- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Rapid Nickel-based Isolation of Extracellular Vesicles from Different Biological Fluids
Published: Vol 10, Iss 3, Feb 5, 2020 DOI: 10.21769/BioProtoc.3512 Views: 6833
Reviewed by: Alba Blesasujan kumar mondalilgen MenderAnonymous reviewer(s)
Original research article
The authors used this protocol in:
May 2019
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Extracellular vesicles (EVs) are membranous structures that cells massively release in extracellular fluids. EVs are cargo of cellular components such as lipids, proteins, and nucleic acids that can work as a formidable source in liquid biopsy studies searching for disease biomarkers. We recently demonstrated that nickel-based isolation (NBI) is a valuable method for fast, efficient, and easy recovery of heterogeneous EVs from biological fluids. NBI exploits nickel cations to capture negatively charged vesicles. Then, a mix of balanced chelating agents elutes EVs while preserving their integrity and stability in solution. Here, we describe steps and quality controls to functionalize a matrix of agarose beads, obtain an efficient elution of EVs, and extract nucleic acids carried by them. We demonstrate the versatility of NBI method in isolating EVs from media of primary mouse astrocytes, from human blood, urine, and saliva processed in parallel, as well as outer membrane vesicles (OMVs) from cultured Gram-negative bacteria.
Keywords: Extracellular vesiclesBackground
Extracellular vesicles (EVs) are heterogeneous membranous structures that cells release in the extracellular fluids. Besides the generation of apoptotic bodies, we now recognize at least two mechanisms that generate EVs, comprising the formation of multivesicular bodies (MVBs) in mature endosomes that fuse with the plasma membrane and discharge exosomes, and the direct release of membrane-budding vesicles known as microvesicles or ectosomes (van Niel et al., 2018). EVs are increasingly referred to as intercellular communicators which are able to influence the metabolism of recipient cells through the horizontal exchange of different components such as lipids, proteins, and nucleic acids (Raposo and Stoorvogel, 2013; Hirshman et al., 2016; Jeppesen et al., 2019). Besides the research efforts aiming to convert EVs into novel carriers of therapeutics, the incredible density and heterogeneity of EVs present in biological fluids hold promise for the interception of pathological conditions such as cancer or neurodegenerative diseases (Schiera et al., 2015; Bandu et al., 2019; Latifkar et al., 2019). Especially in cancer, this perspective is nowadays expanding the field of liquid biopsy tests, where the non-invasive detection of biomarkers through a systematic screening of circulating tumor cells, cell-free DNA, and EVs could advance early detection and monitoring, with novel opportunities for patients’ stratification and prediction of disease progression.
EVs have been detected in blood, urine, cerebrospinal fluid, saliva, tears, breast milk, as well as from cell culture media (Lässer et al., 2011; Rolfo et al., 2014; Salih et al., 2014; Pieragostino et al., 2019; Palviainen et al., 2019). Multiple strategies have been presented to isolate them, with protocols based on differential ultracentrifugation, density gradient, or polymer precipitation, allowing for a massive enrichment of dimensionally heterogeneous vesicles depending on their size, buoyant density, or lipid structure (Chen et al., 2019). Alternatively, immunocapture approaches have been exploited to enrich for EV sub-populations, virtually deriving from the same tissue of origin (Kowal et al., 2016).
In research laboratories, the most widely used method for EV isolation is the differential ultracentrifugation, in which large EVs of about 200-1,000 nm sediment at low speed (10,000 x g), while small EVs of about 50-200 nm pellet at high speed (100,000 x g), as described by Théry et al., 2006. This size-based classification cannot be exploited to infer the intracellular pathways of their biogenesis, as ectosomes generated by the outward budding of the plasma membrane can overlap in size with exosomes derived from the MVBs (multivesicular bodies). Nevertheless, specific tetraspanins (CD9, CD81, CD63) or lumen proteins (Alix, synthenin) are found to be enriched in the small EVs, including exosomes (Jeppesen et al., 2019; Latifkar et al., 2019). Ultracentrifugation has some drawbacks related to reproducibility of results when comparing different instruments/rotors, or to the physical force applied to balance yield versus integrity (Livshits et al., 2015). Typically, this technique is combined with size-exclusion chromatography or floatation in Optiprep gradient to separate EVs from co-sedimenting protein aggregates (Théry et al., 2018). However, these approaches are inapplicable in view of a routine clinical practice, where the recovery of EVs needs to be fast and the biological sample not exposed to chemicals that could contribute experimental variability but interfere in downstream analysis. For these reasons, other microfluidic- or electromagnetic-based technologies that exploit other biophysical features of EVs have been implemented (Hou et al., 2019). The strategy of capturing EVs through electrostatic interactions (negative zeta potential values) has recently shown the advantages of rapidity and versatility in terms of device implementation, combination with other protocols like size-exclusion chromatography, and recovery of heterogeneous suspended vesicles (Petersen et al., 2018; Heath et al., 2018).
