- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Study of Microbial Extracellular Vesicles: Separation by Density Gradients, Protection Assays and Labelling for Live Tracking
Published: Vol 10, Iss 2, Jan 20, 2020 DOI: 10.21769/BioProtoc.3502 Views: 5853
Reviewed by: Kristin L. ShinglerPooja VermaLiwei Chen
Original research article
The authors used this protocol in:

Jan 2019
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Extracellular vesicles (EVs) are produced by all domains of life including Bacteria, Archaea and Eukarya. EVs are critical for cellular physiology and contain varied cargo: virulence factors, cell wall remodeling enzymes, extracellular matrix components and even nucleic acids and metabolites. While various protocols for isolating EVs have been established for mammalian cells, the field is actively developing tools to study EVs in other organisms. In this protocol we describe our methods to perform density gradient purification of EVs in bacterial cells, allowing for separation of EV subpopulations, followed by protection assays for EV cargo characterization. Furthermore, we devised a protocol which incorporates a fluorescent conjugate of fatty acids into EVs, the first to allow live-cell EV tracking to observe release of EVs, including during infection of mammalian cells by pathogenic bacteria. These protocols are powerful tools for EV researchers as they enable the observation of EV release and the study of the mechanisms of their formation and release.
Keywords: Extracellular VesiclesBackground
Microbial extracellular vesicles (EVs) are fundamental for cellular physiology (Deatherage et al., 2012; Brown et al., 2015). For pathogenic microbes, EVs can be harnessed for diagnostics and vaccines. However, the study of EVs has been hampered by limited availability of tools which allow in-depth functional and mechanistic study of these small and extraordinary structures.
The protocol for isolating microbial EVs is well established (Rodrigues et al., 2008) and consists of isolation of culture supernatants followed by several (ultra)-centrifugation cycles to separate EVs from cells and debris. Several other EV isolation techniques have been developed in the last years, but these techniques are usually tested and implemented in the context of mammalian EVs and thus do not readily translate to microbial EVs. Researchers should follow the International Society for Extracellular Vesicles ISEV guidelines for EV isolation (Théry et al., 2018) when implementing such protocols.
In our most recent work (Coelho et al., 2019) we developed a density separation purification, a protection assay and a labelling protocol for the live-cell imaging of EVs from the pathogenic bacteria Listeria monocytogenes. In the protocols described here, growth conditions and media are optimized for L. monocytogenes, but our protocols should be readily adaptable to other microbes by adjusting the media and growth conditions. For example, density gradient purification was first developed for the pathogenic yeast Cryptococcus neoformans (Vij et al., 2018).
In this work we successfully used a multi-omics approach, a single-sample analysis of metabolite, protein, and lipid extraction (MPLEx) (Nakayasu et al., 2016) which provided a very cost effective analysis of EV composition. The MPLEx protocol has been well described elsewhere (Burnum-Johnson et al., 2017; Nicora et al., 2018).
Density separation methods for EVs have been used previously (Horstman and Kuehn, 2000; Elluri et al., 2014) and we adapted it to our specific needs. Density separation methods have two advantages over the use of EV preparations described in Protocol A: i) it allows the study the several subpopulations of EVs and, ii) it increases purity of EV preparations by removing contaminants such as soluble and aggregated proteins. Density separation of EVs extracted from 1 L microbial supernatant from L. monocytogenes (or C. neoformans) generates sufficient EVs for electron microscopy as well as dot blot protocols and we are confident that other analysis, such as proteomics and other -omics, can be performed. We have used variations of this protocol to separate melanin from the pathogenic yeast Cryptococcus neoformans (Camacho et al., 2019). Optiprep was used to prepare a step-density gradient, as we found it more straightforward to remove Optiprep from vesicle samples than other reagents used to prepare density gradients. In our experience, colloidal reagents, such as Percoll, were difficult to remove from preparations and subsequently interfered in downstream analysis of EVs by electron microscopy and western blotting.
