- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Structural Alignment and Covariation Analysis of RNA Sequences
Published: Vol 10, Iss 3, Feb 5, 2020 DOI: 10.21769/BioProtoc.3511 Views: 6618
Reviewed by: Danish MemonGarima KhandelwalJian Wang
Original research article
The authors used this protocol in:

Aug 2019
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
RNA molecules adopt defined structural conformations that are essential to exert their function. During the course of evolution, the structure of a given RNA can be maintained via compensatory base-pair changes that occur among covarying nucleotides in paired regions. Therefore, for comparative, structural, and evolutionary studies of RNA molecules, numerous computational tools have been developed to incorporate structural information into sequence alignments and a number of tools have been developed to study covariation. The bioinformatic protocol presented here explains how to use some of these tools to generate a secondary-structure-aware multiple alignment of RNA sequences and to annotate the alignment to examine the conservation and covariation of structural elements among the sequences.
Keywords: RNABackground
Biological RNA molecules fold into specific secondary (2D) and tertiary (3D) structures that are critical for their function. Therefore, for comparative analysis, which usually requires sequence alignment, it is desirable to take structure information into account in order to obtain a more reliable and meaningful alignment. Numerous computational algorithms and tools have been developed to generate alignments based on secondary structure, such as MAFFT (Katoh and Toh, 2008), TurboFold (Tan et al., 2017), R-Coffee (Wilm et al., 2008), locARNA (Will et al., 2007), ProbCons (Do et al., 2005), MXSCARNA (Tabei et al., 2008), and LaRA (Bauer et al., 2007). In a benchmark comparison of these leading tools (Tan et al., 2017) TurboFold and MAFFT were shown to have comparable and highest accuracies. The running time of TurboFold is considerably longer than that of MAFFT (Katoh and Toh, 2008; Tan et al., 2017), thus in this protocol we use MAFFT because its speed, which can be further augmented by parallel processing (Katoh and Standley, 2013), allows alignment of a large number (>100) of sequences in a limited amount time. Like several of the other tools MAFFT employs an iterative strategy where pairwise structural alignments are first computed and are then progressively combined into a multiple alignment through several rounds of refinement.
Because of the tight structure-function relationship, functional RNAs undergo a selection pressure to maintain their structures (Nowick et al., 2019). This is reflected by the occurrence of covarying consistent or compensatory mutations in paired nucleotides that can be observed in sequence alignments. Covariation data are therefore very valuable and have been used to validate or predict the secondary, and even tertiary, structure of RNAs and to understand their evolution (Michel and Westhof, 1990; Cannone et al., 2002). A number of software tools are available for examining covariation within alignments, such as the structural alignment editors RALEE (Griffiths-Jones, 2005), 4SALE (Seibel et al., 2006), S2S (Jossinet and Westhof, 2005), ConStruct (Wilm et al., 2008b), or SARSE (Andersen et al., 2007), R-chie (Lai et al., 2012), a tool that scores and annotates covariation, and complex programs that include methods for performing statistical analysis of covariation with or without a phylogenetic framework such as R-scape (Rivas et al., 2017) and CoMap (Dutheil, 2012). R-chie highlights basepairs and employs arc diagrams to represent the secondary structure alongside the alignment, and can generate highly customizable figures.
In the protocol below, we explain how to use MAFFT to compute a structural alignment of multiple RNA sequences, and how to use R-chie to annotate the alignment with conservation and covariation information.
Equipment
- Personal computer, preferably with multiple processors (CPUs) to speed up computations
A Unix/Linux operating system is preferred. All software mentioned here, except the optional LaRA program, can also be run under Mac and Windows systems. For Windows, a terminal or Linux emulator such as Cygwin (http://www.cygwin.com/) or Ubuntu (https://www.microsoft.com/store/p/ubuntu/9nblggh4msv6) is needed. In any case, familiarity with the use of command-line-driven applications is required.