Here, we describe the procedure of nickel-based isolation (NBI) we recently developed as a low-cost and convenient method for isolating EVs (Notarangelo et al., 2019). This method is based on nickel cations to capture EVs and on the use of chelating agents that synergistically elute vesicles in a physiological pH (or pH-controlled) solution. We demonstrated that NBI efficiently captures EVs and isolates them as non-aggregated vesicles in solution while preserving their integrity and minimizing the co-elution of exogenous protein contaminants (Notarangelo et al., 2019). Importantly, NBI method is fast, scalable, and easy to apply, offering a valuable option for liquid biopsy investigations and EV preparations that could benefit from increased stability and a non-aggregated distribution in solution. We describe the procedure starting from the functionalization of a matrix of agarose beads with nickel ions, the steps for efficient elution, and the protocol we use to extract nucleic acids from eluted EVs. We show here the versatility of NBI method in isolating EVs from different biological fluids, including media from primary mouse astrocytes, human plasma, urine, and saliva processed in parallel, as well as outer membrane vesicles (OMVs) from cultured Gram-negative bacteria.
Materials and Reagents
- Low-binding protein tubes: LoBind Tubes, Protein LoBind, 2.0 ml, PCR clean (Eppendorf, catalog number: 0030108132)
- Ni Sepharose® High Performance, pack of 25 ml (GE Healthcare, catalog number: 17-5268-01), store at 4 °C
- PVDF Syringe Filters (Euroclone, Primo® Syringe Filters, 0.22 μm pour size, catalog number: EPSPV2230)
- 50 ml tube
- 15 ml Falcon tube
- 1.5 ml centrifuge tube
- Water prepared by ultrafiltration, reverse osmosis, deionization and sterile filtration (e.g., Lonza®, catalog number: 17-724Q)
- Phosphate-buffered saline (PBS 1x, pH 7.4) (Thermo Fisher®, catalog number: 10010023)
- Nickel sulphate powder, NiSO4 (Sigma®, catalog number: 656895)
- NaCl powder (Sigma®, catalog number: 450006)
- EDTA [0.5] M (Thermo Fisher®, UltraPure pH 8.0, catalog number: 15575020)
- Citric acid (Sigma®, catalog number: 251275)
- TRIzolTM Reagent (Thermo Fisher®, catalog number: 15596026)
- Agilent RNA 6000 Pico Kit (Agilent, catalog number: 5067-1513)
- Chloroform (Sigma®, catalog number: C7559)
- Isopropanol Molecular Biology Reagent (Sigma®, catalog number: I9516)
- Ethanol (Sigma®, catalog number: 51976)
- UltraPureTM DNase/RNase-Free Distilled Water (Thermo Fisher®, catalog number: 10977035)
- Glycogen, RNA grade (Thermo Fisher®, catalog number: R0551)
- Buffer S (Stripping buffer) (see Recipe 1)
- Buffer N (see Recipe 2)
- Solution A 5x (see Recipe 3)
- Solution B 5x (see Recipe 4)
Equipment
- Laminar flow hood
- Burker chamber (VWR, MARI0640230)
- Eppendorf® Centrifuge 5702, non-refrigerated, with Swing-bucket Rotor (A-4-38) included adapters for 15/50 ml conical tubes (Eppendorf, catalog number: 5702 000.32) (used in paragraphs A-B-C of the procedure)
- Eppendorf® Thermomixer Comfort (Eppendorf, catalog number: 5355.000.011)
- VWR® Tube Rotator (VWR, catalog number: 10136-084) with interchangeable rotisserie assembly for: 10x 10/15 and 16x 5/7 ml tubes (VWR, catalog number: 444-0504), 6x 50 ml tubes (VWR, catalog number: 444-0505), 36x 1.5/2.0 ml tubes (VWR, catalog number: 444-0503)
- Eppendorf® Centrifuge 5415 R (Eppendorf, model: Centrifuge 5415 R, catalog number: 22 62 140-8), refrigerated, with Rotor (Eppendorf, catalog number: F-45-24-11) (used in Procedure D)
- 2100 Bioanalyzer Instrument (Agilent Technologies, catalog number: G2939BA)
- -80 °C freezer
Procedure
- Stripping and functionalization of agarose beads
Note: Steps A1-A3 are mostly known as part of the standard “stripping/re-charge” protocol well described by manufacturer’s selling nickel-functionalized matrices. We still include these details as these are part of the procedure we follow.- Transfer the beads to a 50 ml tube, centrifuge at 600-800 x g for 2 min at room temperature (RT), and discard the supernatant without disturbing the bead pellet.