Protection Assays in EVs have been performed before by our group (Rodrigues et al., 2008; Brown et al., 2014) as they allow to distinguish EV-internalized cargo from exposed cargo. Protection assays are considered the gold standard to show internalization of cargo versus adsorption or co-purification, and no other method (such as immunostaining and imaging by electron microscopy) is accepted as proof that cargo is internalized. Protection assays consist of subjecting the EV preparation to the action of degradative enzyme and then testing for degradation of EV components. In the specific case of L. monocytogenes, EVs were exposed to a protease (proteinase K or trypsin) that will digest any proteins external to EVs while proteins internalized in EVs are protected from proteases. Since L. monocytogenes secreted listeriolysin O (LLO) in EVs and this virulence factor is very efficient in lysing red blood cells we tested internalization of LLO in EVs by measuring lysis of red blood cells. Variations of these strategies simply use different degradative enzymes (proteases, nucleases, etc.) and test for protection inside EVs by measuring remaining amount of degraded product via immunoblot, activity assays, direct quantification, etc. The key factors for protection assays are gentle handling of the EVs to maintain their integrity, as well as appropriate experimental controls (the EVs from genetic-deleted strains and exogenously added controls such as recombinant proteins).
EV labelling in microbes has proven difficult. Despite wide usage of the lipophilic dyes DiO, DiL and PKH26, in our hands we noticed strong aggregation of EVs after labelling with DiO and DiL dyes (measured via dynamic light scattering). Other authors have reported similar problems (Morales-Kastresana et al., 2017; Dehghani et al., 2019) and we thus strongly discourage the use of DiO, DiL and PKH26 for the study of EVs. Alternatively, authors have used molecular tagging of EV cargo coupled to fluorescent or luminescent proteins. Those approaches are very successful when the protein cargo of EVs has been established, and if the cargo is amenable to molecular tagging. We developed a cargo-independent protocol which allows for live-tracking of bacteria, and EVs released by the bacteria, without altering the size (measured via dynamic light scattering) of EVs (Coelho et al., 2019). In brief, we allowed bacterial membranes to incorporate a conjugate of the fluorescent dye Bodipy558 to a long-chain fatty acid dodecanoic acid (BODIPY® FL C12, Thermo Fisher Scientific) followed by high-resolution live-cell microscopy, allowing us to capture videos of release of EVs (Coelho et al., 2019). The strength of this approach is it can be easily applied to other organisms, from mammalian cells to bacteria and fungi, as other fatty acids-BODIPY dye conjugates exist (hexadecanoic for example). In addition, these conjugates are relatively cheap. For our particular labelling protocol we noted strong staining of bacterial cells as well as EVs. We reason that labelling with other fatty acids conjugates may allow more specific staining of EVs. We would recommend to readers to attempt labelling with other fatty acids- BODIPY conjugates as they optimize for their microbes of choice.