Software
- MAFFT (Katoh and Toh, 2008, https://mafft.cbrc.jp/alignment/software/source.html)
- R (R Development Core Team, 2018, http://www.R-project.org/)
- R-chie (Lai et al., 2012, https://www.e-rna.org/r-chie/)
- (Optional) MXSCARNA (Tabei et al., 2008, https://www.ncrna.org/softwares/mxscarna/)
- (Optional) LaRA (Bauer et al., 2007, http://www.mi.fu-berlin.de/w/LiSA/Lara)
- (Optional) FOLDALIGN (Sundfeld et al., 2016, http://rth.dk/resources/foldalign)
- The MAFFT package is provided in different forms. Be sure to download the bundle that provides support for RNA structural alignment, such as the package with extensions for Unix/Linux, the Standard package for Mac, and the Ubuntu or Cygwin version for Windows.
- It is not necessary to install MXSCARNA separately as it is included within the MAFFT package.
- Install LaRA and/or FOLDALIGN only if you want to use them as alternatives to MXSCARNA. LaRA runs on Linux only. LaRA version 1.3 may need to be used as the later versions 1.31 and 1.32 frequently abort.
Procedure
- Prepare a set of related (homologous) RNA sequences to analyze, either using local sequences or by downloading sequences from a database. Sequences can be retrieved via keyword searches from general-purpose databases such as NCBI GenBank/RefSeq (https://www.ncbi.nlm.nih.gov/), ENA (https://www.ebi.ac.uk/ena), or Ensembl (http://www.ensembl.org/), or from specialized RNA databases such as SILVA (https://www.arb-silva.de/), ncRNA databases (https://ncrnadatabases.org/), RNAcentral (https://rnacentral.org/), the Comparative RNA Website (http://www.rna.icmb.utexas.edu/), or Rfam (https://rfam.org/; e.g., see the Nucleic Acids Research website for a non-exhaustive listing of RNA sequence databases; http://www.oxfordjournals.org/nar/database/cat/2). Some of the RNA databases (e.g., SILVA, Rfam, Comparative RNA Website) provide sequences that are already aligned in the form of structure-aware multiple alignments. Homologs of RNA sequences of interest can also be identified by sequence similarity search, e.g., using the well-known BLASTN tool. All general databases and some of the RNA databases provide a BLAST service. One can also search for related RNA sequences at the structural level with the help of covariance models using INFERNAL (Nawrocki and Eddy, 2013) or CMfinder (Yao et al., 2006).
