- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Approaching RNA-seq for Cell Line Identification

Published: Vol 10, Iss 3, Feb 5, 2020 DOI: 10.21769/BioProtoc.3507 Views: 5147
Reviewed by: Prashanth N SuravajhalaAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Feb 2019
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Cancer cell lines serve as invaluable model systems for cancer biology research and help in evaluating the efficacy of new therapeutic agents. However, cell line contamination and misidentification have become one of the most pressing problems affecting biomedical research. Available methods of cell line authentication suffer from limited access, time-consuming and often costly for many researchers, hence a new and cost-effective approach for cell line authentication is needed. In this regard, we developed a new method called CeL-ID for cell line authentication using genomic variants as a byproduct derived from RNA-seq data. CeL-ID was trained and tested on publicly available more than 900 RNA-seq dataset derived from the Cancer Cell Line Encyclopedia (CCLE) project; including most frequently used adult and pediatric cancer cell lines. We generated cell line specific variant profiles from RNA-seq data using our in-house pipeline followed by pair-wise variant profile comparison between cell lines using allele frequencies and depth of coverage values of the entire variant set. Comparative analysis of variant profiles revealed that they differ significantly from cell line to cell line whereas identical, synonymous and derivative cell lines share high variant identity and their allelic fractions are highly correlated, which is the basis of this cell line authentication protocol. Additionally, CeL-ID also includes a method to estimate the possible cross-contamination using a linear mixture model with any possible CCLE cells in case no perfect match was detected.
Keywords: Cell line authenticationBackground
Cell lines are widely used as simple model systems in biomedical research to study complex biological processes and to test the therapeutic efficacy of new agents. Its application as a model system has contributed to the characterization and understanding of important biological systems, and has made possible enormous progress in the field of biomedical research (ATCC, 2010; Yu et al., 2015), while the International Cell Line Authentication Committee (ICLAC) lists more than 400 misidentified cell lines (https://iclac.org/databases/cross-contaminations/). Contamination and misidentification of laboratory cell lines have plagued the field for more than half a century, either unintentionally or cell line changes due to passage effects, and continues to remain as one of the most prevalent problems affecting biomedical research to date. Not only it affects overall scientific reproducibility and slows down development in the field but estimated to cost billions of dollars on subsequent research work based on those irreproducible publications (Neimark, 2015; Zaaijer et al., 2017). A lot of effort has been made lately to bring attention to this widespread problem and to correct the contamination of laboratory cell lines. The impact of research on two impostor cell lines: Hep2 and INT407 was reported recently and it was estimated that $713 million were spent on the original papers published on Hep2 and INT407 and additional $3.5 billion were spent on subsequent research based on these publications (Neimark, 2015). In addition, over the last two decades, work from Dr. Korch’s group at the University of Colorado has established more than 70 commonly used cell lines, including Hep2 and INT407, to be contaminated with other cells, such as the fast-growing HeLa cancer cells (Neimark, 2015). Other reports documented that approximately 20% of cell lines currently in use are contaminated (Capes-Davis et al., 2010; Neimark, 2015; Fasterius et al., 2017) that includes many cell lines from the large datasets stored in public repositories (Fasterius et al., 2017). Thanks in part to awareness efforts towards the importance of cell line authentication many journals and institutions including NIH now require researchers to ascertain cell line integrity (Yu et al., 2015).
The most commonly used method to check the integrity of cell lines is the profiling of short tandem repeats (STRs) across several loci as recommended by the Standards Development Organization Workgroup ASN-0002 of American Type Culture Collection (ATCC) (ATCC, 2010; Yu et al., 2015; Fasterius et al., 2017; Zaaijer et al., 2017). Additionally single nucleotide polymorphism (SNP)-based assays are also used to ensure the quality and integrity of cell lines, either in combination with STR profiles or alone (Yu et al., 2015; Fasterius et al., 2017; Zaaijer et al., 2017). Both STR- and SNP-based assays have some inherent limitations. STR-based methods are not reliable in cases with microsatellite instability, loss of heterozygosity or aneuploidy as in case of many cancer cell lines, whereas currently there are no standard reference profiles available for SNP genotyping. Typically, both methods collect genomic DNA prior to the lab experiment, rather than RNAs at the beginning (control samples) as well as at the end of experiment (treated samples), most likely when cell line misuses and contaminations occur.
