- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

ChIP-Seq from Limited Starting Material of K562 Cells and Drosophila Neuroblasts Using Tagmentation Assisted Fragmentation Approach
(*contributed equally to this work) Published: Vol 10, Iss 4, Feb 20, 2020 DOI: 10.21769/BioProtoc.3520 Views: 4760
Reviewed by: Imre GáspárPrashanth N SuravajhalaNarendranath Bhokisham
Original research article
The authors used this protocol in:

Jul 2019
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Chromatin immunoprecipitation is extensively used to investigate the epigenetic profile and transcription factor binding sites in the genome. However, when the starting material is limited, the conventional ChIP-Seq approach cannot be implemented. This protocol describes a method that can be used to generate the chromatin profiles from as low as 100 human or 1,000 Drosophila cells. The method employs tagmentation to fragment the chromatin with concomitant addition of sequencing adaptors. The method generates datasets with high signal to noise ratio and can be subjected to standard tools for ChIP-Seq analysis.
Keywords: Low Input ChIP-SeqBackground
Epigenetic state and transcription factors occupancy are critical determinants of gene expression. To understand this regulation, the precise mapping of chromatin features are very important. Chromatin immunoprecipitation coupled with next generation sequencing is a powerful technique that gives valuable insight into genome wide distributions of chromatin features (Ghavi-Helm et al., 2016). Although powerful, this technique is limited in its application owing to the need of significant starting material which in some conditions are hard to meet, if not completely impossible. This limitation has driven many recent efforts to adapt ChIP-Seq approach for low amount starting material or for rare cell types (Adli et al., 2010; Zheng et al., 2015). One of the key limitations of these efforts is the use of sonication or MNAse for fragmenting the chromatin. The former approach is detrimental for the epitope when present in limited amount as it can lead to destruction of the epitope used for subsequent immunoprecipitation (Stathopulos et al., 2004). The MNAse approach on the other hand is hard to control for its efficacy and saturation. The recent application of CUT&RUN approach was successful in generating profiles from samples of 100 cells using antibody-targeted micrococcal nuclease (Skene et al., 2018). However, all these approaches still resort to ligation-based library preparation approach involving multiple steps, potentially leading to increased variability as well as of loss of complexity (Seguin-Orlando et al., 2013). The method described here overcomes these limitations by using commercially available Tn5 enzyme for fragmenting the chromatin with simultaneous preparation of libraries with fewer intermediate steps. The direct comparison of CUT&RUN approach and TAF-ChIP reveals superior signal to noise ratio in the later, making use of standard bioinformatics pipeline amenable to this approach.
Materials and Reagents
- DNA Low binding 1.5 ml tubes (Eppendorf, catalog number: 0030108051)
- Phase Lock tubes (5Prime, catalog number: 2302830)
- Agencourt AMPure XP Beads (Beckmann Coultier, catalog number: A63381)
- Protein G Dynabeads (Thermo Fisher Scientific, catalog number: 10003D)
- BSA (Sigma-Aldrich, catalog number: A4737)
- Proteinase K (Thermo Fisher Scientific, catalog number: E00491)