- Add 1:1 bead volume of pure water and mix by gentle rotation and inversion of the tube until beads are homogeneously resuspended. Then recover beads by centrifugation at 600-800 x g for 2 min and discard the supernatant.
- Add 1:1 bead volume of Buffer S (Stripping buffer, Recipe 1) and resuspend beads by inverting the tube several times. Incubate for 5 min at RT and recover beads again by centrifugation (at 600-800 x g for 2 min). Discard the supernatant.
Note: This step removes nickel ions from the beads, which turn white indicating the nickel stripping from the matrix. Remember to pay attention to appropriate waste disposals for solutions containing nickel, especially after Step A4. - Add 1:1 bead volume of Buffer N (Recipe 2). Gently rotate the tube to obtain a homogeneous solution of beads. Incubate at room temperature for 5 min, then recover beads by centrifugation at 600-800 x g for 2 min.
Note: The concentration of Nickel sulphate (0.4 M) used to functionalize beads confers a positive charge to the matrix able to capture a number of vesicles at least equivalent to that obtained by ultracentrifugation of the same biological sample (Notarangelo et al., 2019). At this step, beads appear in light blue color. - Wash beads with 1:1 bead volume of pure water. Recover them again after centrifugation (at 600-800 x g for 2 min) and discard the supernatant.
Note: We usually perform Steps A5-A7 under the laminar flow hood to reduce the chances of biological contamination. - Wash beads with 1:1 bead volume of PBS 1x. Gently rotate the tube to homogenize beads in solution. Always recover them by centrifugation at 600-800 x g for 2 min. Repeat this step twice and carefully remove the supernatant using a serological pipet with a slow, constant aspiration of the volume to avoid disturbing the bead pellet.
Note: The removal of unbound nickel residues is crucial to avoid contamination of the biological sample. - Resuspend beads with PBS 1x to obtain a 20 mg/mx final concentration, corresponding to a range of 0.7 x 1010-1.8 x 1010 particles/ml. Store at 4 °C.
Note: Figure 1 indicates: i) the concentration of beads, calculated using a Burker chamber (MARI0640230, VWR) under the inverted microscope with 10x, 20x, and 40x objectives; ii) the size-distribution of beads, with a mean diameter of 30 ± 6.2 μm; calculated as the mean diameter of beads with respect to the reference scale bars in 20x and 40x acquisitions; iii) the elemental composition of beads as determined by X-ray photoelectron spectroscopy (XPS) in terms of oxygen, carbon, nitrogen, and nickel; iiii) the differential zeta potential (ZP) of +6 mV in comparison to the stripped beads, as determined by electrophoretic light scattering (ELS).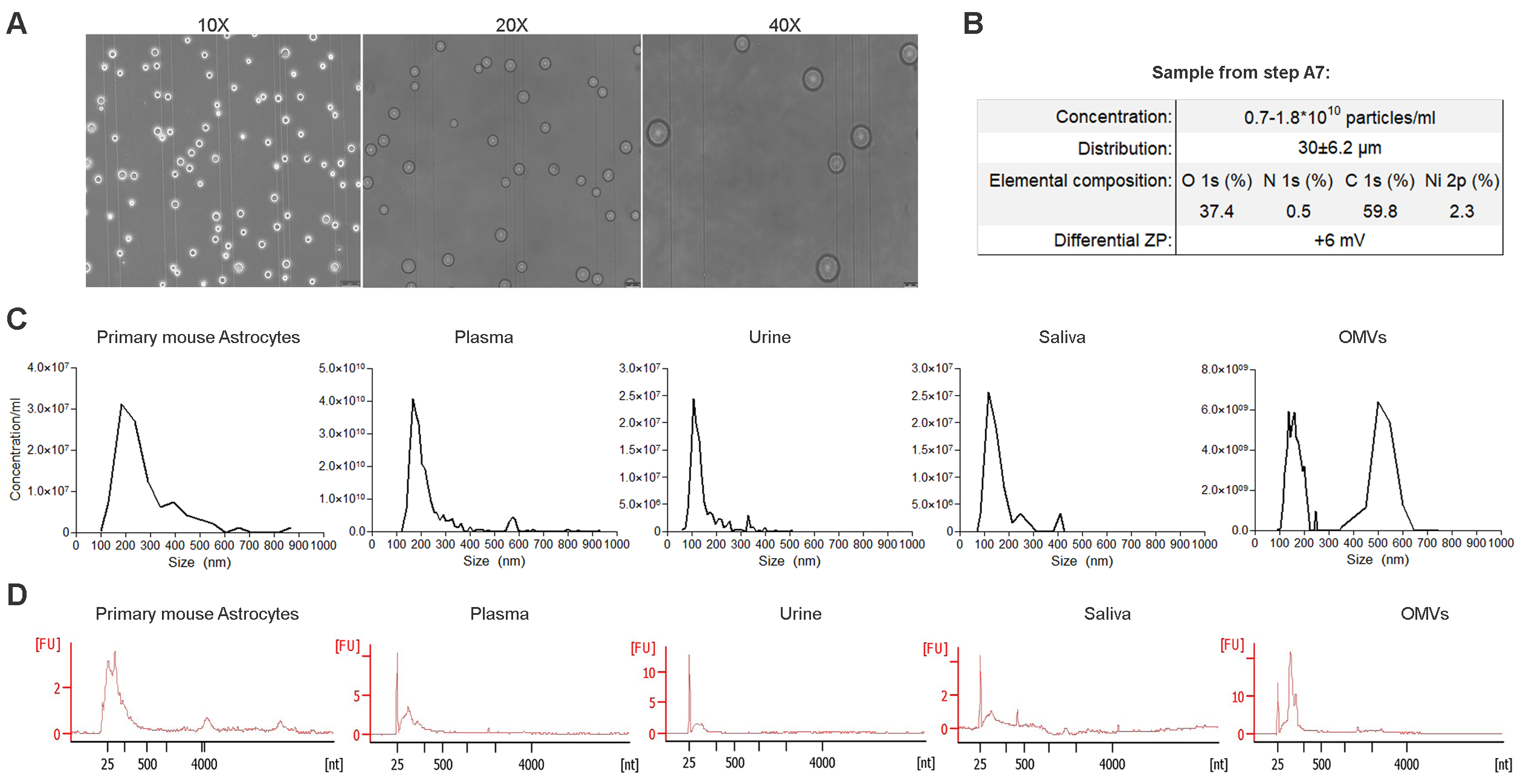