RNA sequencing has become a routine tool for transcriptome profiling in order to gain new biological insights. It is also being used to identify single nucleotide variants in some specific applications. Seven colorectal cell lines were re-identified by comparing their SNP based discovery profiles from RNA-seq data to the mutational profile of these cell lines available in COSMIC database (Fasterius et al., 2017). In this protocol, we present a detailed pipeline for how to avail RNA sequencing data for cell line identification. An RNA-seq based method for cell line authentication is thought to be potentially more accessible to the biomedical community as a significant number of studies involve the use of data from RNA-seq and therefore, the same can also be availed to check the integrity of the cell line without any extra reagent cost. This process includes detecting cell line specific variant profile from RNA-seq data followed by pairwise comparisons between cell lines using depth of coverage (DP) and allele frequency (FREQ) values of each variant. We have shown earlier that variant profiles are unique to each cell line and benchmarking studies including tests on independent datasets revealed that CeL-ID can identify a cell line with high accuracy (Mohammad et al., 2019). Furthermore, we also developed an additional module to identify one possible contaminator in case no perfect match was detected. In brief, we identified a total of 869,826 variants across all 934 cell lines and constructed master matrices for DP and allele frequency values where each row represents a unique variant and each column a cell line (Mohammad et al., 2019). Authentication of a query cell line includes RNA sequencing, bioinformatics analysis to identify genomic variants and subsequent comparison to DP or FREQ values of variants derived from 934 cell lines. Taken together, scientific rigor and reproducibility mandate the documentation of cell line identity and its purity before and after the biological experiment. The protocol provided utilizes the richness of RNA sequencing data to confirm the cell line integrity after each experiment.
The CeL-ID protocol can be summarized as a procedure of following steps (Figure 1). This protocol will provide details of software and program commands to complete the task.
- RNA sequencing
- Quality Assessment
- Alignment to reference genome
- Post-alignment processing
- Variant calling
- Cell line identification
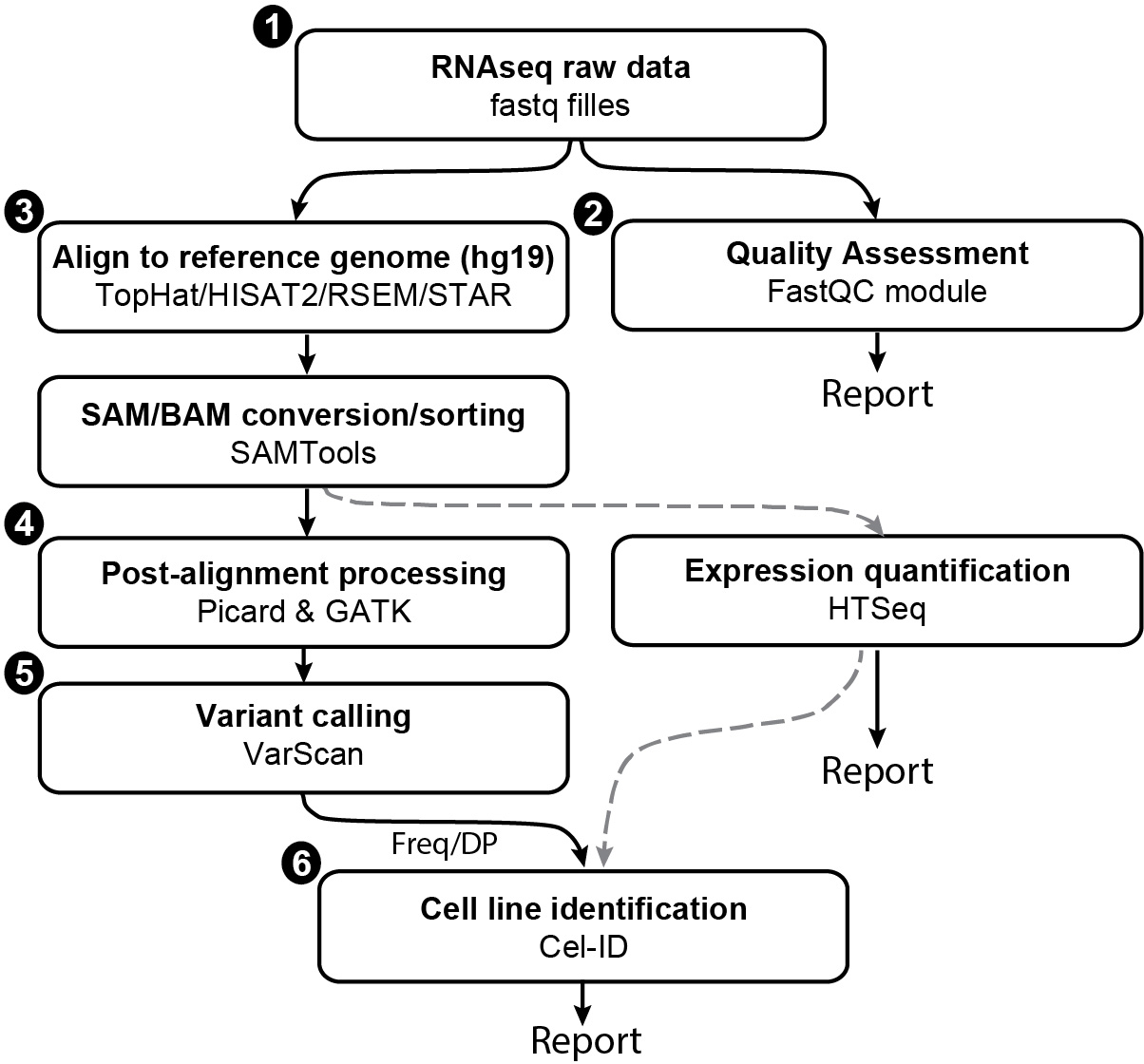
Figure 1. Flowchart showing different steps involved in cell line identification from RNA-seq data
Software
All software mentioned in this paper can be run on any workstation- or server-running UNIX based system like Linux and Mac OS. Alternatively, for Windows machines, Windows subsystem for Linux that comes with Windows 10 can be enabled to run Bash.