- Yeast tRNA (Sigma-Aldrich, catalog number: R8759)
- Anti H3K27Me3 antibody (Active Motif, catalog number: 39155)
- Anti H3 antibody (Abcam, catalog number: ab1791)
- Anti H3K4Me3 antibody (Abcam, catalog number: ab8580)
- Anti H3K9Me3 antibody (Active Motif, catalog number: 39161)
- Collagenase I (Sigma-Aldrich, catalog number: 1148089)
- Papain (Sigma-Aldrich, catalog number: 1495005)
- Primers (Table 1)
Table 1. List of primers with unique barcodesPrimers fw ATAC-seq primer, general, no index AATGATACGGCGACCACCGAGATCTACACTCGTCGGCAGCGTCAGATGT*G rev ATAC-seq primer, Truseq index, 34 CATGGC CAAGCAGAAGACGGCATACGAGATGCCATGGTCTCGTGGGCTCGGAGATG*T rev ATAC-seq primer, Truseq index, 48 TCGGCA CAAGCAGAAGACGGCATACGAGATTGCCGAGTCTCGTGGGCTCGGAGATG*T rev ATAC-seq primer, Truseq index, 22 CGTACG CAAGCAGAAGACGGCATACGAGATCGTACGGTCTCGTGGGCTCGGAGATG*T rev ATAC-seq primer, Truseq index, 15 ATGTCA CAAGCAGAAGACGGCATACGAGATTGACATGTCTCGTGGGCTCGGAGATG*T rev ATAC-seq primer, Truseq index, 46 TCCCGA CAAGCAGAAGACGGCATACGAGATTCGGGAGTCTCGTGGGCTCGGAGATG*T rev ATAC-seq primer, Truseq index, 45 TCATTC CAAGCAGAAGACGGCATACGAGATGAATGAGTCTCGTGGGCTCGGAGATG*T rev ATAC-seq primer, Truseq index, 40 CTCAGA CAAGCAGAAGACGGCATACGAGATTCTGAGGTCTCGTGGGCTCGGAGATG*T rev ATAC-seq primer, Truseq index, 39 CTATAC CAAGCAGAAGACGGCATACGAGATGTATAGGTCTCGTGGGCTCGGAGATG*T rev ATAC-seq primer, Truseq index, 38 CTAGCT CAAGCAGAAGACGGCATACGAGATAGCTAGGTCTCGTGGGCTCGGAGATG*T rev ATAC-seq primer, Truseq index, 37 CGGAAT CAAGCAGAAGACGGCATACGAGATATTCCGGTCTCGTGGGCTCGGAGATG*T rev ATAC-seq primer, Truseq index, 36 CCAACA CAAGCAGAAGACGGCATACGAGATTGTTGGGTCTCGTGGGCTCGGAGATG*T rev ATAC-seq primer, Truseq index, 35 CATTTT CAAGCAGAAGACGGCATACGAGATAAAATGGTCTCGTGGGCTCGGAGATG*T rev ATAC-seq primer, Truseq index, 25 ACTGAT CAAGCAGAAGACGGCATACGAGATATCAGTGTCTCGTGGGCTCGGAGATG*T rev ATAC-seq primer, Truseq index, 26 ATGAG CAAGCAGAAGACGGCATACGAGATGCTCATGTCTCGTGGGCTCGGAGATG*T rev ATAC-seq primer, Truseq index, 27 ATTCCT CAAGCAGAAGACGGCATACGAGATAGGAATGTCTCGTGGGCTCGGAGATG*T rev ATAC-seq primer, Truseq index, 28 CAAAAG CAAGCAGAAGACGGCATACGAGATCTTTTGGTCTCGTGGGCTCGGAGATG*T rev ATAC-seq primer, Truseq index, 23 GAGTGG CAAGCAGAAGACGGCATACGAGATCCACTCGTCTCGTGGGCTCGGAGATG*T rev ATAC-seq primer, Truseq index, 24 GGTAGC CAAGCAGAAGACGGCATACGAGATGCTACCGTCTCGTGGGCTCGGAGATG*T rev ATAC-seq primer, Truseq index, 42 TAATCG CAAGCAGAAGACGGCATACGAGATCGATTAGTCTCGTGGGCTCGGAGATG*T rev ATAC-seq primer, Truseq index, 41 GACGAC CAAGCAGAAGACGGCATACGAGATGTCGTCGTCTCGTGGGCTCGGAGATG*T - NEXTERA XT DNA Library prep Kit (Illumina, catalog number: FC-131-1024)
- NEBNext High-Fidelity 2x PCR Master Mix (New England Biolabs, catalog number: M0541)
- Glycogen (Roche, catalog number: 10901393001)
- 37% Formaldehyde (Sigma-Aldrich, catalog number: F8775)
- Glycine (Sigma-Aldrich, catalog number: 50046)
- Protease Inhibitor Cocktail (Bimake, catalog number: B14001)
- Tris Buffer pH 8.0 (Applichem, catalog number: A4577)
- EDTA (Sigma-Aldrich, EDS catalog number: EDS-500G)
- Sodium Chloride (Sigma-Aldrich, catalog number: S3014)
- Triton X-100 (Sigma-Aldrich, catalog number: T8787)
- 20% sodium dodecyl sulfate (Sigma-Aldrich, catalog number: 05030)
- Nuclease-free water (Qiagen, catalog number: 129115)
- Tris (hydroxymethyl) aminomethane (Sigma-Aldrich, catalog number: 252859)
- N,N-Dimethylformamide (Sigma-Aldrich, catalog number: D4551)
- Qubit dsDNA High Sensitivity Kit (Thermo Fisher Scientific, catalog number: Q33230)
- DNA High Sensitivity Kit for Bioanalyzer (Agilent, catalog number: 5067-4627)
