- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

CRISPR-Cas9 Genome Editing of Plasmodium knowlesi
Published: Vol 10, Iss 4, Feb 20, 2020 DOI: 10.21769/BioProtoc.3522 Views: 7487
Reviewed by: Samantha E. R. DundonWathsala WijayalathAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jun 2019
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Plasmodium knowlesi is a zoonotic malaria parasite in Southeast Asia that can cause severe and fatal malaria in humans. The main hosts are Macaques, but modern diagnostic tools reveal increasing numbers of human infections. After P. falciparum, P. knowlesi is the only other malaria parasite capable of being maintained in long term in vitro culture with human red blood cells (RBCs). Its closer ancestry to other non-falciparum human malaria parasites, more balanced AT-content, larger merozoites and higher transfection efficiencies, gives P. knowlesi some key advantages over P. falciparum for the study of malaria parasite cell/molecular biology. Here, we describe the generation of marker-free CRISPR gene-edited P. knowlesi parasites, the fast and scalable production of transfection constructs and analysis of transfection efficiencies. Our protocol allows rapid, reliable and unlimited rounds of genome editing in P. knowlesi requiring only a single recyclable selection marker.
Keywords: Plasmodium knowlesiBackground
Malaria is an infectious disease caused by Plasmodium parasites worldwide with over 200 million cases yearly (WHO, 2018). Six different Plasmodium species are known to cause significant numbers of human infections, but of these only Plasmodium knowlesi is a zoonotic malaria parasite. Its main hosts are macaque monkeys that are distributed in Southeast Asia, but it can also cause severe and fatal malaria in humans (Rajahram et al., 2019). Due to its morphological similarity to P. malariae, it was often misdiagnosed by microscopy. New PCR-based diagnostic tools reveal increasing numbers of human infections in Malaysia and Indonesia, revealing that up to 80% of overall malaria cases in some regions are caused by P. knowlesi (Lubis et al., 2017; Cooper et al., 2020). After P. falciparum, P. knowlesi is the only other malaria parasite adapted to grow in a continuous human red blood cell in vitro culture system (Moon et al., 2013; Lim et al., 2013; Gruring et al., 2014). P. knowlesi is a close relative of P. vivax, the most widespread human malaria parasite and both rely on Duffy positive (Fy+) red blood cells for invasion.
Compared to P. falciparum, P. knowlesi has a shorter life cycle (27 h in cell culture vs approximately 46 h for P. falciparum 3D7), more balanced AT-content (60% vs 82%), larger merozoites and higher transfection efficiencies (up to 30% with episomal plasmids) giving P. knowlesi some advantages over P. falciparum as an experimental system. The more balanced AT-content alone means that it is easier to design oligonucleotide primers, generate transfection constructs, and find well-positioned PAM sites required for Cas9 binding and cleaving of the target DNA. Successful use of CRISPR-Cas9 genome editing in P. falciparum (Ghorbal et al., 2014) gave promise of a very efficient method for P. knowlesi.
Here we present a marker-free CRISPR gene editing method for P. knowlesi based on a two plasmid system allowing the use and recycling of a single selection marker. The first plasmid contains a Cas9 expression cassette, as well as positive and negative selection markers. The second plasmid contains the repair template which provides the template for repair. This normally contains the modification to be introduced flanked either side by 800 bp regions of homology to the target locus. P. knowlesi readily accepts linearized plasmids for homologous recombination, therefore repair template generation can be carried out with conventional cloning or with a two- or three-step PCR method. This protocol can be used to generate parasite lines with C- and N-terminal tagging, knock-out and orthologue replacement (Mohring et al., 2019). The PCR method for donor generation is much faster and ideally suited for generation of knockout or tagging constructs. For more complex or larger constructs like gene replacement lines we recommend using a conventional cloned and sequenced plasmid construct.
These approaches are well suited to undertaking functional analysis of parasite genes through knockout or by addition of a tag. As the desired modification can be introduced at any position within the gene, including internally, the introduction of N-terminal or internal tags is far easier than when using conventional approaches. The resultant parasite lines are markerless and thus, using iterative modifications, gene knockouts and tagged genes can be combined together to aid phenotype or examine large redundant gene families. The method can also be combined with conditional knockout approaches such as the DiCre recombinase that has recently been implemented to study essential genes in P. knowlesi (Knuepfer et al., 2019). Another major usage for this system is for orthologue replacement (OR) approaches, where a P. knowlesi gene is directly replaced with its orthologue from a different malaria parasite species. It is currently only possible to grow P. falciparum and P. knowlesi in culture with human RBCs, so this provides a mechanism to study genes from the important human malaria pathogen that cannot be maintained in culture, P. vivax–but also neglected parasites such as P. ovale and P. malariae. The gene is placed directly under the control of the endogenous P. knowlesi promoter ensuring appropriate stage specific and stable expression levels–providing significant advantages over the highly variable expression levels of episomal constructs. This has already been used to develop a P. knowlesi PvDBPOR line, which enables scalable screening for inhibitory antibodies targeting P. vivax duffy binding protein (PvDBP), the current lead P. vivax blood stage vaccine candidate (Mohring et al., 2019; Rawlinson et al., 2019). The same approach can readily be applied to studying other vaccine or drug resistance markers from P. vivax, or indeed used to generate a range of tools to study both the basic biology or support translational research into non-falciparum malaria parasites.