- FastQC v0.11.8 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/)
- TopHat v2.1.1 (Trapnell et al., 2009) (http://ccb.jhu.edu/software/tophat/index.shtml)
- Bowtie 2 v2.3.1 (Langmead and Salzberg, 2012) (http://bowtie-bio.sourceforge.net/bowtie2/index.shtml)
- HISAT2 v2.1.0 (Kim et al., 2015) (http://ccb.jhu.edu/software/hisat2/index.shtml)
- RSEM v1.2.31 (Li and Dewey, 2011) (https://github.com/deweylab/RSEM)
- STAR v2.7.3a (Dobin et al., 2013) (https://github.com/alexdobin/STAR)
- SAMtools v0.1.19 (Li et al., 2009) (http://samtools.sourceforge.net/)
- Picard v2.9.0 (https://broadinstitute.github.io/picard/)
- GATK v3.7 (DePristo et al., 2011) (https://software.broadinstitute.org/gatk/)
- VarScan v2.3.9 (Koboldt et al., 2009) (http://varscan.sourceforge.net/)
- HTSeq v0.11.1 (Anders et al., 2015) (https://htseq.readthedocs.io/en/release_0.11.1/)
All abovementioned software comes with their own README page, tutorial and/or manual detailing how to install and make use of the program. Users can choose to install software either locally or globally by installing with root/administrative privileges or just copying program executables to the /usr/bin directory (folder).
Procedure
RNA sequencing: CCLE cell lines were subjected to non-strand specific RNA sequencing performed at the Broad Institute (Tsherniak et al., 2017). RNA sequencing included poly-A selection, cDNA synthesis, library preparation (400 bp insert), sequencing (101 bp paired reads) and a sample identification quality control check (Tsherniak et al., 2017). Samples were prepared using an automated variant of the Illumina TruSeqTM RNA sample preparation protocol (https://www.illumina.com/products/by-type/sequencing-kits/library-prep-kits/truseq-rna-v2.html). An amount of 200 ng of total RNA was used as starting material followed by mRNA selection from the total RNA using oligo dT beads, heat fragmentation, cDNA synthesis and Illumina library preparation (end repair, base “A” addition, adapter ligation and enrichment; please see https://www.illumina.com/documents/products/datasheets/datasheet_truseq_sample_prep_kits.pdf). Sequencing was done using the Illumina HiSeq 2,000 or HiSeq 2,500 platform (Illumina Inc., San Diego, CA).
Data analysis
- Quality Assessment
Checking the quality of the raw sequencing data forms the very first step of any sequencing-based data analysis. Open-source software FastQC and it provides different utility modules to compute per base sequence quality, per base sequence content and sequence duplication
levels. Detailed information on different FastQC mdules is available at https://www.bioinformatics.babraham.ac.uk/projects/fastqc/. Check the quality of the read using the command (if paired end sequencing, you may have to run fastqc for the read2.fastq file as well):
fastqc --extract read1.fastq -o read1_reports/ - Alignment to the reference genome
RNA sequencing reads were mapped to the human reference genome hg19 using TopHat in the CCLE project. Process includes creating bowtie2 indices for hg19 reference genome and mapping of the sequence reads to the indexed genome. Please see https://ccb.jhu.edu/software/tophat/index.shtml for details.