All collected sequences must be put in a single file in the commonly used FASTA format (https://en.wikipedia.org/wiki/FASTA_format; Figure 1).
Note: The composition of the dataset may influence the analysis of covariation, depending on the amount of similarity or dissimilarity between the sequences and their phylogenetic relationships.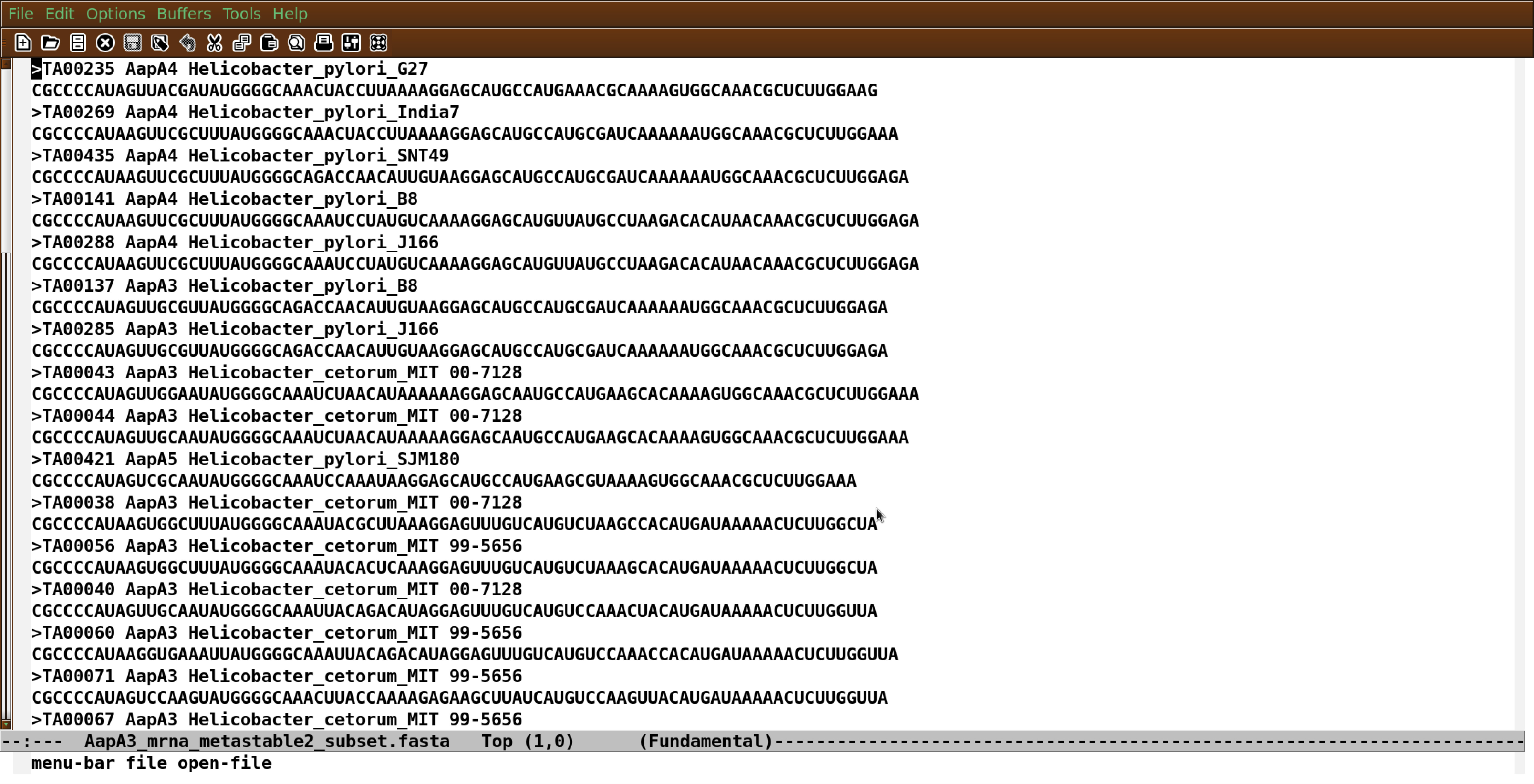
Figure 1. Example of unaligned RNA sequences in FASTA format (visualized in Emacs) - Compute a structural multiple alignment by running MAFFT in the ‘X-INS-I’ mode:
mafft-xinsi --scarnapair --nuc --reorder --maxiterate max_number_of_iterations --thread number_of_CPUs_to_use sequence_file.fasta 1> mafft_alignment.fasta 2> mafft_alignment_details.log
The alignment generated will be in FASTA format (Figure 2).
Notes:- The option “--scarnapair” instructs MAFFT to use MXSCARNA to perform pairwise structural alignments, which is the default option. To use the LaRA aligner instead, invoke the pair of options “--larapair --laraparams parameter_file”, where “parameter_file” is a file with LaRA configuration parameters. A template file “lara.params” is provided with the LaRA software. To use the FOLDALIGN aligner, invoke the option “--foldalignlocalpair” or “--foldalignglobalpair” to perform local or global pairwise alignment, respectively. In benchmarking comparisons (Katoh and Toh, 2008) the accuracy of MXSCARNA was generally higher than that of LaRA, except when the identity among the sequences was low (<40%), in which case LaRA may be preferred. FOLDALIGN is another structural alignment program that is highly accurate and that can carry out structural alignments of sequences with low similarity ( Havgaard et al., 2005; Sundfeld et al., 2016).
- To obtain high accuracy alignments, use a large number of iterative refinements, e.g., set “max_number_of_iterations” to 1,000 for the “--maxiterate” option”.