Materials and Reagents
- 1.5 ml reaction tube (Eppendorf, catalog number: 0030120086)
- 15 ml centrifuge tube (Falcon, Corning, catalog number: 352196)
- 96 flat-bottom plates (CytoOne, Starlab, catalog number: CC7672-7596)
- 6-well plate (CytoOne, Starlab, catalog number: CC7672-7506)
- Plasmodium knowlesi A1-H.1 wild type (Mike Blackman, Francis Crick Institute London) (Moon et al., 2013)
- XL10-Gold Ultracompetent Cells (Agilent, catalog number: 200315), -70 °C
- StellarTM competent cells (Clontech, catalog number: 636766)
- pCas9/sg plasmid (Robert W. Moon, London School of Hygiene and Tropical Medicine, London, UK)
- pKconGFP plasmid (Robert W. Moon, London School of Hygiene and Tropical Medicine, London, UK)
- RPMI-1640 Medium (Sigma, catalog number: R5886)
- L-glutamine (Sigma, catalog number: G7513-100ML), -20 °C
- Horse serum (PAN BIOTECH, catalog number: P30-0711), -20 °C
- Nycodenz (Progen, catalog number: 1002424), RT
- 4-[7-[(dimethylamino)methyl]-2-(4-fluorphenyl)imidazo[1,2-a]pyridin-3-yl]pyrimidin-2-amine; compound 2 (Michael Blackman, Francis Crick Institute, Lonon, UK)
- P3 Primary cell 4D Nucleofector X Kit L (Lonza, calalog number: V4XP-3024)
- CloneAmp HiFi PCR Premix (TaKaRa, catalog number: 639298)
- BtgZI restriction enzyme (New England BioLabs, catalog number: R0703S), -20 °C
- In-Fusion HD Cloning Kit Plus (TaKaRa, catalog number: 638910)
- NEBuffer 2 (New England BioLabs, catalog number: B7002S), -20 °C
- Ampicillin sodium salt (Sigma-Aldrich, catalog number: A0166-5G), -20 °C
- Hoechst 33342 (New England BioLabs, catalog number: 4082), -20 °C
- Agarose, Molecular Grade (Bioline Reagents Ltd, catalog number: BIO-41026)
- TAE Buffer 50x (VWR International, catalog number: A1691.1000)
- HyperLadder 1 kb (Bioline Reagents Ltd, catalog number: BIO-33026)
- QIAquick PCR Purification kit (QIAGEN, calalog number: 28104)
- Sequencing Kit, Mix2Seq (eurofins)
- QIAGEN Plasmid Mini Kit (QIAGEN, catalog number: 12123)
- QIAGEN Plasmid Midi Kit (QIAGEN, catalog number: 12143)
- QIAquick Gel Extraction Kit (QIAGEN, catalog number: 28704)
- LB Broth, Miller (Sigma-Aldrich, catalog number: L3522-250G)
- CH3COONa, Sodium acetate, NaAc (Sigma-Aldrich, catalog number: S2889-250G)
- CaNa2·EDTA (Sigma-Aldrich, catalog number: ED2SC-100G)
- Ethanol (Fisher Chemicals, E/0650DF/17)
- DNeasy blood and tissue kit (QIAGEN, catalog number: 69504)
- GoTaq Green Master Mix (Promega, catalog number: M7122), -20 °C
- Ibidi μ-slide VI 0.4, Poly-L-Lysine (Ibidi, Thistle Scientific Ltd, catalog number: 80604)
- Oligos (IDT, Integrated DNA Technologies)
- Ancotil/ 5-Fluorocytosine 2.5 g/250 ml Solution for Infusion (PubChem CID: 3366), RT
- DMSO (Sigma-Aldrich, catalog number: 276855-250ML)
- Tris HCl, Trizma® hydrocloride solution 1 M (Sigma, catalog number: T-3038-1L)
- KCl (Sigma, catalog number: P9541-500g)
- RPMI-1640 (HEPES Modification, With 25 mM HEPES, without L-glutamine, Merck, catalog number: R558)
- Sodium bicarbonate (Sigma, catalog number: S5761)
- Dextrose (Sigma, catalog number: G7021)
- Hypoxanthine (Sigma, catalog number: H9636)
- Albumax II (Gibco, catalog number: 11560376)
- L-glutamine (Merck, catalog number: 59202C)
- Custom Modified RPMI media w/o glutamine (Life Technology Brand) (see Recipes)
- Nycodenz stock solution (100%) (see Recipes) (Alere Technologies AS, catalog number: 1002424)
- Nycodenz working solution (55%) (see Recipes)
- Pyrimethamine (Sigma, catalog number: P4200000) (see Recipes)
- Ancotil 2.5 g/250 ml Solution for Infusion (see Recipes)
Equipment
- Amaxa 4D-NucleofectorTM Core Unit (Lonza, catalog number: AAF-1002B)
- Amaxa 4D-NucleofectorTM X Unit (Lonza, catalog number: AAF-1002X)
- Centrifuge with cooling function (Eppendorf, catalog number: 5424 R)
- Heat block (USA Scientific, Thermal-Lok 2-Position Dry Heat Bath, catalog number: 2510-1102)
- Gel electrophoresis chamber (Analytik Jena, catalog number: 846-025-100)
- ChemiDoc Imaging System (BioRad, catatog number: 17001401)
- ThermoMixer C (Eppendorf, catalog number: 2231000574)
- Microscope (Invitrogen EVOS Digital Color Fluorescence Microscope, AMEFC4300, catalog number: 15319601)
- Spectrophotometer DS-11 Fx+ (DeNovix)
- Thermal Cycler PCRmax Alpha Cycler (Cole-Parmer, catalog number: SI-93945-12)
Software
- SnapGene (GSL Biotech LLC, www.snapgene.com)
- Protospacer Software (BIHP, Institute Pasteur, Paris, France, www.protospacer.com)
- Image Lab Software (BioRad)
- CRISPR software (e.g., Benchling, Protospacer)
Procedure
- Cloning of pCas9/sg plasmid
- Choose guide sequence in the gene of interest
- Find PAM site (NGG) in the gene region that is going to be deleted/replaced. The positioning of the PAM site will vary depending on the goal. For knock-out or replacement studies it can be placed anywhere within the deleted/replaced region. For tagging studies the guide RNA sequence should cross the position of tag insertion.
- Determine off- and on-target scores of the guide sequence (N20 upstream of the NGG site) with CRISPR software (e.g., Benchling, Protospacer). Choose one or multiple guide sequences with off-target scores < 0.03. The positioning and off-target scores should be your overriding factors when deciding on a guide sequence. We recommend avoiding extremely AT-rich sequences. The on-target scores are normally determined in the context of other species, and in our experience do not correlate well with success, so whilst they should be considered, the other criteria should be prioritized.
- Design In-Fusion oligos for integration of the target sequence into the pCas9/sg plasmid
Forward oligo: 5′-TTACAGTATATTATT(N20)GTTTTAGAGCTAGAA-3′
Reverse oligo: 5′-TTCTAGCTCTAAAAC(N20)AATAATATACTGTAA-3′
The schematic in Figure 1 depicts the insertion of the guide sequence into the pCas9/sg plasmid. - Linearize 2 μg pCas9/sg plasmid with 1 μl BtgZI enzyme and 5 μl cut smart buffer in 50 μl reaction volume for at least 2 h at 60 °C and clean up with QIAquick PCR Purification kit. Confirm linearization by running on 0.8% agarose gel. Dilute linearized plasmid to 50 ng/μl.
- Anneal guide oligos
- Prepare oligo suspensions of 100 μM and mix 10 μl of each (forward and reverse oligo) with 2.2 μl NEB2 buffer in a 1.5 ml reaction tube.
- Incubate in heat block for 10 min at 95 °C. Move heat block on ice and let cool down. (It takes approximately 1 min for the block to cool down.) From now on, always keep the reaction tube on ice.
- Dilute annealed oligos 1:200 to 0.5 μM with cold EB buffer from the QIAGEN kit (1 μl of annealed oligos to 199 μl EB buffer).