Figure 1. Quality controls and parallel recovery of EVs and EV-RNA from different biological fluids. A. Representative images of beads prepared in Step A7, diluted 1:500 in PBS, taken with a microscope Leica DM IL LED with a LAS X software. Scale bar is 50 µm. B. Parameters of beads prepared in Step A7. Distribution was assessed by ImageJ software, elemental composition by X-ray photoelectron spectroscopy (XPS), differential ZP by electrophoretic light scattering (ELS) in stripped (Step A3) and functionalized (Step A7) beads diluted 1:50 in PBS 1x (standard error <8%). C. Distribution of EVs recovered from media incubated with primary mouse astrocytes, 2 ml of plasma, 30 ml of urine, and 3 ml of saliva, all collected from the same healthy volunteer and processed in parallel. Luria-Bertani media incubated with pure culture of E. coli cells. A representative EV distribution was assessed by TRPS using NP250 nanopores (A58835, A59044). D. Bioanalyzer profiles of the relative RNA profile loading 1.5 μl out of 10 in an Agilent Pico RNA Chip.
Annex 1:
XPS analysis: A drop of beads, either stripped (Step A3) or functionalized with nickel sulphate (Step A7) was dried at room temperature on a silicon substrate and measured at 0° take-off angle. Whilst the samples were similar in terms of oxygen (35.3% vs 37.4%), carbon (64.3% vs 59.8%), and nitrogen (present in EDTA; 0.4% vs 0.5%), the nickel content instead varied from stripped beads (undetected) and functionalized beads (2.3%). The standard error did not exceed the 1-2% of the value reported in Figure 1B.
Sample details of Figures 1C and 1D. Primary astrocytes were derived from cortex of neonatal mice (P0-1) and were grown under standard conditions (10% FBS in DMEM) in 6-well plates for two weeks (Basso et al., 2013). Forty-eight hours before the collection, 36 ml of serum-free media were incubated with approximately 4 x 106 cells seeded (2 ml per well). In case of OMVs, we processed 50 ml of LB that fed overnight a BL21 strain already characterized for the secretion of OMVs (kindly provided by Prof. Grandi’s lab) (Fantappiè et al., 2017).