Materials and Reagents
- 96-well Flat Bottom Plate (Millipore Sigma, catalog number: CLS3997)
- 96-well Round Bottom Plate (Millipore Sigma, catalog number: CLS3999)
- 8-well microscopy µ-slides, tissue culture treated (ibidi, catalog number: 80826-90)
- Steritop Vacuum Filter Sterilizer, 0.22 µm, GP Millipore Express Plus Membrane, 150 ml Funnel, 45 mm neck size (Millipore, catalog number: SCGPT01RE)
- Amicon Ultra-4 Centrifugal Filter Unit, 10 kDa (Millipore, catalog number: UFC801096) for buffer exchange
- Pipette tips (USA Scientific TipOne, catalog number: 1111-0006)
- 1.5 ml Eppendorf tube
- 3 ml ultracentrifuge tube
- 100% Sheep Red Blood Cells (Store at 2-8 °C for up to one month, Innovative Research, IC100-0210-26568)
- Human epithelial MCF-7 breast cancer cells (ATCC, catalog number: HTB-22)
- 70% ethanol
- Brain-Heart Infusion (BHI) broth (Gibco, Fisher Scientific, catalog number: 211059) and agar (Fisher Scientific, catalog number: B11065)
- Vegitone Infusion broth (Millipore, Merck, catalog number: 41960-500G-F)
- Optiprep Density Gradient Medium (Sigma, catalog number: D1556-250ML)
- BODIPYTM FL C12 (4,4-Difluoro-5,7-Dimethyl-4-Bora-3a,4a-Diaza-s-Indacene-3-Dodecanoic Acid) (Molecular Probes, Thermo Fisher Scientific, catalog number: D-3822)
- Bovine Serum Albumin (BSA) Fraction V, pH 7.0 (store at 2-8 °C, MP Biomedicals, catalog number: 810034)
- Dithiothreitol (DTT), use stock dilutions kept at -20 °C for no more than a couple of months (store at -20 °C, Invitrogen, catalog number: 15508-013)
- Recombinant LLO (rLLO) (store at -20°C in small aliquots to avoid freez-thaw cycles, Abcam, catalog number: ab83345)
- Trypsin-EDTA (0.05%), Phenol Red (stored -20 °C for up to 2 years, and prior to use thawed and kept at 2-8 °C, Gibco, Thermo Fischer Scientific, catalog number: 25300054)
- Dulbecco's Modified Eagle Medium (DMEM; Corning, catalog number: 10-013-CV), supplemented with 10% FBS (Millipore Sigma, catalog number: TMS-016-B), GlutaMAX (Thermo Fisher Scientific, catalog number: 35050061), non-essential amino acids (Thermo Fisher Scientific, catalog number: 11140076), and Gibco Antibiotic-Antimycotic (Thermo Fisher Scientific, catalog number: 15240096)
- DMEM without phenol red (Thermo Fisher Scientific, catalog number: 11054020) with 10% FBS (Millipore Sigma, catalog number: TMS-016-B), GlutaMAX (Thermo Fisher Scientific, catalog number: 35050061), and non-essential amino acids (Thermo Fisher Scientific, catalog number: 11140076)
- Hoechst 33342 (1 µg/ml; Thermo Fisher Scientific, catalog number: H3570)
- TrypLE (Thermo Fisher Scientific, catalog number: 12604013)
- Phosphate buffered saline (PBS), pH 7.4, cell culture grade (Corning, catalog number: 21-031-CV)
- 4% paraformaldehyde (PFA; Electron Microscopy Sciences, catalog number: 15710)
- Gentamicin (Thermo Fisher Scientific, catalog number: 15710064)
- 0.02% sodium azide (Sigma, catalog number: S2002-5G)
- HCl (Fisher Scientific, catalog number: A144-212)
- HEPES (Sigma, catalog number: H3375)
- NaCl (Fisher Scientific, catalog number: S271)
- DMSO (Sigma, catalog number: D2650)
- RBC Lysis buffer (see Recipes)
Equipment
- Ultracentrifuge (Beckman, model: Optima-XL) with SW Ti 55 rotor
- Sorvall Centrifuge (Sorvall RC 5C Plus)
- Sorvall GSA fixed-angle rotor
- Polycarbonate Ultracentrifuge Tubes, 3 ml (Beckman Coulter, catalog number: 355635)
- Ultra-Clear Tube, 25 x 89mm, 38.5 ml (Beckman Coulter, catalog number: 344058)
- Multi-Angle Particle Sizing analyzer (Brookhaven Instruments Corp, 90Plus/ BI-MAS)
- Microbiology shaking incubator, heated water bath
- For EV isolation of large volumes cultures, ultrafiltration or centrifugation devices [Amicon Stirred Cell Model 8200, 200 ml (Millipore, catalog number: WW-29968-16) with Ultracel 100 kDa Ultrafiltration Disks (Millipore, catalog number: PLHK07610) with a N2 source]
- Nalgene 250 ml Centrifuge bottles (Thermo Fischer Scientific, catalog number: 3120-0250)