- Prepare In-Fusion reaction
- Mix 1 μl of linearized plasmid (50 ng), 1 μl of diluted annealed oligos, 2 μl H2O and 1 μl In-Fusion buffer on ice.
- Incubate for 15 min at 50 °C.
- Transformation of pCas9/sg_guide
- Add 5 μl of In-Fusion reaction to 50 μl of XL10 gold ultracompetent cells or alternative cells, for example StellarTM competent cells (Clontech).
- Incubate on ice for 30 min.
- Apply heat shock according to the manufacturer’s instructions. For XL10 gold cells this is 30 s in a heat bath at 42 °C.
- Incubate reaction on ice for 2 min.
- Add 200 μl of LB-media and incubate for 20 min at 37 °C with 350 rpm shaking.
- Plate reaction out on agar plates with 0.1 μg/ml ampicillin and incubate overnight at 37 °C.
- Confirm insertion of N20 guide sequence into pCas9/sg plasmid
- Inoculate a colony in 5 ml LB-media with 0.1 μg/ml ampicillin and incubate overnight at 37 °C.
- Purify plasmid with Mini-prep kit.
- Sequence at least two plasmids (efficiency typically between 50 and 100%) with oligo 5′-CATTGTTCCCCCCTTTGTTTTGCAAG-3′ to confirm insertion.
- Purify plasmid for parasite transfection
- Inoculate 250 ml LB-media with 0.1 μg/ml ampicillin and incubate overnight at 37 °C.
- Purify plasmid with Midi-prep kit. The yield should be at least 30 μg in order to have enough plasmid for one parasite transfection.
- Precipitate plasmid by mixing with 0.1 volume of 3 M NaAc and 2 volumes of ice-cold 100% ethanol and incubate for at least 30 min at -20 °C.
- Spin down plasmid for 30 min at 4 °C and 15,871 rcf. Wash pellet twice with 700 μl 70% ethanol, each time spin down pellet for 15 min at 4 °C and 15,871 rcf.
- Dry plasmid pellet in sterile laboratory cabinet and resuspend with 10 μl sterile TE buffer.
- Measure DNA concentration.
- Keep plasmid at 4 °C until transfection.
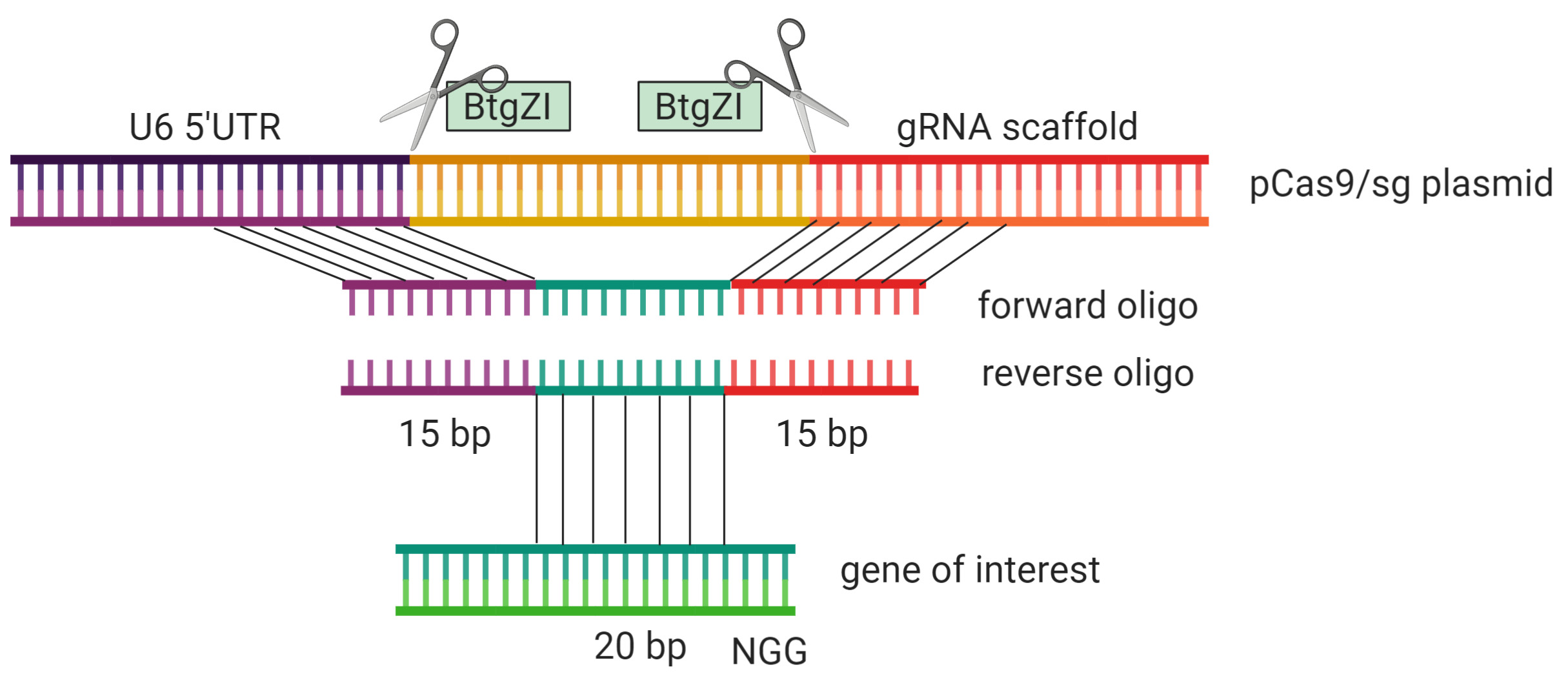
Figure 1. In-Fusion cloning of guide sequence into pCas9/sg plasmid. BtgZI cleaves outside of its recognition site. Two BtgZI sites (green) are used to linearize the plasmid and remove the recognition sites. Oligos for In-Fusion cloning are generated by using 15 bp homology regions of either side of the integration site, flanking the 20 bp upstream of the NGG site within the target gene. Gray lines indicate sequence homology.
- Choose guide sequence in the gene of interest
- Generating PCR template DNA
- Generating oligos
- Choose oligos to amplify homology regions (HRs) up- (product 1, oligo5 + oligo2) and downstream (product 2, oligo3 + oligo6) of the guide sequence (double-strand break) of at least 500 bp length each, but ideally 800 bp. If possible, avoid long poly-A or T tracts.
- Add an adaptor sequence of the DNA to be integrated (this could be a tag or a whole expression cassette) to oligo 2 and oligo 3 of ~20 bp (Tm of at least 56 °C).
- Choose oligos at least 50 bp upstream of the 5′HR and 50 bp downstream of the 3′HR (oligo1 and oligo4), these will allow you to carry out the fusion reaction as a semi-nested PCR, this is critical to ensure minimal background products and sufficient yield.