- Schneider’s medium (Thermo Fisher Scientific, catalog number: 21720-001)
- Phenol:Chloroform (Sigma-Aldrich, catalog number: P3803)
- 3 M sodium acetate (Thermo Fisher Scientific, catalog number: AM9740)
- Ethanol (Sigma-Aldrich, catalog number: 51976)
- Hoechst stain (Thermo Fisher Scientific, catalog number: 62249)
- MgCl2 (Sigma-Aldrich, catalog number: 63068)
- Dissociation Solution (see Recipes)
- Blocking Buffer (see Recipes)
- RIPA (140 mM) (see Recipes)
- RIPA (250 mM) (see Recipes)
- TE Buffer (see Recipes)
- Tagmentation Buffer (Home-made) (see Recipes)
Equipment
- DiaMag 0.2 ml-magnetic rack (Diagenode, catalog number: B04000001)
- DynaMag-2 magnetic rack (Thermo Fisher Scientific, catalog number: 12321D)
- Bioruptor plus (Diagenode, catalog number: B01020001)
- Thermomix compact (Eppendorf, catalog number: T1317)
- Bio-Rad PCR Cycler (Bio-Rad, catalog number: C1000)
- Mini tube rotator (Biotools, AG, R2001)
- Tabletop minicentrifuge (Thermo Fisher Scientific, catalog number: 75002478)
- Agilent Bioanalyzer 2100 (Agilent, catalog number: G2939BA)
- Qubit Fluorometer 3 (Thermo Fisher Scientific, catalog number: Q33216)
- Flow cytometer (BD FACS Aria)
Software
- MACS2 (version 2.1.1.20160309, https://github.com/taoliu/MACS)
- Deeptools (version 3.1.3, https://github.com/deeptools/deepTools)
- Samtools (version 1.09, https://github.com/samtools/samtools)
- bowtie2 (version 2.3.5, https://github.com/BenLangmead/bowtie2)
- FastQC (version 0.10.1, https://github.com/s-andrews/FastQC)
Procedure
- Cell fixation and sorting (human cells)
- Harvest the cells in a 1.5 ml Eppendorf tube, and resuspend them in PBS.
- Fix the cells for 10 min at room temperature with 1% formaldehyde (in PBS).
- Quench the crosslink with 125 mM glycine and incubate for 5 min at room temperature.
- Stain the cells with Hoechst stain and use the flow cytometer to sort 100 cells into 200 μl of 140 mM RIPA buffer (Recipe 3).
Note: Sorting small number of cells will have no effect on buffer composition owing to small volume.
- Cell isolation, fixation and sorting (Drosophila neuroblasts)
- Dissect the required number of larval brains in PBS (for example: transgenic flies expressing GFP-tagged deadpan (Dpn) protein under the control of its endogenous enhancer).
- Wash once with PBS and fix the brains with 1% formaldehyde (in PBT) for 10 min at room temperature.
- Quench the crosslink with 125 mM glycine and incubate for 5 min at room temperature.
- Remove the quenching solution and wash twice with PBS.
- Add the dissociation solution (Recipe 1) to the brain tissue and leave it for 1 h at 30 °C with constant shaking at 650 rpm. Stir the brains at every 15 min interval by pipetting them up and down.
- Pellet the cells by centrifuging them at 100 x g and resuspend in PBS.
- Sort cells on flow cytometer under GFP channel, and directly sort 1,000 cells into 200 μl of 140 mM RIPA buffer.
Note: Sorting small number of cells will have no effect on buffer composition owing to small volume.
- Immunoprecipitation
- Bind 1 μg of antibody to 15 μl of protein G dynabeads in 200 μl blocking buffer (Recipe 2).
- Incubate at 4 °C for 2-3 h.
- Break the nuclei of the cells collected in RIPA for 3 cycles at low power setting in a Bioruptor sonicator, 30 sec “ON”/“OFF”.
- Remove the blocking buffer by putting the tubes on DynaMag-2 magnetic rack, and add the lysate from Step C1.
- Incubate at 4 °C for overnight with head over tail rotations.