- NBI: EV capture
- Add at least 1:40 volume of beads prepared in Step A7, drop by drop, on the surface of the biological fluid containing EVs/exosome-like liposomes/OMVs and incubate for 30 min at RT in gentle shaking (using the VWR Tube Rotator) to avoid sedimentation of beads.
Note: The biological fluids include: cell culture medium, plasma, cerebrospinal fluid (CFS), urine, saliva, as well as LB medium to recover OMVs from E. coli strains. We typically use 25 μl of beads per ml of:- Cell culture medium (the presence of more than 2% FBS in the medium will cause bead aggregation during incubation, affecting the yield of EVs; in the presence of 5% FBS for example, we usually dilute it 1:5 in PBS to efficiently recover EVs). Importantly, refer to FBS characterization (Wei et al., 2016) as it normally contains small vesicles of non-human origin;
- Human plasma diluted 1:3 in PBS 1x (Figure 1C);
- Human urine, un-diluted (Figure 1C);
- Human saliva diluted 1:3 in PBS 1x (Figure 1C).
- Centrifuge the mixture at 600-800 x g for 2 min at RT.
Note: To avoid disturbing the bead pellet, it is advisable to use 15 ml Falcon tubes during incubation of cell supernatant//biological fluid with beads. Moreover, it is advisable to use swing-bucket rotors during centrifugation steps, to better locate the bead-pellet on the bottom of the tube and allow gentle sedimentation of beads. - Carefully remove (transfer or discard) the supernatant.
Note: We usually call this supernatant as “EV-depleted medium” and use it in internal control experiments for EV enumeration or downstream RNA analyses, especially in case we compare the potential enrichment of specific EV-transcripts.
- Add at least 1:40 volume of beads prepared in Step A7, drop by drop, on the surface of the biological fluid containing EVs/exosome-like liposomes/OMVs and incubate for 30 min at RT in gentle shaking (using the VWR Tube Rotator) to avoid sedimentation of beads.
- NBI: EV elution
- Prepare the elution buffer 1x by diluting Solution A concentrated 5x (Recipe 3) and Solution B concentrated 5x (Recipe 4) in 0.2 µm filtered PBS 1x. Gently mix the solution obtained.
Note: This buffer must be freshly prepared at RT prior usage and should be used within 15-20 min to obtain an efficient release of polydisperse EVs from the beads. - Add 0.5 to 2 bead-volumes of elution buffer 1x to the bead-pellet and transfer them in a low-binding tube. Incubate the tube in a Thermomixer for 15 min, in orbital shaking at 600 rpm and 28 °C, to keep beads in suspension and favor the dissociation of bound vesicles. During the incubation, flick the tube every 2-3 min to avoid sedimentation of beads.
Note: Low-binding tubes are designed for proteins, but are useful for minimizing interaction with vesicles. The volume of elution buffer 1x determines the concentration of EVs obtained. - Pellet beads at 600-800 x g for 30 s and carefully transfer the EV-containing supernatant in a new labeled 1.5 ml centrifuge tube.
Note: Optionally, we perform an additional centrifugation step to pellet any residual beads. - Optional: Repeat Step C2 to recover EVs in case you have used less than two bead-volumes of elution buffer 1x.
Note: In addition to Step C2, it ensures an efficient elution of bead-captured EVs. Figure 1C shows a distribution of EVs recovered from media of primary mouse astrocytes, from 2 ml of plasma, 30 ml of urine, and 3 ml of saliva all collected from the same healthy volunteer and processed in parallel, as well as from E. coli cells.