- For the “--thread” option, increase “number_of_CPUs_to_use” to speed up alignment computation. Runtime increases with the number and length of the sequences. For example, for a set of 100 sequences of length 50-200 nt calculations can take 3-10 min on a single CPU, and under a minute when using at least 8 CPUs.
- In some environments, MAFFT may abort with the following error: “mafft-xinsi: line 2369: /dev/stderr: Not a directory”. The error can be solved by replacing “/dev/stderr” by “/dev/null” in the mafft-xinsi bash script.
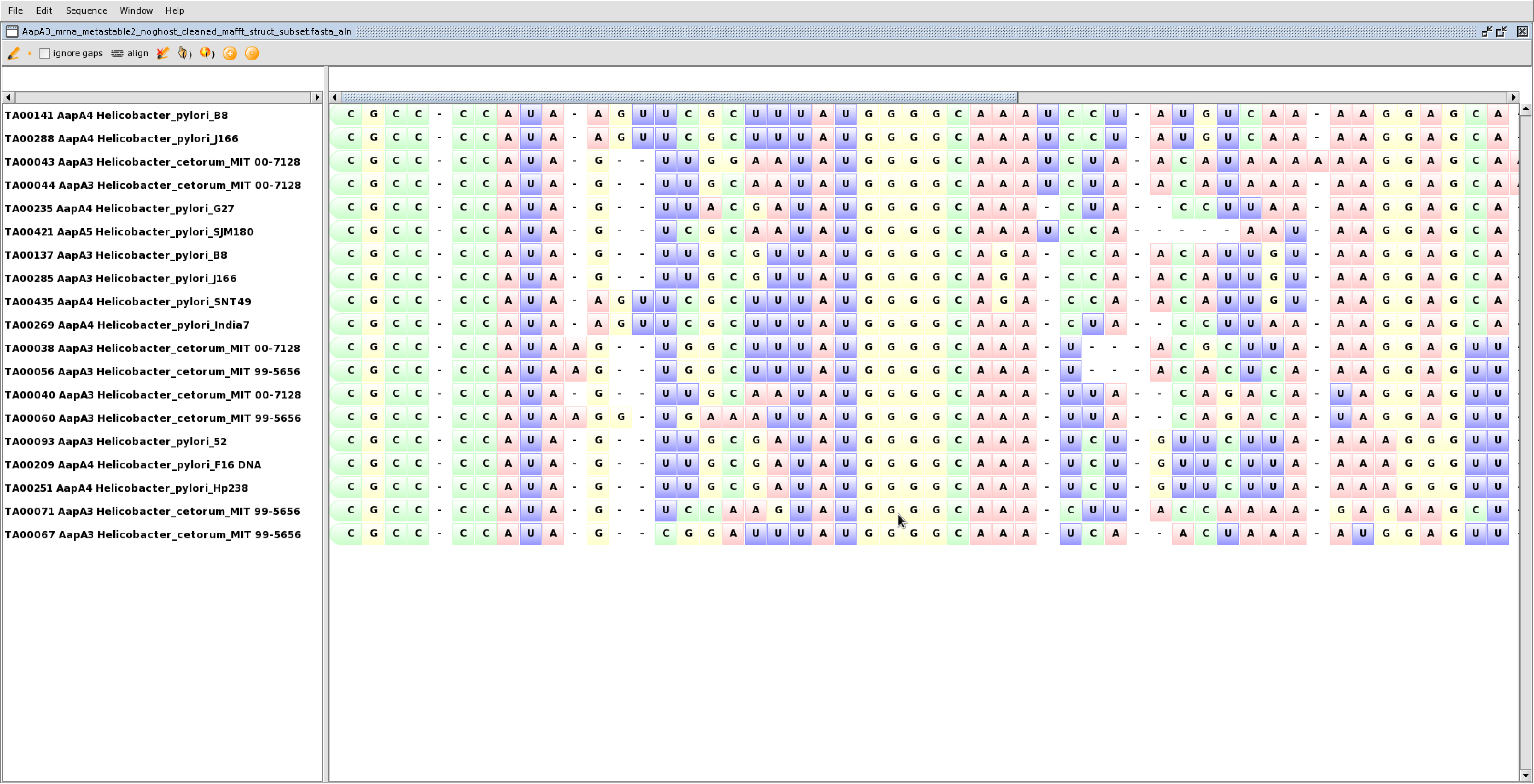
Figure 2. Structural alignment produced by MAFFT visualized in 4SALE - (Optional) Predict a reference secondary structure:
In order to reveal covariation, a reference secondary structure is needed. It can be the structure of one of the RNA sequences included in the analysis, a consensus structure inferred from the alignment, or an external structural model. In lack of a known or experimentally-determined model, the structure needs to be predicted. Prediction of the 2D structure of a single RNA sequence can be done with widely used tools such as MFOLD (Zuker, 2003), http://unafold.rna.albany.edu/?q=mfold), RNAfold from the ViennaRNA package (Lorenz et al., 2011), https://www.tbi.univie.ac.at/RNA/), Fold or MaxExpect from the RNAStructure package (Reuter and Mathews, 2010), https://rna.urmc.rochester.edu/RNAstructure.html), or RNAshapes from the RNA shapes studio (Janssen and Giegerich, 2015; https://bibiserv.cebitec.uni-bielefeld.de/rnashapesstudio). All these tools can be run on-line at dedicated webservers or installed locally as command-line programs. For example, standard commands to run MFOLD or RNAfold with default parameters on an RNA sequence in FASTA format would be:
mfold SEQ=sequence_file.fasta (structures will be output in files named “sequence_file*.ct”)
RNAfold < sequence_file.fasta > structure_file.b
The above-mentioned packages have numerous options to tune the folding computation (e.g., by changing the algorithm, temperature, ionic conditions), and most of them provide the possibility to impose constraints on the structure.