Build bowtie index from the human genome FASTA file hg19.fa as follows:
bowtie2-build hg19.fa hg19
Alignment is done using resulting index as follows:
tophat -p 30 -r 80 --mate-std-dev 40 --no-sort-bam --no-convert-bam --no-coverage-search --library-type fr-unstanded -o tophat_out --GTF genes-hg19.gtf --transcriptome-index anno hg19 read1.fastq read2.fastq
Here we use 30 CPU threads (-p 30) to complete this alignment request. While we recommend using TopHat aligner to match the aligner used for CCLE RNA-seq, our earlier results indicated the comparable results using RSEM in terms of variant calling (Mohammad et al., 2019). It may be noted here that TopHat has entered a low maintenance stage and has been superseded by a more accurate and efficient program called HISAT2 with the same core functionality. HISAT2 is similar to that of TopHat and more details about HISAT2 are available at http://ccb.jhu.edu/software/hisat2/index.shtml.
Build genome index for HISAT2:
hisat2-build hg19.fa hg19
Alignment to the genome:
hisat2 -x hg19 -1 read1.fastq -2 read2.fastq -S align.sam
Additionally, in our previous study, we also tested RSEM aligner to rule out the effect of aligner and observed that RSEM gave a comparative performance. The detailed documentation of RSEM is available at http://deweylab.biostat.wisc.edu/rsem/README.html. Alignment process includes creating genome indexing followed by mapping. Index of the reference genome can be prepared using command:
rsem-prepare-reference --gtf hg19-genes.gtf hg19.fa hg19
Sequence reads are then mapped to the indexed genome using command:
rsem-calculate-expression --paired-end read1.fastq read2.fastq hg19 sample_name
In our experience, all three abovementioned aligners give comparative performances as far as the current task is concerned. A separate benchmarking study showed that STAR aligner achieves the highest sensitivity for both SNPs and, importantly, indels (Engstrom et al., 2013). Therefore, users can use their own discretion and employ their preferred aligner. - Post-alignment processing:
The alignment file produced from above steps is now processed using usual SAMtools, Picard and GATK (Genome Analysis Toolkits) and the processing steps includes adding read group information, sorting, marking duplicates and indexing. Post-processing can be performed in one of two ways: (a) first only involves sorting and indexing whereas; (b) the second method involves more processing to take care of PCR duplicates, local realignment and base recalibration using Picard and GATK. In general, the latter results in better overall variant calling but when applied to good quality data even the first one leads to pretty good variant calling.- Simple alignment post-processing: This just includes alignment sorting and indexing to create analysis-ready alignment (bam) files. Perform the sorting and indexing of the aligned file using SAMtools utilities. SAMtools sort will generate sorted bam file and SAMtools index will generate .bai file. This sorted bam file will be used for variant calling in step 5.
samtools sort -o align_sorted.bam align.bam
samtools index -b align_sorted.bam - Advanced alignment post-processing: This involves more intricate alignment post-processing, including local realignment, base recalibration, and alignment sorting and indexing to make analysis-ready bam files.
- Alignment filtering, sorting, and indexing
This command removes non-primary alignment
samtools view -hS -F 0x100 align.sam > align_filtered.sam
The following 3 Picard commands sort BAM (alignment) according to chromosomal coordinate, remove duplicates, and add read group IDs.
java -Xmx4g -Djava.io.tmpdir=temp/ -jar picard.jar SortSam SORT_ORDER=coordinate CREATE_INDEX=true INPUT=align_rmContam.sam OUTPUT=align_sorted.bam VALIDATION_STRINGENCY=LENIENT
java -Xmx4g -Djava.io.tmpdir=temp/ -jar picard.jar MarkDuplicates INPUT=align_sorted.bam OUTPUT=align_sorted_marked.bam METRICS_FILE=metrics CREATE_INDEX=true VALIDATION_STRINGENCY=LENIENT REMOVE_DUPLICATES=true
java -Xmx4g -Djava.io.tmpdir=temp/ -jar picard.jar AddOrReplaceReadGroups INPUT=align_sorted_marked.bam OUTPUT=align_sorted_marked_readGroup.bam RGID=2 RGLB=Unknown RGPL=Illumina RGPU=sampleIndex RGSM=sampleName VALIDATION_STRINGENCY=SILENT
The command to reorder SAM into numerical chromosomal 1, 2, 3, etc., order, instead of typical alphabet order 1, 10, 11, 12, etc. The “sorted_hg19” shall be provided with the correct numerical chromosomal order, if your default hg19.fa is not organized in the same order. User shall make sure the disk space where the temporary directory (temp/) shall be sufficiently large, considering many sequencing data are relatively larger than your typical linux system storage allocation to the default “temp” directory.