- Separate the beads with magnetic rack and wash twice with 300 μl of home-made tagmentation buffer (Recipe 6) by resuspending the beads with 1,000 μl pipette. Pulse-spin the tubes on micro centrifuge and separate again with DynaMag-2 magnetic rack, remove the buffer as much as possible without disturbing the beads.
- Tagmentation, reverse cross linking, and Phenol:Chloroform extraction
- Resuspend the beads in 20 μl of 1x tagmentation buffer containing 1 μl of Tn5 transposase (from NEXTERA XT DNA Library prep Kit).
- Incubate the resuspended beads at 37 °C for 40 min in a thermoblock with constant shaking at 500 rpm.
- Remove tagmentation buffer by separating the beads on DynaMag-2 magnetic rack and wash the beads as following:
- Once with 140 mM RIPA;
- Four times with 250 mM RIPA (Recipe 4);
- Twice with TE buffer (Recipe 5).
- Resuspend the washed beads in 100 μl of TE buffer, and add 5 μl of 20 mg/ml proteinase K. Incubate the beads for at least 6 h at 60 °C in thermoblock with shaking at 500 rpm.
- Add 100 μl of TE buffer, followed by 300 μl of Phenol:Chloroform. Vortex the tubes briefly and transfer the content to the phase lock tube (pre-spun at 14,000 rpm for 30 s). Centrifuge the tubes at 20,000 x g for 5 min at room temperature.
- Transfer the upper aqueous phase to fresh DNA low bind tube, add 5 μl of 20 mg/ml of glycogen and 20 μl of 3 M sodium acetate (pH 5.2) in the exact order, vortex briefly. Add 700 μl of absolute ethanol and vortex briefly. Incubate overnight at -80 °C to precipitate.
- Centrifuge the tubes at full speed (20,000 x g ) at 4 °C for 30-45 min. Wash the pellet once with 75% ethanol by centrifuging at 20,000 x g at 4 °C for 10 min and resuspend the pellet in 30 μl of TE buffer.
- PCR amplification and purification
- Set up the PCR reaction as per the following reaction mix.
Forward primer (Universal) 20 μM: 2 μl
Reverse primer (with index) 20 μM: 2 μl
Reverse cross-linked material: 30 μl
Nuclease free water: 16 μl
2x NEBNext High-Fidelity PCR Mix: 50 μl - Run the PCR reaction in a thermal cycler with heated lid set at 105 °C, according to following cycling parameter:
72 °C for 3 min
{98 °C for 10 s
63 °C for 30 s
72 °C for 30 s}, 12 cycles
72 °C for 5 min
Hold at 4 °C. - In the meantime, when PCR reaction is running, remove the Ampure XP beads from 4 °C to room temperature. The Ampure beads should be left at room temperature for at least 30 min to equilibrate, prior to use.
- Add 0.2x volume (relative to PCR reaction volume) of Ampure XP beads to the PCR reaction and incubate at room temperature for 5 min. Separate the beads on DiaMag 0.2 ml magnetic rack and carefully transfer the solution without carrying over the beads to a new PCR tube. Discard the Ampure XP beads.
- Add 0.8x volume of Ampure XP (relative to PCR reaction volume) beads to the solution and incubate at room temperature for 5 min. Separate the beads on DiaMag 0.2 ml magnetic rack and carefully discard the solution without disturbing the beads.
- Wash the beads twice with 200 μl freshly prepared of 80% ethanol, without disturbing the beads.
- Dry the beads until the appearance of the bead surface turns from glossy to matte.
- Elute the finished library in 10-15 μl of nuclease free water.
- Set up the PCR reaction as per the following reaction mix.
- Quality control of libraries
- Measure the concentration of the library with Qubit dsDNA High Sensitivity Kit, following the manufacture’s protocol.
- Run the library on a DNA High Sensitivity Chip with Agilent Bioanalyzer, to check for size distribution of the DNA fragments. Follow the protocol provided with the DNA High Sensitivity Kit for loading and running the sample.
- Determine the molar concentration of the library and dilute it to 10 nM concentration or per the requirement of the sequencing facility.
Note: The concentration of the library and average size fragment of the library (as revealed by Bioanalyzer) is used to calculate the molar concentration. The formula employed to calculate the concentration is, concentration in nM = (concentration in ng/μl from Qubit measurement)/ (660 g/mol x average size of the library) x 106.