- Multichannel pipettes (BrandTech Scientific Inc., Transferpette S-12 Multichannel Pipettes, catalog number: 703730)
- Dot blot apparatus (such as Bio-Dot® Microfiltration System (Bio-Rad, catalog number: 1703938) or apply samples to membrane manually)
- DeltaVision Elite High-Resolution Deconvolution Microscope (GE Healthcare) equipped with:
- A Scientific CMOS camera (Chip size: 2560 x 2160 pixels)
- An UltraFast solid-state illumination
- An environmental chamber for live cell imaging
- A 60x (N.A. 1.42) oil immersion objective
- UltimateFocus module
Software
- SoftWoRx (Applied Precision, www.gelifesciences.com)
- ImageJ/FIJI (http://fiji.sc/)
Procedure
Isolate EV using protocol A and keep them in a buffer that preserves their stability. In our experience we can keep EVs for 1-2 days at 4 °C without loss of yield, but we endeavor to use them as quickly as possible. If downstream analysis allows it, we add 0.02% azide at the concentration step in ultrafiltration devices to prevent microbial growth. Buffer exchange can be performed by ultracentrifugation or dialysis with 10-50 KDa membranes.
For L. monocytogenes EVs we used PBS at pH 5.5 to maximize activity of the LLO protein. For any other use a physiological relevant buffer, such as PBS pH 7.4, is appropriate. For the protection assays and activity consider the need to perform buffer exchange to maximize enzymatic activity.
- Prepare Extracellular Vesicles from L. monocytogenes culture (Figure 1)
- Inoculate overnight (16 h) culture of L. monocytogenes strains, both wild type and Δhly, from BHI agar plates in 1 L of BHI broth in a 4 L flask at 37 °C, while shaking at 180 RPM.
- The next day (approximately 16-18 h later), pour cultures from the 4 L flask into 250 ml Nalgene centrifugation bottles, and centrifuge cultures at 10,000 RPM (8,800 x g for a Sorvall GSA fixed-angle rotor) for 30 min at 4 °C.
- Pour out supernatant from bottles, careful to not disrupt the pellet. Pour supernatants through 0.22 µm Steritop vacuum filter into a sterile bottle and keep them on ice. There is no need to keep it fully sterile but we minimize contaminations by using sterile material when possible.
- If downstream application allows it, add a final concentration of 0.02% sodium azide to each filtered supernatant to keep samples free of bacterial growth.
- Clean the ultrafiltration systems by thorough rinses with distilled water (aprox. ¼ of the volume of the system) followed by a rinse with ethanol 70% (aprox. 1/8 of the volume of the system) to minimize cross contaminations and bacterial growth.
- Pour the supernatant into Amicon ultrafiltration system 300 ml at a time (with a N2 stream) until all of the supernatant has passed through the 100 kDa cutoff filter.
- Turn off Amicon filtration system when there is approximately 3-9 ml of “vesicle-enriched” supernatant remaining unpassed through the filter.
- Gently pipette up and down on top of the filter to resuspend extracellular vesicles, and transfer this vesicle-enriched supernatant into 3 ml ultracentrifuge tubes. Any tubes that are not completely filled should be filled to the top with PBS pH 5.5 (1x PBS that is adjusted to pH 5.5 using HCl). Make sure the tubes are balanced before centrifugation and that you followed the instructions to prevent tube collapse during centrifugation.
- Ultracentrifuge the preparations at 100,000 x g for 1 h in Beckman SW Ti 55 rotor at 4 °C with brake set to low (slow brake improves EV yield).
- Decant supernatant in one smooth continuous motion, careful not to allow supernatant to flow back and disturb EV pellet.
- Repeat the ultracentrifugation, by resuspending the pellet in PBS pH 5.5. Then fill the tubes to the top with PBS pH 5.5 (1x PBS that is adjusted to pH 5.5 using HCl). Make sure the tubes are balanced before centrifugation. Ultracentrifuge the preparations at 100,000 x g for 1 h in Beckman SW Ti 55 rotor at 4 °C with brake set to low.