There exists also several pieces of software for predicting consensus structures from alignments, such as RNAalifold (Bernhart et al., 2008) from the ViennaRNA package, the graphical tool ConStruct (Wilm et al., 2008b); http://www.biophys.uni-duesseldorf.de/construct3/), and RNAalishapes from the RNA shapes studio (Voß, 2006); https://bibiserv.cebitec.uni-bielefeld.de/rnaalishapes). Notably, RNAalifold does not weight the sequences and is highly sensitive to the particular sample of sequences under study and the prediction can be affected by the varying amount of similarity between the sequences as well as by sequences containing insertions. ConStruct incorporates custom sequence weighting for optimal consensus prediction while RNAalishapes is based on the concept of abstract structural shapes and also includes gap-aware energy evaluation which makes it outperform RNAalifold when sequences contain insertions and/or alignment is of low quality (Voß, 2006). In our experience RNAalishapes performed quite well. RNAalishapes relies on older Linux libraries and may not install on current architectures, thus the on-line version may have to be used. - Analyze the covariation in the alignment and generate an image of the covariation-annotated alignment using R-chie:
Covariation is revealed by mapping the reference secondary structure onto the multiple alignments. The structure can be provided to R-chie in one of the common formats used by structure prediction and analysis software, including dot-bracket (or Vienna; “.b”), connect-table (“.ct”), as well as bpseq (from The Comparative RNA Website [Cannone et al., 2002], http://www.rna.icmb.utexas.edu/). The alignment must be in FASTA format.
rchie.R --msafile=mafft_alignment.fasta --pdf --output=alignment_covariation_figure.pdf --format1=vienna --rule1=7 --group1=4 --legend1 --legend3 --msaspecies --msagrid --msatext reference_structure.b &> rchie.log
This will create a figure of the alignment in PDF format, in which the paired regions in the reference secondary structure are represented by arcs colored according to the covariation score and nucleotides in the sequences are colored according to their base-pairing status (Figure 3).
Notes:- The above command runs with a reference structure in dot-bracket (“.b”) format, which is indicated by the option “--format1=vienna”. For a structure in connect (“.ct”) or bpseq format “--format1=connect” or “--format1=bpseq” would be used, respectively.