Data analysis
- Sequence quality check
Use FastQC to assess the sequencing quality, using fastqc files. Alternatively, one can also use other equivalent QC tools such as MAPQC. - Generate index for reference genomes using bowtie2 (download the fasta files from Ensemble database)
bowtie2-build -f $FASTA_PATH/genome.fa $INDEX_PATH/bowtie2_index - Mapping the reads either to dm6 or hg38 assembly using bowtie2 (Langmead, 2010)
bowtie2-x $INDEX_PATH/genome -p 24 -1$SEQ_PATH/*file_1*.gz -2 $SEQ_PATH/*file_2*.gz | samtools view -bS > file.bam - Sorting mapped reads
Samtools sort file.bam > file.sorted.bam - Indexing bam files
Samtools index file.bam file.bam.bai - Generating coverage files using DeepTools (Ramirez et al. , 2016)
bamCoverage–b file.bam–normalizeUsing RPKM-bs 50–smoothLength 175-o file.bw
A representative track example after generating coverage files is shown in Figure 1. - Peak calling using MACS2, and H3 as control (Zhang et al. , 2008)
Macs2 callpeak-t $File-c $File -f BAMPE-g d–outdir–name
Macs2 callpeak-t $File-c $File -f BAMPE-g hs–outdir–name - Heatmaps using DeepTools
ComputeMatrix scale-regions–S file.bw-R genes.bed–b 600–a 600-o matrix.mat.gz
plotHeatmap-m matrix.mat.gz-out Heatmap.pdf
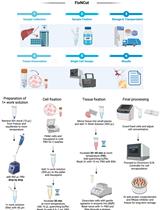