- Choose oligos to amplify the DNA sequence to be integrated (product 3, oligo7 + oligo8).
- Amplify 5′HR (product 1, oligo1 + oligo2), 3’HR (product 2, oligo3 + oligo4) and DNA sequence to be integrated (product 3, oligo7 + oligo8). Run each PCR in duplicate with a total volume of 25 μl according to the user manual. For CloneAmp Hifi PCR Premix this is 32 cycles of:
Run on agarose gel and gel purify.98 °C 10 s Denaturation 55 °C 15 s Annealing 72 °C 5 s/kb Extension - Combine product 1 and product 3 by PCR (oligo5 + oligo8). Allow 15 s/kb during the extension step. Run on agarose gel and gel purify.
Example: for 25 μl PCR use: 12.5 μl CloneAmp Premix, 0.5 μl fwd oligo, 0.5 μl rev oligo, 1 μl product 1, 1 μl product 2 and 10 μl H2O. - Combine product 1/3 and product 2 by PCR (oligo5 + oligo6). Again allow 15 s/kb during the extension step. Run at least 6x 50 μl reactions of this final PCR to yield 40 μg of DNA. Note that whilst it sometimes is possible to simultaneously fuse all three products in a single reaction, splitting this into two steps increases robustness and yield so we recommend this as default.
- Without purification (to avoid loss of DNA) precipitate DNA, wash and dry as described for pCas9/sg plasmid. A schematic of the 3-step PCR for generating template DNA is shown in Figure 2.
Notes:- If the inserted DNA sequence is a small tag (like hemagglutinin tag) the whole tag sequence can be added as adaptors in oligos 2 and 3 and only two PCR steps are needed to fuse 5′HR and 3′HR.
- If the aim is to knock-out a gene, homology regions targeting the 5′ and 3′ UTRs can simply be fused together (or with a short intervening “barcode” or linker) to create a PCR knockout construct.
- If there is no PAM site located close to the C- or N-term of the gene of interest, a PAM site further away can be chosen but part of the sequence needs to be recodonized to avoid repair of the locus without modification (see example in Figure 3).
- If larger volumes of the template DNA is needed, or if the sequence needs to be confirmed by sequencing, the final PCR can also be integrated into a plasmid (e.g., by TA cloning into pGEM).

Figure 2. Schematic of 3-step PCR for generating template DNA. The genomic sequence of the gene of interest is used to design oligos for amplification of homology regions flanking the sequence to be deleted/replaced (red). Separately, oligos are designed to amplify the sequence to be introduced (green). In three PCR steps the template DNA for homogous repair is produced. In the first PCR step, three products are generated. In the second PCR step, products 1 and 3 are fused and in the third PCR, products 1/3 and 2 are fused. ol = oligo.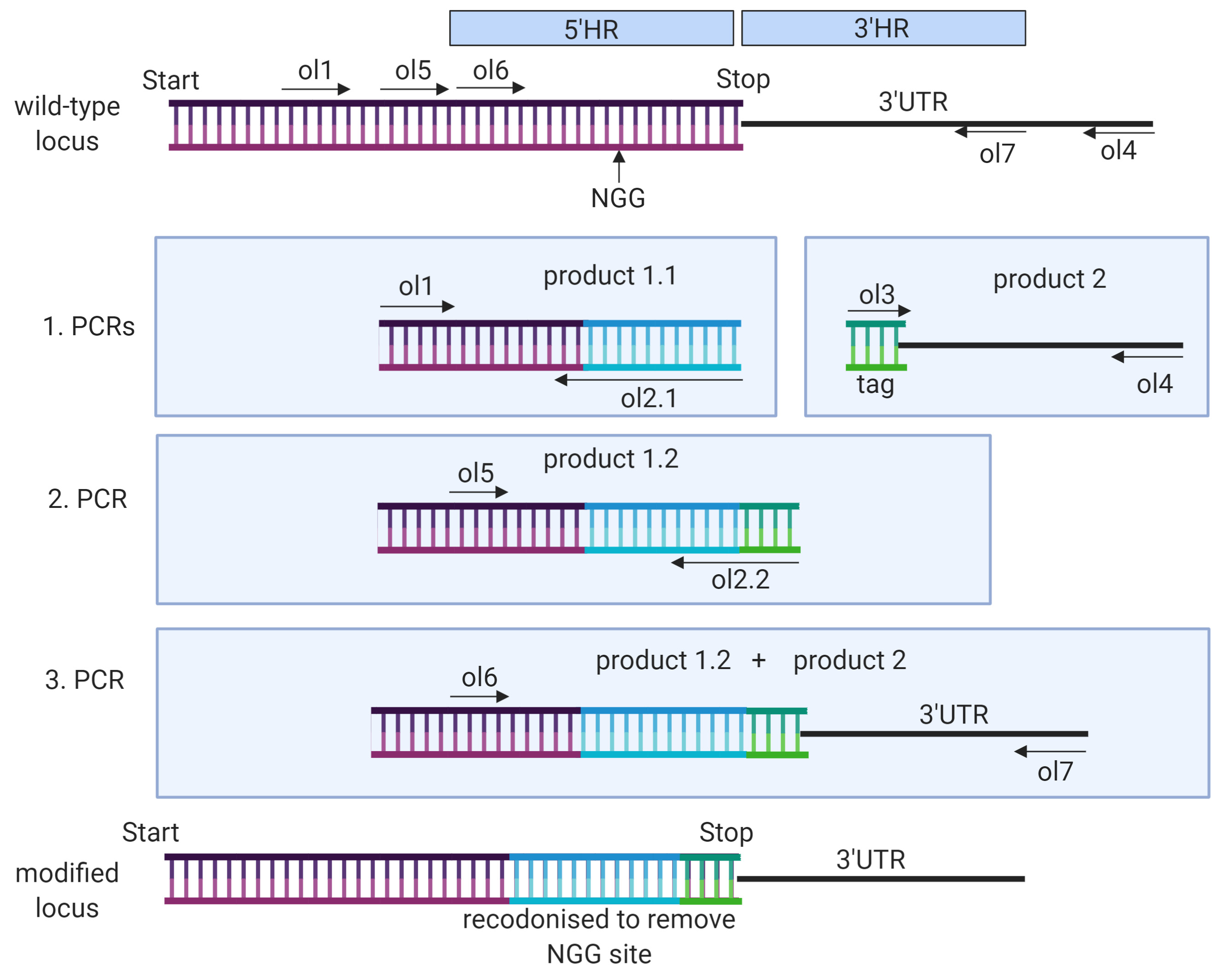
Figure 3. Schematic showing generation of template DNA with recodonization. In this example the aim is to tag the C-terminal end of a gene of interest (purple), while the chosen PAM site is within the 3′ end of the gene and the 20 bp guide sequence would not be disrupted by introduction of a tag sequence. The PAM site must therefore be deleted by introducing a recodonized sequence (blue) that runs from the PAM site up until the tag sequence (to ensure there is no chance of integration between mutated PAM site and tag). In this case the tag is a short sequence like a hemagglutinin-tag or spot-tag (green). The PCR oligos are designed as described before with the difference that at least two reverse oligos for HR1 are needed. In the first reverse oligo (oligo 2.1), the 5′ end contains the recodonized sequence (blue), the second oligo (oligo 2.2) binds within the recodonized sequence and contains the tag sequence in its 5′ end. Template DNA for the first PCR is genomic DNA, the template for second PCR is product 1.1 and the template for the final PCR are product 1.2 and product 2. - If the inserted DNA sequence is a small tag (like hemagglutinin tag) the whole tag sequence can be added as adaptors in oligos 2 and 3 and only two PCR steps are needed to fuse 5′HR and 3′HR.