java -Xmx4g -Djava.io.tmpdir=temp/ -jar picard.jar ReorderSam INPUT=align_sorted_marked_readGroup.bam OUTPUT=align_sorted_marked_ordered.bam R=sorted_hg19
samtools index -b align_sorted_marked_ordered.bam - Local realignment and base quality recalibration
Though the overall effect of local realignment is marginal, performing local realignment around indels helps in rescuing a few indels that would otherwise be missed. Therefore, this step is only recommended when spare compute and time is available.
java -Xmx4g -Djava.io.tmpdir=temp/ -jar GenomeAnalysisTK.jar -T RealignerTargetCreator -R sortedGenome -I align_sorted_marked _ordered.bam –o realignment_targets.list
java -Xmx4g -Djava.io.tmpdir=temp/ -jar GenomeAnalysisTK.jar -I align_sorted_marked_ordered.bam -R sortedGenome -T IndelRealigner -targetIntervals realignment_targets.list -o aligned_sorted_marked_realigned.bam
java -Xmx4g -Djava.io.tmpdir=temp/ -jar picard.jar FixMateInformation INPUT=align_sorted_marked_realigned.bam OUTPUT=align_sorted_marked_realigned_fixed.bam SO=coordinate VALIDATION_STRINGENCY=LENIENT CREATE_INDEX=true
Similarly, base calibration has limited effect when applied to good quality data but significantly helps in cases where the qualities have systematic error modes.
java -Xmx4g -Djava.io.tmpdir=temp/ -jar GenomeAnalysisTK.jar -T BaseRecalibrator -R sortedGenome -I aligned_sorted_marked_realigned_fixed.bam -knownSites vcf_dbSNP135 -o recal_data.csv
java -Xmx4g -Djava.io.tmpdir=temp/ -jar GenomeAnalysisTK.jar -T PrintReads -R sortedGenome -I align_sorted_marked_realigned_fixed.bam -BQSR recal_data.csv -o align_sorted.bam
- Alignment filtering, sorting, and indexing
- Simple alignment post-processing: This just includes alignment sorting and indexing to create analysis-ready alignment (bam) files. Perform the sorting and indexing of the aligned file using SAMtools utilities. SAMtools sort will generate sorted bam file and SAMtools index will generate .bai file. This sorted bam file will be used for variant calling in step 5.
- Variant calling
Finally, the variants were called from the sorted and aligned file using VarScan and the resulting call set were filtered to exclude clusters of at least 3 SNPs that are within a window of 35 bases.
samtools mpileup -B -C 50 -q 10 -d 10000 -f hg19.fa align_sorted.bam | java –jar VarScan.v2.3.9.jar mpileup2cns --min-avg-qual 10 --mpileup 1 --min-coverage 10 --min-var-freq 0.05 --output-vcf 1 --p-value 0.05 --variants 1 > outVCF.vcf
This will generate the Variant Call Format (VCF) file with all discovered variants in the cell line. The output .vcf file will be used to extract DP and frequency values in the next step.
Note: There are many variant calling programs that fit for this task and they all have their own strengths and weaknesses as reviewed in several earlier publications (Wang et al., 2013; Xu, 2018). The supplied CCLE RNA-seq FREQ and DP matrices were derived from VarScan, while we recommend matching the variant calling program; we believe the difference between different variant callers should be minimum since we only select variants detected in CCLE samples. For others who would like to know the function of these variants, many variant annotation programs, such as ANNOVAR (Wang et al., 2010) may be selected (more choices may be found in this recent review paper (Bailey et al., 2018)). - Cell line identification: From the resultant variant call file we first select all the variants present in 934 cell lines and then extract DP and FREQ values for each variant using in-house Perl code (vcfFilter_DP_Freq.pl). The output files from the above step contain the variant information (chromosome number, position, REF and ALT allele) and their DP or FREQ values discovered in 934 cell lines. These files are available for download from links provided at CeL-ID GitHub page (https://github.com/chenlabgccri/CeL-ID/blob/master/README.md) and users are not supposed to repeat the variant calling for 934 CCLE cell lines. Users only need to do variant calling for their cell line of interest (Query cell line) and use abovementioned Perl code - vcfFilter_DP_Freq.pl to extract DP and FREQ values for all the variants discovered in query cell line.