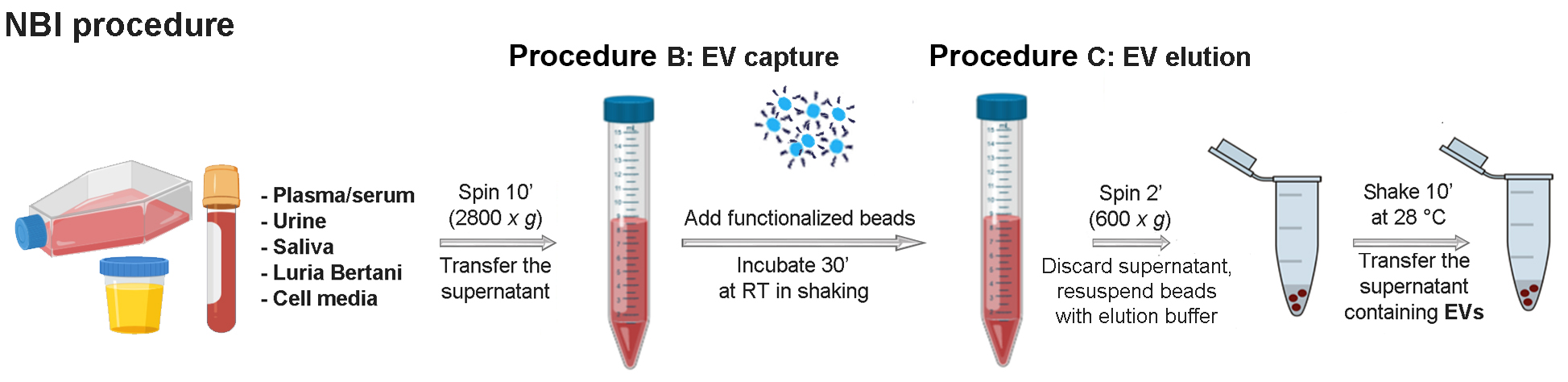
Figure 2. Schematic representation of the NBI method described in Procedures B and C. The NBI procedure it is entirely performed at room temperature, except the Step C2. Functionalized agarose beads prepared in Procedure A are stable for at least four months at 4 °C. The procedures described in Procedures B and C are routinely performed to isolate EVs and last approximately 45 min, depending on the number of samples to be processed. - Prepare the elution buffer 1x by diluting Solution A concentrated 5x (Recipe 3) and Solution B concentrated 5x (Recipe 4) in 0.2 µm filtered PBS 1x. Gently mix the solution obtained.
- RNA extraction from EVs
To extract nucleic acids preserving the majority of RNA classes enclosed in EVs, we typically use the TRIzolTM Reagent protocol (Rio et al., 2010) with minor modification in the procedure and following the general guidelines to avoid/minimize RNase contamination.- Add 0.5 ml of TRIzolTM Reagent per 1 ml of sample (a suspension of EVs in the elution buffer 1x) and briefly vortex to homogenize. Incubate at room temperature for 2-3 min.
- Add 0.2 ml of chloroform per 1 ml of TRIzolTM Reagent used, then homogenize by inverting the tube several times and incubate for 2-3 min.
- Centrifuge the sample for 10 min, at 13,000 x g at 4 °C, to obtain a lower red phenol-chloroform, an interphase, and a colorless upper aqueous phase.
Note: In case of EVs the interphase appears as a slightly visible ring. - Carefully transfer the aqueous phase to a new tube, avoiding any contamination from the interphase or organic layer into the pipette.
- Add 0.6 ml of isopropanol to the aqueous phase, per 1 ml of TRIzolTM Reagent used in Step D1. Briefly vortex and incubate 5 min on ice.
- Centrifuge for 15 min, at 13,000 x g and 4 °C, to precipitate nucleic acids.
Note: A nucleic acid pellet derived from EVs is hardly visible. Optionally, with glycogen you can facilitate visualization of gel-like pellet at the bottom of the tube. - Wash the pellet with 70% Ethanol and air dry for 5-10 min.
Note: We optionally skip the step of 70% Ethanol to avoid the potential waste of the precipitated material in case of a recovery of EVs < 107 particles/ml. - Resuspend the pellet in 7-15 μl of RNase-free water, by pipetting several times, and keep it on ice until stored at -80 °C.
Note: Transfer 1.5 μl of the solution in a new 0.2 ml tube for incubation at 70 °C and ice-killing to profile the nucleic acid fragment distribution by Bioanalyzer Pico Chip. Figure 1D shows the size distribution of nucleic acid fragments recovered from the different biological fluids indicated in Figure 1C.
Data analysis
Image analysis of Figure 1A was performed using ImageJ software. TRPS data displayed in Figure 1C were analyzed by Izon Control Suite software v3 and CSV data were plotted using GraphPad Prism Software v5.1. Nucleic acid profile of samples in Figure 1D was examined with Agilent 2100 Expert Software.
Recipes
- Buffer S (Stripping buffer)
PBS 1x
Under the biological hood, filter with a 0.22 μm disposal unit
Store at room temperature
PBS 1x + 0.5 M NaCl + 50 mM EDTA pH 8.0
Filter with 0.2 µm disposals
Store at room temperature - Buffer N
0.4 M of NiSO4 in pure water
Filter with 0.2 µm disposals
Store at 4 °C. - Solution A 5x
PBS 1x + 16 mM EDTA
Filter with 0.2 µm disposals
Store at 4 °C
Note: This storage ensures stability for months. A pH of 7.8 is a quality control. - Solution B 5x
PBS 1x + 10 mM NaCl + 225 μM citric acid
Filter with 0.2 µm disposals
Note: This storage ensures stability for months. A pH of 7.2 is a quality control.