- Decant PBS in one smooth continuous motion, again careful not to allow supernatant to flow back.
- Add 100-500 µl of pH 5.5 PBS (or other physiological buffer) to the EV pellet, very gently resuspend pellet and transfer to a 1.5 ml Eppendorf. Use as soon as possible.
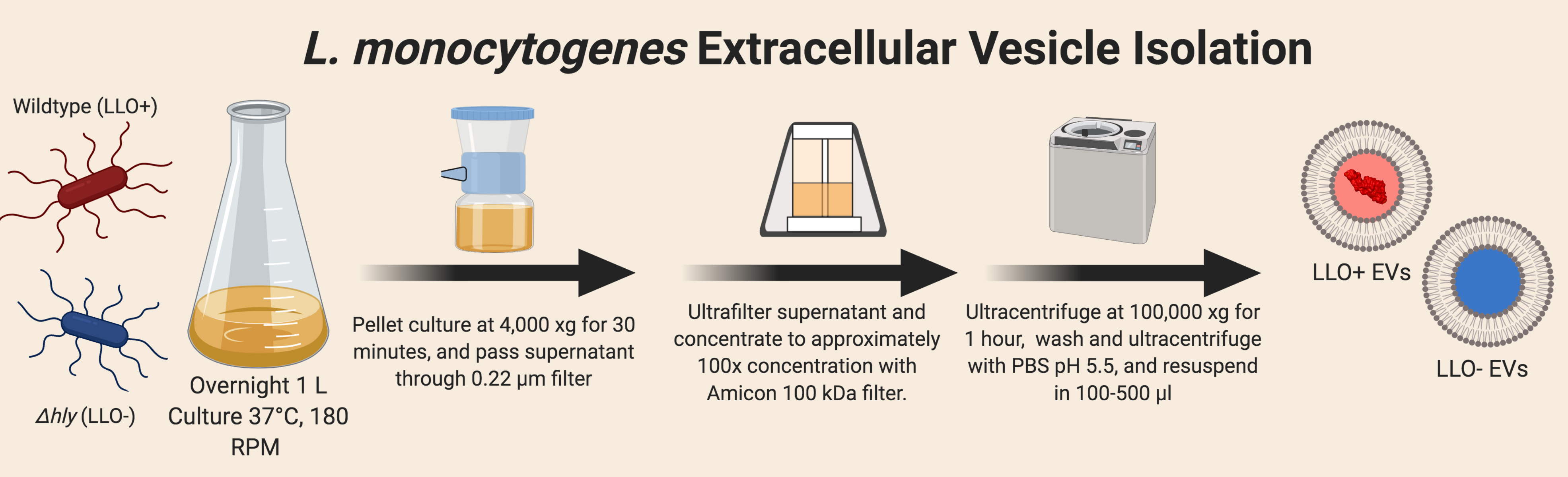
Figure 1. Diagram of Extracellular Vesicle (EV) Isolation
- Density Gradient using Optiprep (Figure 2)
- For density gradients, prepare Optiprep solutions in 10 mM HEPES and 0.85% NaCl, with concentrations of 60%, 35%, 30%, 25%, 20%, 15% and 10%.
- Mix 75 µl of EVs with 225 µl 60% Optiprep to a final concentration of 45% Optiprep and place at the bottom of 3 ml ultracentrifuge tube. Layer the Optiprep solutions in a step-wise gradient by pipetting into the wall of the tube drop by drop the following: 450 µl 35%, 450 µl 30%, 450 µl 25%, 450 µl 20%, 300 µl 15% and 300 µl 10%.
- Centrifuge gradient for 3 h at 180,000 x g in Beckman SW Ti 55 rotor at 4 °C with slow break.
- Collect 10 fractions of equal volume, approximately 270 µl (starting from the top of the tube).