- Generating oligos
- Synchronize P. knowlesi parasites via Nycodenz purification
- Transfer 5 ml of 55% Nycodenz working solution to a 15 ml conical tube and warm up to RT.
Note: 1 Nycodenz tube is needed for 1 ml of red blood cells (50 ml culture with 2% hematocrit). - Centrifuge down high parasitemia (4-10%) P. knowlesi culture at 900 x g for 4 min at RT.
- Resuspend parasite pellet at 50% hematocrit in RPMI.
- Carefully layer 2 ml of this culture above the 5 ml Nycodenz.
- Centrifuge at 900 x g for 12 min with low brake/acceleration. Uninfected red cells and ring-stage parasites will sink to the bottom and schizonts form a layer on top of the Nycodenz.
- Transfer top layer schizonts (brownish color) to a new conical tube and wash with RPMI to remove Nycodenz (see Figure 4).
- Incubate schizonts with 1 μM Compound 2 for 2-3 h. Compound 2 is a PKG inhibitor that reversibly blocks merozoite egress. This step is optional but will help to maximize yield of late schizonts and also provides the user with some flexibility in timing for subsequent steps. Viability of parasites will dramatically decline for incubations longer than 3 h.
- Wash off Compound 2 and transfer schizonts back to culture (with 2% hematocrit red blood cells).
Note: To get parasites even more synchronous undertake the steps above and then let them invade red blood cells for 30 to 60 min and Nycodenz purify again, only this time keep ring stage parasite pellet and remove schizonts. You can slow down the maturation of the parasites by leaving them at RT for several h in order to get them to the schizont stage at a convenient time for the next purification/ transfection.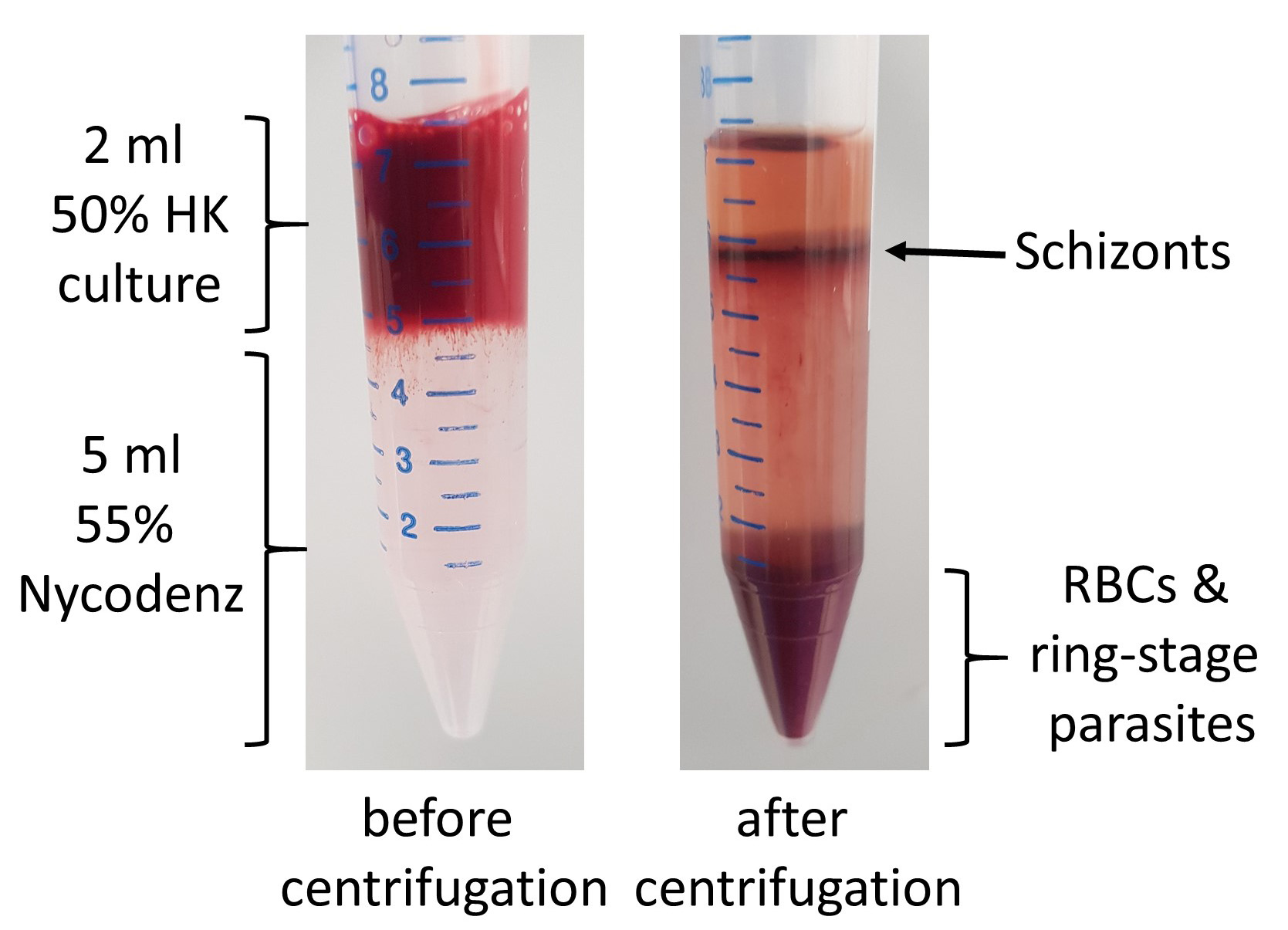
Figure 4. Schizont enrichment with Nycodenz
- Transfer 5 ml of 55% Nycodenz working solution to a 15 ml conical tube and warm up to RT.