A flow-chart displaying the major steps in the protocol is shown in Figure 1. A representative track example after generating coverage files is shown in Figure 2. The comparison of TAF-ChIP approach performed with 100 cells and recently published CUT&RUN datasets, together with the ENCODE datasets, is shown in Figure 3.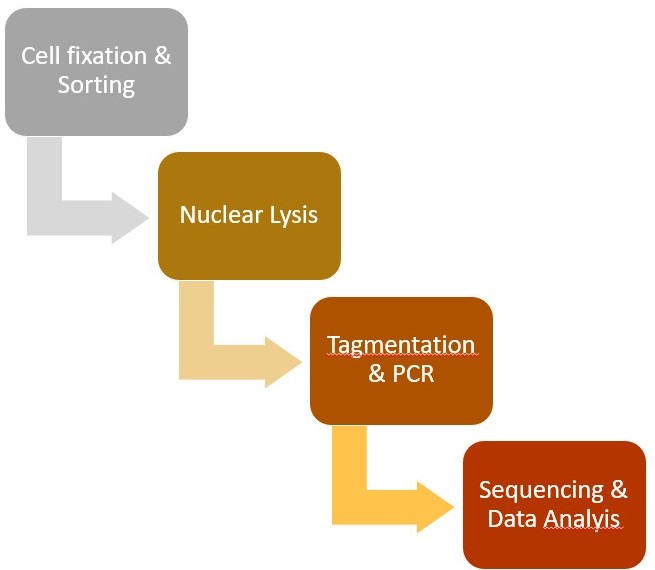
Figure 1. Steps involved in the Tagmentation Assisted Fragmentation (TAF)-ChIP protocol and subsequent data processing. Steps involved in the TAF-ChIP workflow in chronological order represented as a flow-chart.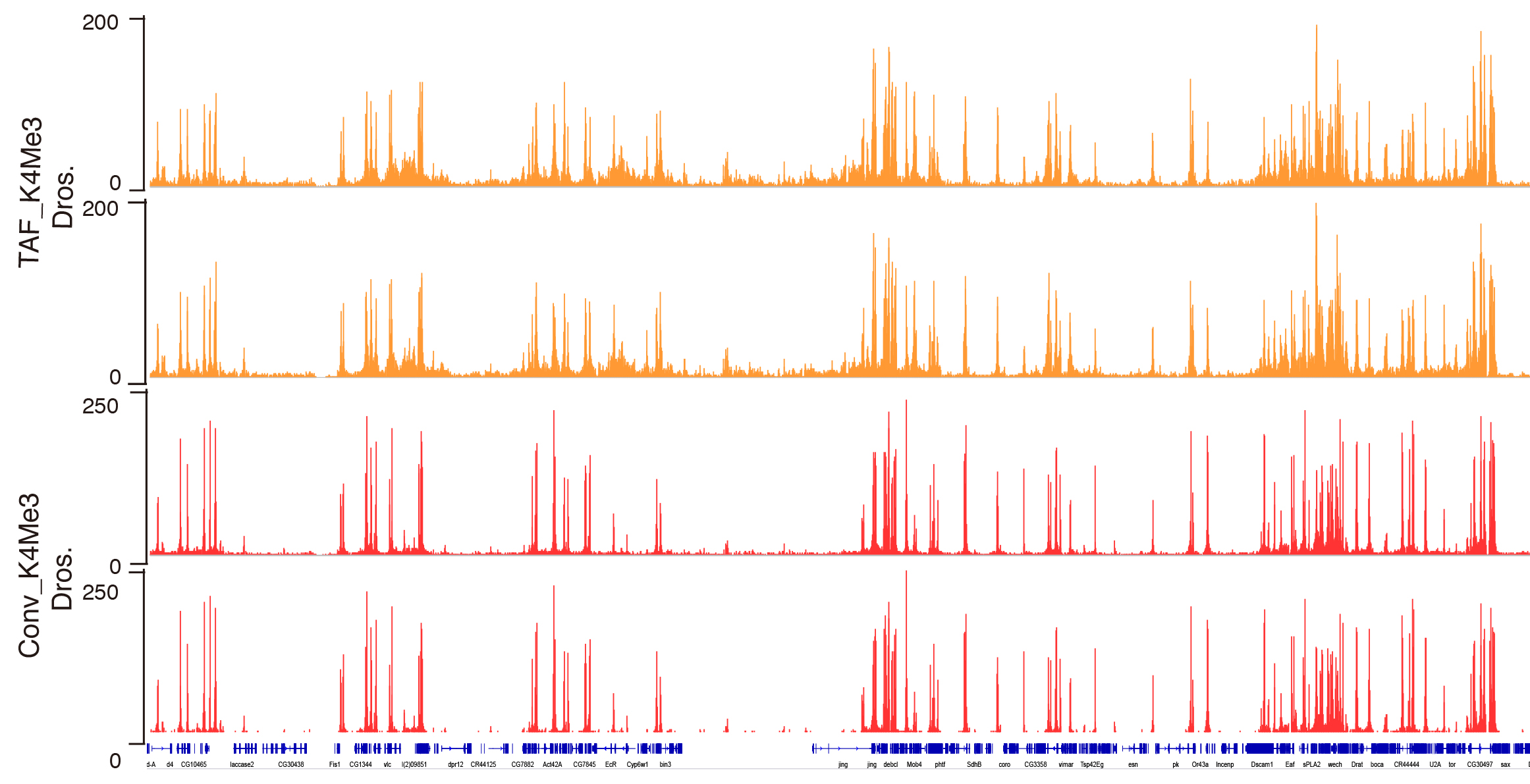
Figure 2. Genome browser track example of H3K4Me3 generated by TAF-ChIP approach and conventional ChIP-Seq approach. Genome browser tracks of ChIP performed in 1,000 FACS sorted Drosophila neural stem cells (NSCs) with TAF-ChIP approach (TAF_K4Me3) and corresponding conventional ChIP-Seq datasets from 1.2 million FACS sorted NSCs (Conv_K4Me3) with independent biological duplicates. The label below the tracks shows the gene model and the y-axis represents normalized read density in reads per million.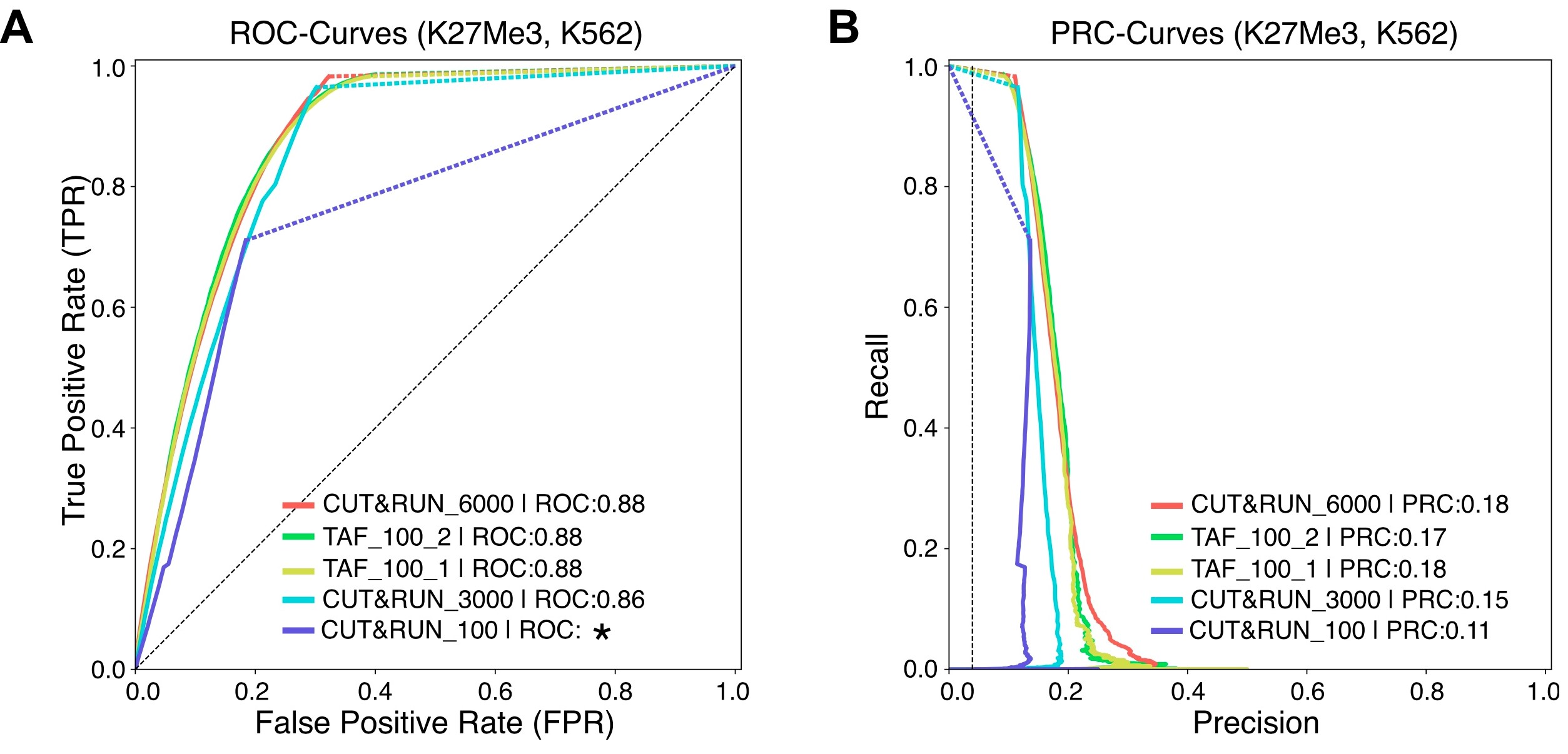
Figure 3. Comparison of TAF-ChIP with conventional ChIP-Seq and with another contemporary low amount method. A. Comparison of TAF-ChIP and CUT&RUN for H3K27Me3 in K562 cells using ROC curves. The plots were generated using the conventional ChIP-Seq ENCODE dataset as reference with 5% FDR cutoff. The peaks for TAF-ChIP replicates