Acknowledgments
We acknowledge the previous work (Notarangelo et al., 2019) describing the novelty and the application of NBI procedure. We are grateful to Prof. G. Grandi’s lab (University of Trento, CIBIO) for providing us the E. coli strain that we used in this work to isolate OMVs and OMV-nucleic acids.
This project was supported by Fondazione Cassa di Risparmio Trento e Rovereto (CARITRO) to VGD and AQ, and Horizon 2020 research and innovation program under the Marie Sklodowska-Curie (n. 752470) to MB.
Competing interests
MN, AP, AQ, and VGD declare a potential conflict of interest for a patent application regarding the NBI method (P019950WO).
Ethics
Human specimens involved in this study derived from healthy subjects who gave informed consent before blood and urine sampling in the frame of a study approved by the Local Ethics Committee of the University of Trento. Mice were used to obtain astrocytes in the frame of a study approved by the Italian Ministry of Health (project number 603/2017-PR).
References
- Bandu, R, Oh, J. W. and Kim, K. P. (2019). Mass spectrometry-based proteome profiling of extracellular vesicles and their roles in cancer biology. Exp Mol Med 15;51(3): 30.
- Basso, M., Pozzi, S., Tortarolo, M., Fiordaliso, F., Bisighini, C., Pasetto, L., Spaltro, G., Lidonnici, D., Gensano, F., Battaglia, E., Bendotti, C. and Bonetto, V. (2013). Mutant copper-zinc superoxide dismutase (SOD1) induces protein secretion pathway alterations and exosome release in astrocytes: implications for disease spreading and motor neuron pathology in amyotrophic lateral sclerosis. J Biol Chem 288(22): 15699-15711.
- Chen, B. Y., Sung, C. W., Chen, C., Cheng, C. M., Lin, D. P., Huang, C. T. and Hsu, M. Y. (2019). Advances in exosomes technology. Clin Chim Acta 493: 14-19.
- Fantappiè, L, Irene, C., De, Santis., M., Armini, A., Gagliardi, A., Tomasi, M., Parri, M., Cafardi, V., Bonomi, S., Ganfini, L., Zerbini, F., Zanella, I., Carnemolla, C., Bini, L., Grandi, A. and Grandi, G. (2017). Some gram-negative lipoproteins keep their surface topology when transplanted from one species to another and deliver foreign polypeptides to the bacterial surface. Mol Cell Proteomics 16:1348-1364.
- Heath, N., Grant, L., De, Oliveira., TM, Rowlinson, R., Osteikoetxea, X., Dekker, N. and Overman, R. (2018). Rapid isolation and enrichment of extracellular vesicle preparations using anion exchange chromatography. Sci Rep 8(1):5730.
- Hirshman, B. R., Kras, R. T., Akers, J. C., Carter, B. S. and Chen, C. C. (2016). Extracellular vesicles in molecular diagnostics: an overview with a focus on CNS diseases. Adv Clin Chem 76: 37-53.
- Hou, R., Li, Y., Sui, Z., Yuan, H., Yang, K., Liang, Z., Zhang, L. and Zhang, Y. (2019). Advances in exosome isolation methods and their applications in proteomic analysis of biological samples. Anal Bioanal Chem 411(21):5351-5361
- Jeppesen, D. K., Fenix, A. M., Franklin, J. L., Higginbotham, J. N., Zhang, Q., Zimmerman, L. J., Liebler, D. C., Ping, J., Liu, Q., Evans, R., Fissell, W. H., Patton, J. G., Rome, L. H., Burnette, D. T. and Coffey, R. J. (2019). Reassessment of Exosome Composition. Cell 177(2): 428-445 e418.
- Kowal, J., Arras, G., Colombo, M., Jouve, M., Morath, J. P., Primdal-Bengtson, B., Dingli, F., Loew, D., Tkach, M. and Thery, C. (2016). Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc Natl Acad Sci U S A 113(8): E968-977.
- Lässer, C., Alikhani, V. S., Ekström, K., Eldh, M., Paredes, P. T., Bossios, A., Sjöstrand, M., Gabrielsson, S., Lötvall, J. and Valadi, H. (2011). Human saliva, plasma and breast milk exosomes contain RNA: uptake by macrophages. J Transl Med 9:9.