The DP and FREQ values from the output vcf file can be obtained using:
perl vcfFilter_DP_Freq.pl --input=outVCF.vcf --list=sampleList.txt --DP=30 --AD=5 --FREQ=25
Subsequently, we compare DP or frequency values from the query cell line with values from each cell line in the 934 cell line cohort and compute correlation coefficients using R scripts (tested on R version 3.5 and higher). The query is assigned to be the most correlated cell line. All the codes required to perform cell line identification step are available from https://github.com/chenlabgccri/CeL-ID. Once users have DP and FREQ values for the query cell line, the cell line identification step can be performed by executing following commands in R:
#Install packages data.table and corrplot, if not already installed
if(!requireNamespace("data.table", quietly = TRUE)){
install.packages("data.table")}
library(data.table)
if(!requireNamespace("corrplot", quietly = TRUE)){
install.packages("corrplot")}
library(corrplot)
# Cel-ID_functions.R see https://github.com/chenlabgccri/CeL-ID
# contains functions to do cell line identification
source("Cel-ID_functions.R")
## Read DP and FREQ file for 934 cell lines (see GitHub page)
ccle_dp <- read.delim( "CCLE_DP.txt", sep = "\t", header = TRUE,
row.names = 1, stringsAsFactors = FALSE)
ccle_freq <- read.delim( "CCLE_FREQ.txt", sep = "\t", header = TRUE,
row.names = 1, stringsAsFactors = FALSE)
# Read DP and FREQ file for the query cell line
#(MCF7 cell line from GEO (GSE86316), see GitHub page)
query_dp <- read.delim( "Query_DP.txt", sep = "\t", header = TRUE,
row.names = 1, stringsAsFactors = FALSE)
query_freq<- read.delim( "Query_FREQ.txt", sep = "\t", header = TRUE,
row.names = 1, stringsAsFactors = FALSE)
# Find the best match, sdx codes the sorted match order based on
# correlation coefficient
rt = CCLE_Identification( ccle_freq, ccle_dp, query_freq, query_dp )
# Return with correlation coefficient, sorted order index,
# and top/bottom 5 correlation matrix
rho = rt[[1]]
sdx = rt[[2]]
corMatrixTop5Bottom5 = rt[[3]]
# Plot correlation matrix
corrplot( corMatrixTop5Bottom5, method = "pie", type = "lower")
The standard output contains the names, correlation coefficients and P-values for the three best matched cell lines. Typical output for given example query cell line (MCF7) looks like:
Total of 17730 variants selected
1st match cell-line is G41726.MCF7.5 with rho = 0.906 p = 8.592e-21 <<< matched cell-line (> L0.001)
2nd match cell-line is G28020.KPL.1.1 with rho = 0.891 p = 1.743e-19
3rd match cell-line is G25206.NCI.H1694.1 with rho = 0.434 p = 0.781
Additionally, the program also provides a correlation plot for top five and bottom five matches and the results for the example query cell line are depicted in Figure 2. It should be noted that MCF7 and KPL1 are known identical cell lines, share a high SNP identity and hence both came as best matches with high correlation coefficients and significant P-values but the next best match shows poor correlation coefficient and a high P-value.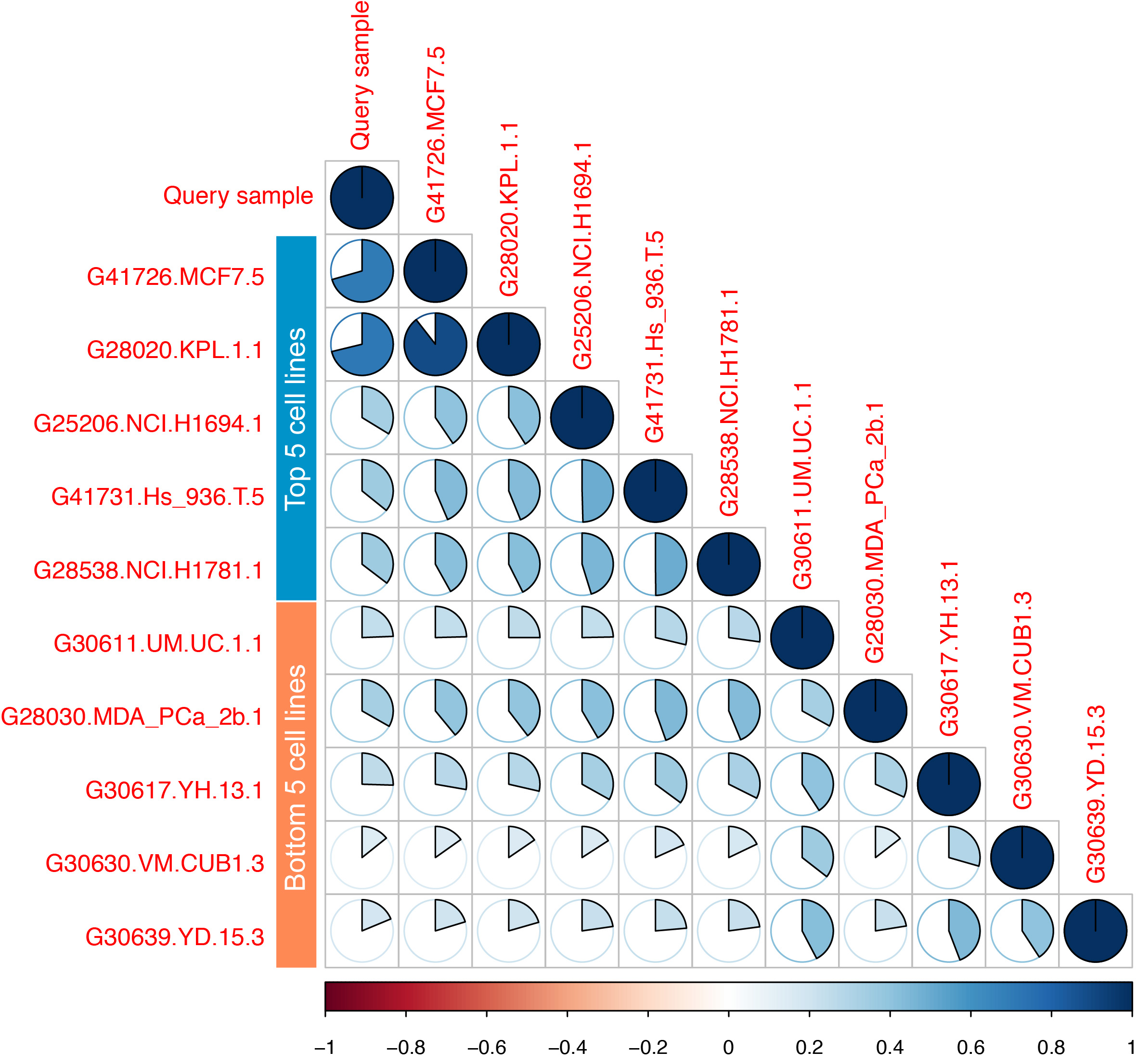
Figure 2. Correlation plot for the best top 5 and bottom 5 matches for the Query sample