- Option “--rule1=7” is used to group base-pairs on covariation scores and option “--group1=4” is used to set the number of groups to 4. Various other criteria can be chosen for grouping; run “rchie.R --help” for details.
- R-chie is highly customizable. Options “--legend1”, “--legend3”, “--msaspecies”, “--msagrid”, and “--msatext” can be invoked or omitted at will in order to turn on or off legends, sequence names, and other graphical settings. Colors for every element in the image can be specified by additional options such as “--colour1”, “--palette1”, or “--msacol”; run “rchie.R --help” for details.
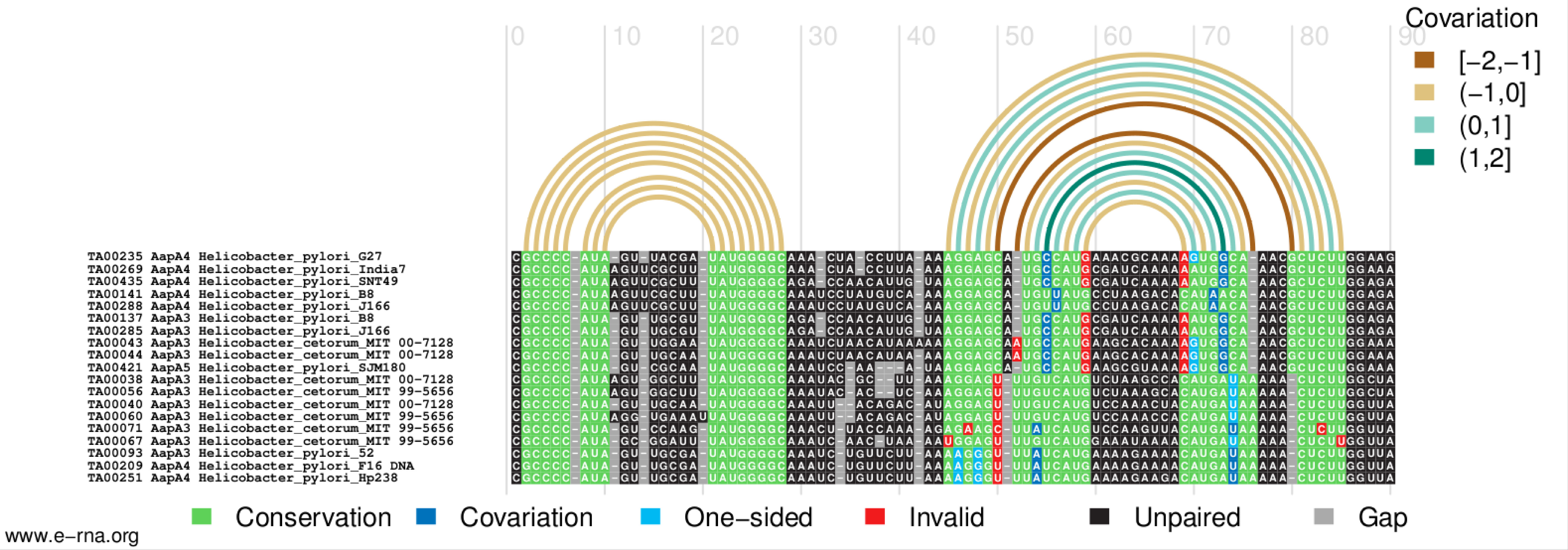
Figure 3. Structure- and covariation-annotated alignment drawn by R-chie - (Optional) If necessary, manually edit the alignment produced by MAFFT to correct errors or adjust the alignment using a structure-aware editor such as RALEE (Griffiths-Jones, 2005), http://sgjlab.org/ralee/), 4SALE (Seibel et al., 2006); http://4sale.bioapps.biozentrum.uni-wuerzburg.de/), S2S (Jossinet and Westhof, 2005); http://bioinformatics.org/assemble/), ConStruct (Wilm et al., 2008b); http://www.biophys.uni-duesseldorf.de/construct3/), or SARSE (Andersen et al., 2007). Then, regenerate the covariation-annotated figure using R-chie.