- For electron microscopy (or applications that require removing Optiprep), remove Optiprep by washing samples. Add PBS pH 5.5 to a new ultracentrifuge tube followed by centrifugation at 120,000 x g for 45 min. Repeat this wash. Resuspend samples in PBS in the volume required for further analysis (usually approximately 200 µl). For some applications include an Optiprep-only sample (for example imaging background) to discard any artifacts.
- Estimate LLO content (or other protein detectable by immunoblot) in each of the fractions by applying them directly to a dot-blot apparatus (no need to remove Optiprep for dot blot) (Coelho et al., 2019).

Figure 2. Diagram of Density Gradient separation of Extracellular Vesicles (EV) and subsequent analysis
- Protection assays (Figure 3)
- Prepare 500 mg/L trypsin in pH 5.5 PBS buffer
- Exchange the buffer of Gibco cell-culture grade trypsin (0.05% trypsin (500 mg/L)) with PBS pH 5.5 using 10 kDa cutoff centricon filtration units. Centrifuge units for 30 min at 4 °C. Based on the volumes, controls, and technical replicates described in this protocol, approximately 70 µl of trypsin will be needed for each replicate of this assay.
- Add more PBS pH 5.5 to the top of the filter unit, and centrifuge again. Continue buffer exchange with pH 5.5 PBS until the pink solution is colorless (indicative of cell culture buffer removed), and resuspend the concentrated trypsin in the original volume of cell culture trypsin added.
- Add 22.5 µl of trypsin to 300 µl of wild-type EV and Δhly EV preparations. For a non-digested control, 22.5 µl of pH 5.5 PBS is added to 200 µl of each EV preparations instead of the trypsin. Prepare two additional controls by combining Δhly-EVs with recombinant rLLO to a final concentration of 13 nM of LLO with and without trypsin addition. These rLLO-added samples are positive control for unprotected/non-vesicle enclosed LLO, which is thus susceptible to proteolytic degradation. In samples with added trypsin, trypsin will digest all extravesicular rLLO while the extravesicular rLLO in PBS-only sample should retain hemolytic activity.
Prepare technical duplicates (or triplicates) for all samples. The same ratios and concentrations worked for larger volumes, such that the final trypsin concentration is approximately 35 mg/ml, or that 75 µl of 0.05% trypsin could be added to 1,000 µl of vesicle or supernatant preparations. - Incubate samples at 37 °C for 30 min to an hour while on an orbital shaker (or any other form of gentle agitation). Incubation with trypsin longer than 1 his not recommended, as the EVs stability might be compromised by extended trypsin exposure.
- During incubation for digestion, prepare red blood cells (RBC) and RBC lysis buffer for the activity assay.
- Prepare RBC Lysis buffer (Recipe 1), and keep refrigerated when not in use. If sterile will keep at 4 °C for several months.
- Wash RBC in pH 5.5 PBS + 0.1% BSA by adding 100 µl of 100% Sheep RBC to 10 ml of buffer and centrifuging at 500 x g for 10 min at 4 °C.
- Resuspend in 10 ml RBC Lysis buffer to make a 1% RBC solution. Add DDT to a concentration of 5 mM (maximum activity of LLO), such that final concentration during lysis will be 2.5 mM of DTT.
- After incubation, serially dilute samples 1:3 in RBC Lysis Buffer in a 96 round-bottom plate. Add 150 µl of sample to the first row, and pipette 50 µl into the second row (containing 100 µl of buffer). Each well should have a final volume of 100 µl of diluted sample in it. Do not continue dilution into the bottom row, and leave that as a blank for the RBC lysis buffer. Round bottom plates allow better separation of RBC pellet for the next steps.
- Add 100 µl of 1% RBC solution to each well, and incubate at 37 °C for one hour on an orbital rocker with gentle shaking for lysis to occur.
- Centrifuge 96-well plate for 5 min at 500 x g to sediment unlysed RBC.
- Transfer 100 µl of supernatant from each well into a new 96-well flat bottom plate, while being careful to not disrupt RBC pellet.