- Transfect P. knowlesi parasites with CRISPR constructs
- For transfection of the parasites, DNA and blood need to be prepared. This can be done simultaneously.
- Blood: Spin down fresh Duffy positive red blood cells (stored for less than 1 week) and transfer 200 μl of packed cells to a new 1.5 ml reaction tube. Add 500 μl of complete media and incubate at 37 °C with shaking at 550 rpm (in, for example, an Eppendorf Thermomixer).
- DNA:
- Combine ~20 μg of the pCas9/sg_guide plasmid with ~40 to 60 μg of template DNA (~1:20 molar ratio) and adjust to a final volume of 10 μl with sterile TE buffer.
- Mix 10 μl DNA with 100 μl of supplement P3 transfection buffer (from the Lonza kit).
- Parasites:
- Purify P. knowlesi parasites with Nycodenz from a synchronous late-stage schizont (24-26 h old) culture and incubate for 2 h with 1 μM Compound 2. The best indicator of schizont age in P. knowlesi is the presence of a single compact pigment granule and full segregation of merozoites. The compact pigment granule forms approximately 30-45 min prior to egress. Maximizing the proportion of these stages will drastically improve transfection efficiency.
- After Compound 2 incubation of schizonts, wash and transfer 10 to 20 μl schizonts (∼1-2 x 108 cells) to a new 1.5 ml reaction tube. Leave them with at least 100 μl of complete media at 37 °C for 15 to 20 min.
- Add program to Amaxa 4D-Nucleofector X or choose existing experiment with the following setting: Pulse code: FP 158, Solution: Primary Cell P3, Volume: 100 μl.
- Immediately after the incubation from Step D1c.ii spin down schizonts for 1 min at 845 rcf and remove culture media.
- Transfer 100 μl of the DNA/transfection buffer to the schizont pellet and gently mix.
- Transfer 100 μl of the schizont/buffer mix to a nucleofection cuvette and move to the Amaxa 4D-Nucleofector X. Press start and wait until the screen shows green signal to indicate successful transfection. Loading more than 100 μl of mix to the cuvettes can increase probability of error messages indicating partial (“yellow”) or complete (“red”) pulse failure.
- Immediately after transfection, transfer schizonts with pastette (included in Lonza kit) to the prepared warm red blood cells. Rinse out cuvette with blood/media to avoid loss of schizont material.
- Incubate schizonts for 20-30 min at 37 °C and shaking at 550 rpm to allow invasion, before moving the culture to a 6-well plate and add 4.5 ml of complete media.
Note: In order to determine the transfection efficiency an episomal eGFP plasmid (e.g., PkconGFP) should be transfected as a control. - The day after transfection change media daily for 5 days with the addition of positive selection drug Pyrimethamine (100 nM final concentration).
- For transfection of the parasites, DNA and blood need to be prepared. This can be done simultaneously.
- Confirm transfection efficie
- Microscopy (if fluorescent episomal plasmid was transfected)
- Around 24 h post-transfection stain 30 μl of culture with 30 μl of 4 μg/ml Hoechst in PBS for 15 min at 37 °C.
- Dilute with 100 μl PBS and apply 100 μl of the dilution to a microscopy channel slide (ibidi).
- Count 20 to 50 parasites with defined Hoechst signal and check how many of these also show fluorescence of the episomal plasmid. Transfection efficiencies are usually between 5 and 40%.
- Example fluorescent microscopy images are shown in Figure 5.
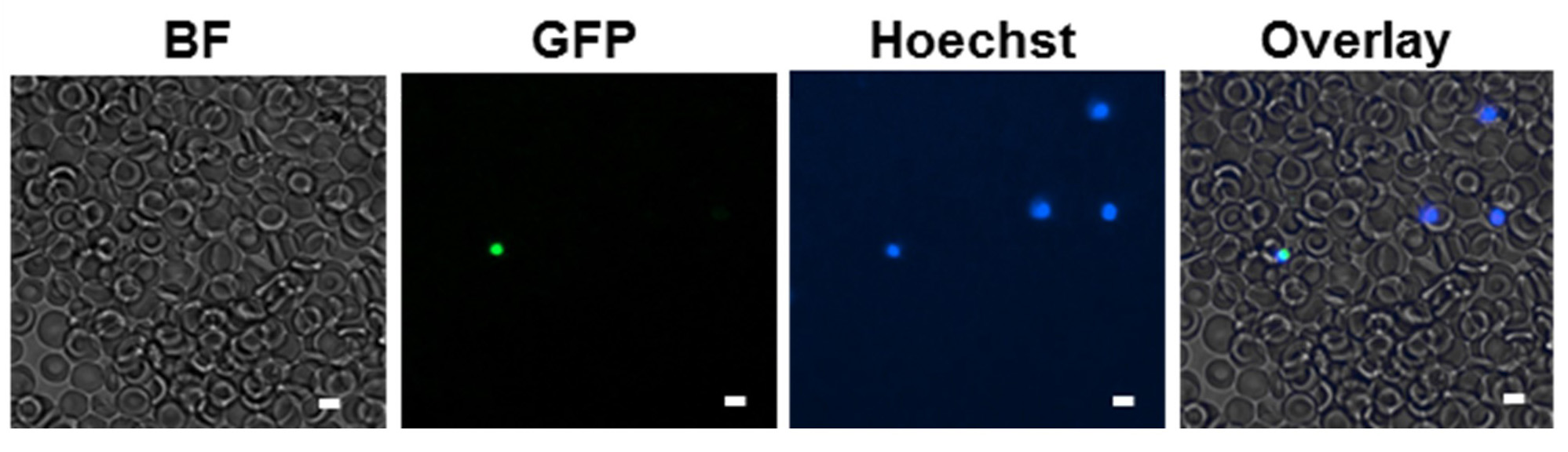
Figure 5. Microscopy image of eGFP positive parasites - Diagnostic PCR
- Choose the forward oligos at least 100 bp outside the repair template HRs in the genome. Choose one reverse oligos inside the wild-type locus and one reverse oligo inside the modified locus.
Note: The product must be unique to integrated parasites and should not also amplify out the transfected template DNA. As large quantities are used in transfections the construct can remain detectable for some time after transfection resulting in a potential false-positive PCR result. Additional control PCRs should be designed which provide a product only in wild type parasites and also in an unrelated control locus (to demonstrate there are no issues with the gDNA). - Spin down 1 ml of parasite culture, wash blood pellet with RPMI or PBS and store pellet at -20 °C or immediately use for gDNA extraction (Dneasy blood and tissue kit).
- Set up six PCR reactions to determine wild-type locus, modified locus and an independent locus of wild-type parasites and transfected parasites. Run on agarose gel as shown in Figure 6.