- Latifkar, A., Hur, Y. H., Sanchez, J. C., Cerione, R. A. and Antonyak, M. A. (2019). New insights into extracellular vesicle biogenesis and function. J Cell Sci 132(13).
- Livshits, M. A., Khomyakova, E., Evtushenko, E. G., Lazarev, V. N., Kulemin, N. A., Semina, S. E., Generozov, E. V. and Govorun, V. M. (2015). Isolation of exosomes by differential centrifugation: Theoretical analysis of a commonly used protocol. Sci Rep 5: 17319.
- Notarangelo, M., Zucal, C., Modelska, A., Pesce, I., Scarduelli, G., Potrich, C., Lunelli, L., Pederzolli, C., Pavan, P., la Marca, G., Pasini, L., Ulivi, P., Beltran, H., Demichelis, F., Provenzani, A., Quattrone, A. and D'Agostino, V. G. (2019). Ultrasensitive detection of cancer biomarkers by nickel-based isolation of polydisperse extracellular vesicles from blood. EBioMedicine 43: 114-126.
- Palviainen, M., Saari, H., Kärkkäinen, O., Pekkinen, J., Auriola, S., Yliperttula, M., Puhka, M., Hanhineva, K. and Siljander, PR-M. (2019). Metabolic signature of extracellular vesicles depends on the cell culture conditions. J Extracell vesicles 8(1):1596669.
- Petersen, K. E., Shiri, F., White, T., Bardi, G. T., Sant, H., Gale, B. K. and Hood, J. L. (2018). Exosome isolation: cyclical electrical field flow fractionation in low-ionic-strength fluids. Anal Chem 90(21): 12783-12790.
- Pieragostino, D., Lanuti, P., Cicalini, I., Cufaro, M. C., Ciccocioppo, F., Ronci, M., Simeone, P., Onofrj, M., van der Pol, E., Fontana, A., Marchisio, M. and Del Boccio, P. (2019). Proteomics characterization of extracellular vesicles sorted by flow cytometry reveals a disease-specific molecular cross-talk from cerebrospinal fluid and tears in multiple sclerosis. J Proteomics 204: 103403.
- Raposo, G. and Stoorvogel, W. (2013). Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol 200(4): 373-383.
- Rio, D. C., Ares, M. Jr., Hannon, G. J. and Nilsen, T. W. (2010). Purification of RNA using TRIzol (TRI reagent). Cold Spring Harb Protoc (6): pdb.prot5439.
- Rolfo, C., Castiglia, M., Hong, D., Alessandro, R., Mertens, I., Baggerman, G., Zwaenepoel, K., Gil-Bazo, I., Passiglia, F., Carreca, A. P., Taverna, S., Vento, R., Santini, D., Peeters, M., Russo, A. and Pauwels, P. (2014). Liquid biopsies in lung cancer: the new ambrosia of researchers. Biochim Biophys Acta 1846(2): 539-546.
- Salih, M., Zietse, R. and Hoorn, E. J. (2014). Urinary extracellular vesicles and the kidney: biomarkers and beyond. Am J Physiol Renal Physiol 306(11): F1251-1259.
- Schiera, G., Di Liegro, CM. and Di Liegro., I. (2015). Extracellular membrane vesicles as vehicles for brain cell-to-cell interactions in physiological as well as pathological conditions. Biomed Res Int 2015:152926.
- Théry, C., Amigorena, S., Raposo, G. and Clayton, A. (2006). Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc cell Biol Chapter 3: Unit 3.22.
- Théry, C., Witwer, K. W., Aikawa, E., Alcaraz, M. J., Anderson, J. D., et al. (2018). Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles 7(1): 1535750.
- van Niel, G., D'Angelo, G. and Raposo, G. (2018). Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol 19(4): 213-228.
- Wei, Z., Batagov, A. O., Carter, D. R. and Krichevsky, A. M. (2016). Fetal bovine serum RNA interferes with the cell culture derived extracellular RNA. Sci Rep 6: 31175.
Article Information
Copyright
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Notarangelo, M., Ferrara, D., Potrich, C., Lunelli, L., Vanzetti, L., Provenzani, A., Basso, M., Quattrone, A. and D'Agostino, V. G. (2020). Rapid Nickel-based Isolation of Extracellular Vesicles from Different Biological Fluids. Bio-protocol 10(3): e3512. DOI: 10.21769/BioProtoc.3512.
Category
Cell Biology > Organelle isolation > Extracellular vesicle
Cancer Biology > Genome instability & mutation > Tumor microenvironment
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.