Moreover, CeL-ID also includes a function to estimate the level of possible cell line contamination in case no perfect match was detected. This can be executed using the following commands in R:
rt = CCLE_MixtureEstimate(ccle_freq, ccle_dp, mixFreq, mixDP, matched_ccleID )
Where, mixFreq and mixDP are FREQ and DP files from the contaminated (mixed) cell line, and matched_ccleID is the CCLE cell line ID that best matches query cell-line (or in this case the mixture cell to be tested). For the sake of illustration, we created a fake mixture (70% targeted cell, 30% contaminator) sample (please see cellAuthentication.R available from CeL-ID GitHub page for details) and CeL-ID mixture model can rightly estimate the level of contamination. Shown below is the output of contamination estimate using mixFreq and mixDP derived from abovementioned fake mixture, as input:
The possible mixture contains G20469.JHOS.2.2, with proportion = 26.9%, t-stat = 53.6, p-value 6.734348e-231
This indicates that fake mixture contains 26.9% of the contaminator cell line (G20469.JHOS.2.2) which is pretty close to the actual number 30%. For more details about the methodology and benchmarking process, readers are encouraged to refer to our previous publication (Mohammad et al., 2019). It is worth mentioning that expression values of genes obtained using HTSeq (as shown by dashed line arrow in Figure 1) can also be used to compute the correlation but the difference of correlation coefficients of the best match and the next-to-best match is much smaller in comparison to those derived from other variant profiles. The gene expression data for the CCLE cell lines are available from https://portals.broadinstitute.org/ccle.
Notes
All software mentioned in this protocol are freely available and can be easily installed and run on workstations with 64 GB of RAM, decent processor and 50-100 GB of disk space. Moreover, online platforms like Galaxy (https://usegalaxy.org/) and Illumina BaseSpace (https://www.illumina.com/products/by-type/informatics-products/basespace-sequence-hub/apps.html) also provide abovementioned software and many more for easy data analysis. It may be noted here that indices generated for one aligner cannot be used for another aligner.
Acknowledgments
Funding for this work was provided partially by grants from the NCI Cancer Center Shared Resources (NIH-NCI P30CA54174) and CPRIT (RP160732 and RP120685-C2) to TAM and YC. This protocol was adapted from Mohammad et al. (2019). TAM and YC designed the study, drafted and finalized the protocol. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
References
- Anders, S., Pyl, P. T. and Huber, W. (2015). HTSeq - A Python framework to work with high-throughput sequencing data. Bioinformatics 31(2): 166-169.
- ATCC, A. (2010). Cell line misidentification: the beginning of the end. Nat Rev Cancer 10(6): 441-448.
- Bailey, M. H., Tokheim, C., Porta-Pardo, E., Sengupta, S., Bertrand, D., Weerasinghe, A., Colaprico, A., Wendl, M.C., Kim, J. and Reardon, B., et al. (2018). Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 173: 371-385 e318.