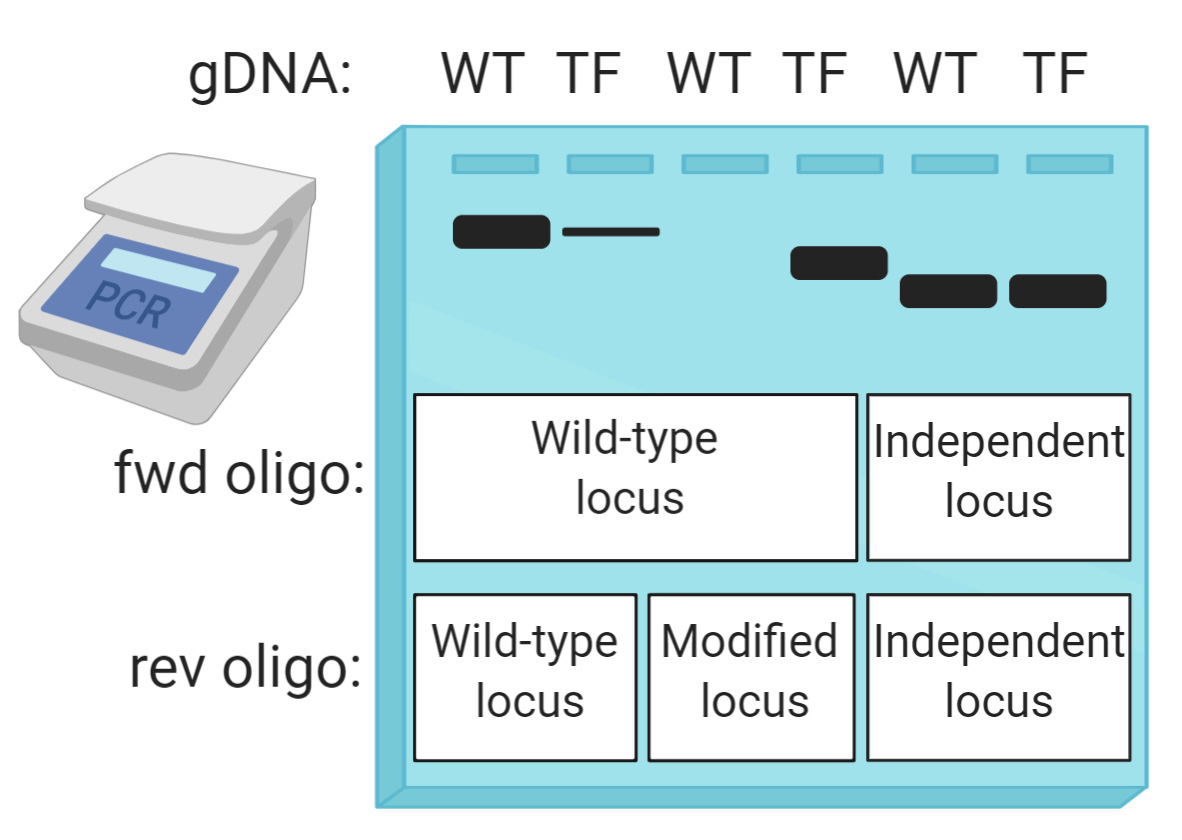
Figure 6. Diagnostic PCRs to confirm integration. The agarose gel is an example of a diagnostic PCR in order to confirm wild-type locus, integration locus and of an unrelated/independent locus. The wild-type forward oligo should be outside of the repair template (~100 bp upstream of the 5′ homology region or downstream of the 3′ homology region). The reverse oligos should be specific for wild-type or integration. The genomic DNA is from wild-type parasites (WT) before transfection or when parasites reappeared after transfection (TF).
Note: Integration can be detectable as early as two days after transfection, but PCR works more reliable with higher DNA concentrations, therefore samples should be taken when drug resistant parasites are easily detectable by Giemsa smear in the cultures (> 0.2% parasitemia).
- Choose the forward oligos at least 100 bp outside the repair template HRs in the genome. Choose one reverse oligos inside the wild-type locus and one reverse oligo inside the modified locus.
- Sequencing
Amplify the modified gene locus by PCR and confirm by sequencing.
- Microscopy (if fluorescent episomal plasmid was transfected)
- Remove Cas9/sg_guide plasmid containing parasites with negative selection
- Add 1 μM Ancotil (5-Fluorocytosine) for 7 days to parasite culture of ~0.5% (parasite should be confirmed integration positive and are stably growing in culture).
Note: Without positive selection, parasites lose plasmid very quickly, therefore no big drop in parasitemia is expected. - Clone out parasites by limiting dilution (Moon et al., 2013).
- Confirm integration in several clonal lines by PCR or microscopy as described above.
- Confirm removal of Cas9/sg_guide plasmid in integration positive clonal lines by treating parasites for 5 days with Pyrimethamine as described before. Keep culture for another week to test if parasites reappear.
Note: Prepare cryo stocks of transfected parasites (bulk culture) and clonal lines.
- Add 1 μM Ancotil (5-Fluorocytosine) for 7 days to parasite culture of ~0.5% (parasite should be confirmed integration positive and are stably growing in culture).
Notes
All parasite work should be carried out in a class II microbiological safety hood for both sterility and user protection. As P. knowlesi is a human infective species all work must be carried out under appropriate approved safety and containment conditions. P. knowlesi cultures can be grown in the presence of gentamycin (25 mg/L) antibiotics, however with good sterile technique this should not be necessary, nor is it recommended.
The transfection and integration efficiencies can be variable if not fully optimized. High efficiencies can be achieved by:
- Using fresh blood (7 days old or younger).
- Making sure you have at least 10 μl of schizonts, lower amounts will disproportionately affect efficiency.
- Ensuring that you are using very late and highly synchronous schizonts (with a large proportion fully segmented with a condensed pigment granule).
- Ensuring that your P. knowlesi parasites are growing reliably 3-4 fold per day (for human RBCs) prior to transfection.
- Ensuring you have sufficient DNA.
- Using long homology regions (500-1,000 bp).
- Comparing several guide sequences.