and the CUT&RUN datasets were generated with MACS2 without FDR thresholding. Peaks were mapped to 5 kb non-overlapping genomic windows to calculate true-positive rate, false-positive rate and precision for a changing P-Value threshold. Area under the curve (AUC) is indicated in the legend and the* indicates the failure to calculate the AUC. B. Precision-recall curve for TAF-ChIP and CUT&RUN datasets for H3K27Me3 in K562 cells. The figure is adapted from Akhtar et al. (2019).
Recipes
- Dissociation Solution
Schneider’s medium
1 mg/ml collagenase I
1 mg/ml papain - Blocking Buffer
10 mM Tris-Cl pH 8.0
140 mM NaCl
0.5 mM EDTA pH 8.0
1% Triton X-100
0.1% SDS
0.2 mg/ml BSA
0.05 mg/ml Glycogen
0.2 mg/ml Yeast tRNA - RIPA (140 mM)
10 mM Tris-Cl pH 8.0
140 mM NaCl
0.5 mM EDTA pH 8.0
1% Triton X-100
0.1% SDS - RIPA (250 mM)
10 mM Tris-Cl pH 8.0
140 mM NaCl
0.5 mM EDTA pH 8.0
1% Triton X-100
0.1% SDS - TE Buffer
10 mM Tris-Cl pH 8.0
0.5 mM EDTA pH 8.0 - Tagmentation Buffer (Home-made)
20 mM Tris(hydroxymethyl)aminomethane pH 7.6
10 mM MgCl2
20% (vol/vol) dimethyl formamide
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft (DFG) grant DFG BE 4728 1-1 and 3-1. The International PhD Programme (IPP) of the Institute of Molecular Biology, Mainz for supporting the PhD work of S.A. and P.M. We would also like to thank IMB FACS core facility for helping us in sorting.