- Capes-Davis, A., Theodosopoulos, G., Atkin, I., Drexler, H. G., Kohara, A., MacLeod, R. A., Masters, J. R., Nakamura, Y., Reid, Y. A., Reddel, R. R. and Freshney, R. I. (2010). Check your cultures! A list of cross-contaminated or misidentified cell lines. Int J Cancer 127(1): 1-8.
- DePristo, M. A., Banks, E., Poplin, R., Garimella, K. V., Maguire, J. R., Hartl, C., Philippakis, A. A., del Angel, G., Rivas, M. A., Hanna, M., McKenna, A., Fennell, T. J., Kernytsky, A. M., Sivachenko, A. Y., Cibulskis, K., Gabriel, S. B., Altshuler, D. and Daly, M. J. (2011). A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43(5): 491-498.
- Dobin, A., Davis, C.A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., Batut, P., Chaisson, M., and Gingeras, T.R. (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15-21.
- Engstrom, P.G., Steijger, T., Sipos, B., Grant, G.R., Kahles, A., Ratsch, G., Goldman, N., Hubbard, T.J., Harrow, J., Guigo, R., et al. (2013). Systematic evaluation of spliced alignment programs for RNA-seq data. Nat Methods 10: 1185-1191.
- Fasterius, E., Raso, C., Kennedy, S., Rauch, N., Lundin, P., Kolch, W., Uhlen, M. and Al-Khalili Szigyarto, C. (2017). A novel RNA sequencing data analysis method for cell line authentication. PLoS One 12(2): e0171435.
- Kim, D., Langmead, B. and Salzberg, S. L. (2015). HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12(4): 357-360.
- Koboldt, D. C., Chen, K., Wylie, T., Larson, D. E., McLellan, M. D., Mardis, E. R., Weinstock, G. M., Wilson, R. K. and Ding, L. (2009). VarScan: variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics 25(17): 2283-2285.
- Langmead, B. and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9(4): 357-359.
- Li, B. and Dewey, C. N. (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12: 323.
- Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., Marth, G., Abecasis, G. and Durbin, R. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25(16): 2078-2079.
- Mohammad, T. A., Tsai, Y. S., Ameer, S., Chen, H. H., Chiu, Y. C. and Chen, Y. (2019). CeL-ID: cell line identification using RNA-seq data. BMC Genomics 20(Suppl 1): 81.
- Neimark, J. (2015). Line of attack. Science 347(6225): 938-940.
- Trapnell, C., Pachter, L. and Salzberg, S. L. (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25(9): 1105-1111.
- Tsherniak, A., Vazquez, F., Montgomery, P. G., Weir, B. A., Kryukov, G., Cowley, G. S., Gill, S., Harrington, W. F., Pantel, S., Krill-Burger, J. M., Meyers, R. M., Ali, L., Goodale, A., Lee, Y., Jiang, G., Hsiao, J., Gerath, W. F. J., Howell, S., Merkel, E., Ghandi, M., Garraway, L. A., Root, D. E., Golub, T. R., Boehm, J. S. and Hahn, W. C. (2017). Defining a Cancer Dependency Map. Cell 170(3): 564-576 e516.
- Wang, K., Li, M., and Hakonarson, H. (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38: e164.
- Wang, Q., Jia, P., Li, F., Chen, H., Ji, H., Hucks, D., Dahlman, K.B., Pao, W., and Zhao, Z. (2013). Detecting somatic point mutations in cancer genome sequencing data: a comparison of mutation callers. Genome Med 5: 91.
- Xu, C. (2018). A review of somatic single nucleotide variant calling algorithms for next-generation sequencing data. Comput Struct Biotechnol J 16: 15-24.
- Yu, M., Selvaraj, S. K., Liang-Chu, M. M., Aghajani, S., Busse, M., Yuan, J., Lee, G., Peale, F., Klijn, C., Bourgon, R., Kaminker, J. S. and Neve, R. M. (2015). A resource for cell line authentication, annotation and quality control. Nature 520(7547): 307-311.
- Zaaijer, S., Gordon, A., Speyer, D., Piccone, R., Groen, S. C. and Erlich, Y. (2017). Rapid re-identification of human samples using portable DNA sequencing. Elife 6: e27798.
Article Information
Copyright
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Mohammad, T. A. and Chen, Y. (2020). Approaching RNA-seq for Cell Line Identification. Bio-protocol 10(3): e3507. DOI: 10.21769/BioProtoc.3507.
Category
Cancer Biology > General technique > Genetics > Genome-wide analysis
Cell Biology > Cell-based analysis > Gene expression
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.