23(4): 736-746.
- Unruh, J. R., Slaughter, B. D. and Jaspersen, S. L. (2018). Functional analysis of the Yeast LINC complex using fluctuation spectroscopy and super-resolution imaging. Methods Mol Biol 1840: 137-161.
- Wang, Y., Yu, Z., Cahoon, C. K., Parmely, T., Thomas, N., Unruh, J. R., Slaughter, B. D. and Hawley, R. S. (2018). Combined expansion microscopy with structured illumination microscopy for analyzing protein complexes. Nat Protoc 13(8): 1869-1895.
- Weisman, L. S., Bacallao, R. and Wickner, W. (1987). Multiple methods of visualizing the yeast vacuole permit evaluation of its morphology and inheritance during the cell cycle. J Cell Biol 105(4): 1539-1547.
- Zimmermann, T., Rietdorf, J. and Pepperkok, R. (2003). Spectral imaging and its applications in live cell microscopy. FEBS Lett 546(1): 87-92.
Article Information
Copyright
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Chen, J., Yu, Z., Unruh, J. R., Slaughter, B. D. and Jaspersen, S. L. (2020). Super-resolution Microscopy-based Bimolecular Fluorescence Complementation to Study Protein Complex Assembly and Co-localization. Bio-protocol 10(4): e3524. DOI: 10.21769/BioProtoc.3524.
- Chen, J., Gardner, J. M., Yu, Z., Smith, S. E., McKinney, S., Slaughter, B. D., Unruh, J. R. and Jaspersen, S. L. (2019). Yeast centrosome components form a noncanonical LINC complex at the nuclear envelope insertion site. J Cell Biol 218(5): 1478-1490.
Category
Molecular Biology > Protein > Protein-protein interaction
Cell Biology > Cell-based analysis > Protein interaction
Biochemistry > Protein > Fluorescence
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.