This work is a detailed and adapted version of our previously published work (Akhtar et al. , 2019). The FACS sorting workflow was adapted and modified from an earlier work (Berger et al. , 2012).
Competing interests
The authors declare no financial and non-financial competing interest.
Ethics
The described experiments in Drosophila were performed according to the guidelines (Invertebrates are not under animal welfare/ethics laws in Germany). None of the experiments were performed on human subjects.
References
- Adli, M., Zhu, J. and Bernstein, B. E. (2010). Genome-wide chromatin maps derived from limited numbers of hematopoietic progenitors. Nat Methods 7(8): 615-618.
- Akhtar, J., More, P., Albrecht, S., Marini, F., Kaiser, W., Kulkarni, A., Wojnowski, L., Fontaine, J. F., Andrade-Navarro, M. A., Silies, M. and Berger, C. (2019). TAF-ChIP: an ultra-low input approach for genome-wide chromatin immunoprecipitation assay. Life Sci Alliance 2(4).
- Berger, C., Harzer, H., Burkard, T. R., Steinmann, J., van der Horst, S., Laurenson, A. S., Novatchkova, M., Reichert, H. and Knoblich, J. A. (2012). FACS purification and transcriptome analysis of Drosophila neural stem cells reveals a role for Klumpfuss in self-renewal. Cell Rep 2(2): 407-418.
- Ghavi-Helm, Y., Zhao, B. and Furlong, E. E. (2016). Chromatin immunoprecipitation for analyzing transcription factor binding and histone modifications in Drosophila . Methods Mol Biol 1478: 263-277.
- Langmead, B. (2010). Aligning short sequencing reads with Bowtie. Curr Protoc Bioinformatics Chapter 11: Unit 11.7.
- Ramirez, F., Ryan, D. P., Gruning, B., Bhardwaj, V., Kilpert, F., Richter, A. S., Heyne, S., Dundar, F. and Manke, T. (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44(W1): W160-165.
- Seguin-Orlando, A., Schubert, M., Clary, J., Stagegaard, J., Alberdi, M. T., Prado, J. L., Prieto, A., Willerslev, E. and Orlando, L. (2013). Ligation bias in illumina next-generation DNA libraries: implications for sequencing ancient genomes. PLoS One 8(10): e78575.
- Skene, P. J., Henikoff, J. G. and Henikoff, S. (2018). Targeted in situ genome-wide profiling with high efficiency for low cell numbers. Nat Protoc 13(5): 1006-1019.
- Stathopulos, P. B., Scholz, G. A., Hwang, Y. M., Rumfeldt, J. A., Lepock, J. R. and Meiering, E. M. (2004). Sonication of proteins causes formation of aggregates that resemble amyloid. Protein Sci 13(11): 3017-3027.
- Zhang, Y., Liu, T., Meyer, C. A., Eeckhoute, J., Johnson, D. S., Bernstein, B. E., Nusbaum, C., Myers, R. M., Brown, M., Li, W. and Liu, X. S. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9(9): R137.
- Zheng, X., Yue, S., Chen, H., Weber, B., Jia, J. and Zheng, Y. (2015). Low-Cell-Number epigenome profiling aids the study of lens aging and hematopoiesis. Cell Rep 13(7): 1505-1518.
Article Information
Copyright
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Akhtar, J., More, P. and Albrecht, S. (2020). ChIP-Seq from Limited Starting Material of K562 Cells and Drosophila Neuroblasts Using Tagmentation Assisted Fragmentation Approach. Bio-protocol 10(4): e3520. DOI: 10.21769/BioProtoc.3520.
Category
Cell Biology > Organelle isolation > Nuclei
Molecular Biology > DNA > DNA sequencing
Systems Biology > Genomics > ChIP-seq
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.