Data analysis
The protocol described here has been used to examine the covariation within metastable regions in a structural alignment of 107 mRNAs of type I toxin-antitoxin systems from bacteria of the genus Helicobacter (Masachis et al., 2019, Figure 8, figure supplement 1 and 2 therein). All files including the unaligned sequences (in FASTA format), the unedited alignment produced by MAFFT (run with the “--scarnapair” option; in FASTA format), the manually-edited alignment (in FASTA format), the reference secondary structure (in dot-bracket/Vienna format), and the covariation-annotated alignment drawn by R-chie (in PDF format) for both datasets (metastable 1 and 2) analyzed in Masachis et al. (2019) are included as supplementary material, to allow the user to learn and reproduce the analyses.
Notes
In this protocol, we employ MAFFT to compute structural alignments, but the procedure may be performed similarly using any other tools that produce structure-aware sequence alignments.
Acknowledgments
This protocol was derived from our original study of metastable structures in mRNAs of type I toxin-antitoxin systems in the bacterium Helicobacter pylori (Masachis et al., 2019).
Competing interests
The authors declare that there are no conflicts of interest or competing interests.
References
- Andersen, E. S., Lind-Thomsen, A., Knudsen, B., Kristensen, S. E., Havgaard, J. H., Torarinsson, E., Larsen, N., Zwieb, C., Sestoft, P., Kjems, J. and Gorodkin, J. (2007). Semiautomated improvement of RNA alignments. RNA 13(11): 1850-1859.
- Bauer, M., Klau, G. W. and Reinert, K. (2007). Accurate multiple sequence-structure alignment of RNA sequences using combinatorial optimization. BMC Bioinformatics 8: 271.
- Bernhart, S. H., Hofacker, I. L., Will, S., Gruber, A. R. and Stadler, P. F. (2008). RNAalifold: Improved consensus structure prediction for RNA alignments. BMC Bioinformatics 9: 474.
- Cannone, J. J., Subramanian, S., Schnare, M. N., Collett, J. R., D’Souza, L. M., Du, Y., Feng, B., Lin, N., Madabusi, L. V., Müller, K. M., Pande, N., Shang, Z., Yu, N. and Gutell, R. R. (2002). The Comparative RNA Web (CRW) Site: An online database of comparative sequence and structure information for ribosomal, intron, and other RNAs. BMC Bioinformatics 3: 2.
- Do, C. B., Mahabhashyam, M. S. P., Brudno, M. and Batzoglou, S. (2005). ProbCons: Probabilistic consistency-based multiple sequence alignment. Genome Res 15(2): 330-340.
- Dutheil, J. Y. (2012). Detecting coevolving positions in a molecule: Why and how to account for phylogeny. Brief Bioinform 13(2): 228-243.
- Griffiths-Jones, S. (2005). RALEE - RNA alignment editor in Emacs. Bioinformatics 21(2): 257-259.
- Havgaard, J. H., Lyngsø, R. B., Stormo, G. D. and Gorodkin, J. (2005). Pairwise local structural alignment of RNA sequences with sequence similarity less than 40%. Bioinformatics 21(9): 1815-1824.
- Janssen, S. and Giegerich, R. (2015). The RNA shapes studio. Bioinformatics 31(3): 423-425.
- Jossinet, F. and Westhof, E. (2005). Sequence to Structure (S2S): Display, manipulate and interconnect RNA data from sequence to structure. Bioinformatics 21(15): 3320-3321.
- Katoh, K. and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol Biol Evol 30(4): 772-780.
- Katoh, K. and Toh, H. (2008). Improved accuracy of multiple ncRNA alignment by incorporating structural information into a MAFFT-based framework. BMC Bioinformatics 9: 212.
- Lai, D., Proctor, J. R., Zhu, J. Y. A. and Meyer, I. M. (2012). R-CHIE: A web server and R package for visualizing RNA secondary structures. Nucleic Acids Res 40(12): e95.