- Read plate absorbances at 492 nm and 562 nm. High absorbances at these wavelengths corresponds to the presence of hemoglobin in the supernatant. The more hemoglobin present, the higher the hemolytic activity of the LLO sample. Samples that are protected from protease degradation (i.e., within EVs vesicles), will retain their hemolytic activity even when incubated with trypsin. Extracellular LLO will be degraded and therefore lower hemoglobin will be released.
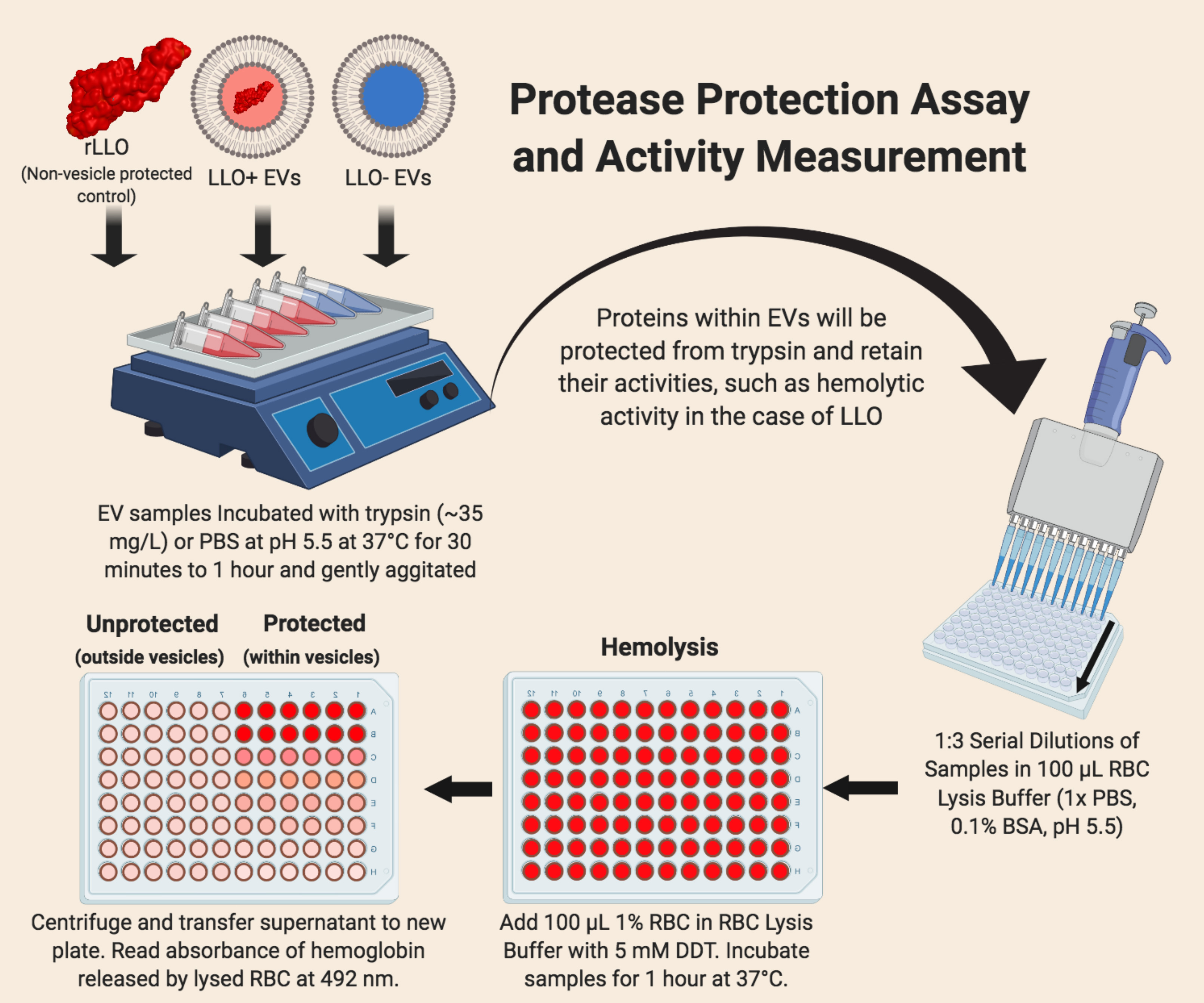
Figure 3. Diagram of the Protection Asssay and Lytic Activity measurements for Extracellular Vesicles (EV)
- Prepare 500 mg/L trypsin in pH 5.5 PBS buffer
- BODIPYTM FL C12 Labelling Protocol for High-Resolution Live-Cell Microscopy (Figure 4)
- Initiate a starter culture of bacteria from plates or glycerol stocks. Grow in appropriate media. For L. monocytogenes we used Brain Heart Infusion broth or Vegitone broth.