Recipes
- Nycodenz stock solution (100%)
Adjust to 100 ml and pH 7.5, sterile filer and store at 4 °CCompound Final concentration For 100 ml Notes Nycodenz 27.6% (w/v) 27.6 g Add to 50 ml warm H2O and dissolve by heating (60 °C) and stirring Tris HCl 5 mM 2.5 ml of 1 M stock KCl 3 mM 22.37 mg CaNa2·EDTA 0.3 mM 11.23 mg - Nycodenz working solution (55%)
Mix 55 ml Nycodenz stock solution with 45 ml RPMI. Store at 4 °C - Pyrimethamine
- Prepare a 10 mM stock solution in DMSO, prepare 60 μl aliquots and store at -20 °C
- Prepare a 100 μM working solution by adding 50 μl of the stock solution to 50 ml of RPMI
- Sterile filter and store at 4 °C. Don’t keep the working solution for more than 2 weeks
- Add 5 μl of working solution to 5 ml of parasite culture (100 nM final concentration in culture)
- Ancotil
- Prepare a 1 mM working solution by diluting the sterile stock solution (2.5 g/250 ml = 77 mM = 1%) 1/77 in RPMI
- Keep at room temperature
- Add 5 μl to 5 ml parasite culture (1 μM)
- Complete media
RPMI-1640 (HEPES Modification, With 25 mM HEPES, without L-glutamine) with the following additions:
2.3 g/L sodium bicarbonate
2 g/L dextrose
0.05 g/L Hypoxanthine
5 g /L Albumax II
0.3 g/L L-glutamine (10 ml of 200 mM solution pro 1 L media)
10% (vol/vol) horse serum
Sterile filter and store at 4 °C
Acknowledgments
This work was supported by an MRC Career Development Award (MR/M021157/1) jointly funded by the UK Medical Research Council and Department for International Development (R.W.M, F.M), by a Bloomsbury Colleges research studentship and a Wellcome Trust Senior Investigator Award (106240/Z/14/Z). The protocol is adapted from Ecker et al. (2006), Ghorbal et al. (2014) and Mohring et al. (2019).
Competing interests
The authors declare no competing financial interests.
Ethics
The project, consent and protocol were approved by the LSHTM Observational Research Ethics Committee under project reference 5520-1.
References
- Cooper, D. J., Rajahram, G. S., William, T., Jelip, J., Mohammad, R., Benedict, J., Alaza, D. A., Malacova, E., Yeo, T. W., Grigg, M. J., Anstey, N. M. and Barber, B. E. (2020). Plasmodium knowlesi malaria in Sabah, Malaysia, 2015-2017: ongoing increase in incidence despite near-elimination of the human-only Plasmodium species. Clin Infect Dis 70(3):361-367.
- Ecker, A., Moon, R., Sinden, R. E. and Billker, O. (2006). Generation of gene targeting constructs for Plasmodium berghei by a PCR-based method amenable to high throughput applications. Mol Biochem Parasitol 145(2): 265-268.
- Ghorbal, M., Gorman, M., Macpherson, C. R., Martins, R. M., Scherf, A. and Lopez-Rubio, J. J. (2014). Genome editing in the human malaria parasite Plasmodium falciparum using the CRISPR-Cas9 system. Nat Biotechnol 32(8): 819-821.
- Gruring, C., Moon, R. W., Lim, C., Holder, A. A., Blackman, M. J. and Duraisingh, M. T. (2014). Human red blood cell-adapted Plasmodium knowlesi parasites: a new model system for malaria research. Cell Microbiol 16(5): 612-620.
- Knuepfer, E., Wright, K. E., Prajapati, S. K., Rawlinson, T. A., Mohring, F., Koch, M., Lyth, O. R., Howell, S. A., Villasis, E., Snijders, A. P., Moon, R. W., Draper, S. J., Rosanas-Urgell, A., Higgins, M. K., Baum, J. and Holder, A. A. (2019). Divergent roles for the RH5 complex components, CyRPA and RIPR in human-infective malaria parasites. Plos Pathog 15(6).
- Lim, C., Hansen, E., DeSimone, T. M., Moreno, Y., Junker, K., Bei, A., Brugnara, C., Buckee, C. O. and Duraisingh, M. T. (2013). Expansion of host cellular niche can drive adaptation of a zoonotic malaria parasite to humans. Nat Commun 4: 1638.
- Lubis, I. N. D., Wijaya, H., Lubis, M., Lubis, C. P., Divis, P. C. S., Beshir, K. B. and Sutherland, C. J. (2017). Contribution of Plasmodium knowlesi to multispecies human malaria infections in north Sumatera, Indonesia. J Infect Dis 215(7): 1148-1155.
- Mohring, F., Hart, M. N., Rawlinson, T. A., Henrici, R., Charleston, J. A., Benavente, E. D., Patel, A., Hall, J., Almond, N., Campino, S., Clark, T. G., Sutherland, C. J., Baker, D. A., Draper, S. J. and Moon, R. W. (2019). Rapid and iterative genome editing in the malaria parasite Plasmodium knowlesi provides new tools for P. vivax research. Elife 8: e45829.
- Moon, R. W., Hall, J., Rangkuti, F., Ho, Y. S., Almond, N., Mitchell, G. H., Pain, A., Holder, A. A. and Blackman, M. J. (2013). Adaptation of the genetically tractable malaria pathogen Plasmodium knowlesi to continuous culture in human erythrocytes. Proc Natl Acad Sci U S A 110(2): 531-536.
- Rajahram, G. S., Cooper, D. J., William, T., Grigg, M. J., Anstey, N. M. and Barber, B. E. (2019). Deaths from Plasmodium knowlesi malaria: case series and systematic review. Clin Infect Dis 69(10):1703-1711.
- Rawlinson, T. A., Barber, N. M., Mohring, F., Cho, J. S., Kosaisavee, V., Gerard, S. F., Alanine, D. G. W., Labbe, G. M., Elias, S. C., Silk, S. E., Quinkert, D., Jin, J., Marshall, J. M., Payne, R. O., Minassian, A. M., Russell, B., Renia, L., Nosten, F. H., Moon, R. W., Higgins, M. K. and Draper, S. J. (2019). Structural basis for inhibition of Plasmodium vivax invasion by a broadly neutralizing vaccine-induced human antibody. Nat microbial 4(9):1497-1507.
- WHO. (2018). World Malaria Report. World Health Organization.
Article Information
Copyright
![]() Mohring et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Mohring et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Mohring, F., Hart, M. N., Patel, A., Baker, D. A. and Moon, R. W. (2020). CRISPR-Cas9 Genome Editing of Plasmodium knowlesi. Bio-protocol 10(4): e3522. DOI: 10.21769/BioProtoc.3522.
- Mohring, F., Hart, M. N., Rawlinson, T. A., Henrici, R., Charleston, J. A., Benavente, E. D., Patel, A., Hall, J., Almond, N., Campino, S., Clark, T. G., Sutherland, C. J., Baker, D. A., Draper, S. J. and Moon, R. W. (2019). Rapid and iterative genome editing in the malaria parasite Plasmodium knowlesi provides new tools for P. vivax research. Elife 8: e45829.
Category
Cell Biology > Cell engineering > CRISPR-cas9
Microbiology > Microbial genetics > CRISPR-Cas9
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.