- Lorenz, R., Bernhart, S. H., Höner zu Siederdissen, C., Tafer, H., Flamm, C., Stadler, P. F. and Hofacker, I. L. (2011). ViennaRNA Package 2.0. Algorithms Mol Biol 6: 26.
- Masachis, S., Tourasse, N. J., Lays, C., Faucher, M., Chabas, S., Iost, I. and Darfeuille, F. (2019). A genetic selection reveals functional metastable structures embedded in a toxin-encoding mRNA. Elife 8: e47549.
- Michel, F. and Westhof, E. (1990). Modelling of the three-dimensional architecture of group I catalytic introns based on comparative sequence analysis. J Mol Biol 216(3): 585-610.
- Nawrocki, E. P. and Eddy, S. R. (2013). Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29(22): 2933-2935.
- Nowick, K., Walter Costa, M. B., Höner zu Siederdissen, C. and Stadler, P. F. (2019). Selection pressures on RNA sequences and structures. Evol Bioinforma 15: 117693431987191.
- R Development Core Team. (2018). R: A language and environment for statistical computing. Vienna, Austria.
- Reuter, J. S. and Mathews, D. H. (2010). RNAstructure: Software for RNA secondary structure prediction and analysis. BMC Bioinformatics 11: 129.
- Rivas, E., Clements, J. and Eddy, S. R. (2017). A statistical test for conserved RNA structure shows lack of evidence for structure in lncRNAs. Nat Methods 14(1): 45-48.
- Seibel, P. N., Müller, T., Dandekar, T., Schultz, J. and Wolf, M. (2006). 4SALE - A tool for synchronous RNA sequence and secondary structure alignment and editing. BMC Bioinformatics 7: 498.
- Sundfeld, D., Havgaard, J. H., De Melo, A. C. M. A. and Gorodkin, J. (2016). Foldalign 2.5: Multithreaded implementation for pairwise structural RNA alignment. Bioinformatics 32(8): 1238-1240.
- Tabei, Y., Kiryu, H., Kin, T. and Asai, K. (2008). A fast structural multiple alignment method for long RNA sequences. BMC Bioinformatics 9: 33.
- Tan, Z., Fu, Y., Sharma, G. and Mathews, D. H. (2017). TurboFold II: RNA structural alignment and secondary structure prediction informed by multiple homologs. Nucleic Acids Res 45(20): 11570-11581.
- Voß, B. (2006). Structural analysis of aligned RNAs. Nucleic Acids Res 34(19): 5471-5481.
- Will, S., Reiche, K., Hofacker, I. L., Stadler, P. F. and Backofen, R. (2007). Inferring noncoding RNA families and classes by means of genome-scale structure-based clustering. PLoS Comput Biol 3(4): 680-691.
- Wilm, A., Higgins, D. G. and Notredame, C. (2008a). R-Coffee: A method for multiple alignment of non-coding RNA. Nucleic Acids Res 36(9): e52.
- Wilm, A., Linnenbrink, K. and Steger, G. (2008b). ConStruct: Improved construction of RNA consensus structures. BMC Bioinformatics 9: 219.
- Yao, Z., Weinberg, Z. and Ruzzo, W. L. (2006). CMfinder - A covariance model based RNA motif finding algorithm. Bioinformatics 22(4): 445-452.
- Zuker, M. (2003). Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31(13): 3406-3415.
Article Information
Copyright
![]() Tourasse and Darfeuille. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Tourasse and Darfeuille. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Tourasse, N. J. and Darfeuille, F. (2020). Structural Alignment and Covariation Analysis of RNA Sequences. Bio-protocol 10(3): e3511. DOI: 10.21769/BioProtoc.3511.
- Masachis, S., Tourasse, N. J., Lays, C., Faucher, M., Chabas, S., Iost, I. and Darfeuille, F. (2019). A genetic selection reveals functional metastable structures embedded in a toxin-encoding mRNA. Elife 8: e47549.
Category
Molecular Biology > RNA > RNA structure
Molecular Biology > RNA > RNA sequencing
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.