- Prepare two identical cultures of bacteria in 1 μM of BODIPYTM FL C12 (or other BODIPY-fatty acid conjugate) diluted in bacterial culture media by inoculating from single plated colony, or 1 μL starter culture, in 1 ml Vegitone broth. Grow for 16-20 h or until they reach early stationary phase. For imaging experiments, we used small volumes of bacteria (up to 5 ml of bacteria) and grew with agitation at 30 °C to repress LLO production. For other studies we grew larger volumes (up to 1 L) at 37 °C and found the experiments readily scaled up. Consider the need to include an unlabeled bacteria control using appropriate volume of vehicle (DMSO).
- Wash away non-incorporated BODIPYTM FL C12 by centrifugation at 2,000 x g for 2 min at room temperature, to separate cells and media, and wash twice with ice-cold PBS of approximately 10x the volume of the cell pellet for high volume preparations, and with 1 ml of PBS for the low-scale required for imaging. Cell pellet should be colored pink. For imaging, resuspend in 1 mL fully-supplemented phenol red-free DMEM, without antibiotics. Determine the optical density (OD) at a wavelength of 600 nm.
- Take only one of the bacteria-containing tubes and use it to prepare a killed bacteria control (for L. monocytogenes we used 70 °C for 10 min-2 h depending on culture and cell pellet volumes). Killed bacteria serve as a control for passive release of lipids or debris from dead cells versus active secretion of EVs by healthy cells. Other methods of killing are appropriate as long as they keep cells relatively intact. Keep the healthy bacterial cells in the second tube on ice to arrest growth during this period. After killing the cells, you now can now either grow and collect BODIPY-labelled EVs (see Step D5) or proceed to imaging (see Step D6). Alternatively proceed to make BODIPY-labelled EVs.
- For collection of BODIPY-labelled EVs: grow the cells for 6 h (or more) in 500 ml to 1 L of fresh media, including the killed cells control and the unlabelled cellls. After an appropriate time, proceed to EV isolation procedure protocol A. Note that the EV pellet should be faint pink due to successful incorporation of BODIPY-fatty acid conjugate into EVs. We confirmed incorporation of BODIPYTM FL C12 into EVs by subjecting EVs from healthy bacterial cells to density gradient separation and found that in live cells BODIPYTM FL C12 signal was found in the same fractions with observable EVs (Coelho et al., 2019).
- The cells are now ready for imaging: add BODIPY-labelled bacteria to mammalian cells for infection studies or other experimental conditions being tested.
- For MCF-7 imaging experiments, 24 h prior to infection, evenly plate 30,000 MCF-7 cells in each well of an 8-well ibidi imaging chamber.
- From OD measurements, calculate the volume of bacterial cells needed for multiplicity of infection (MOI) of 10-50, assuming a doubling of MCF-7 cells until time of infection (approx. 60,000 cells). Assume OD=1 is 1 x 109 L. monocytogenes/ml (this ratio may change if using other organisms).
- Calculate the number of bacteria needed using MOI and cell density. 60,000 MCF-7 cells at time of infection to be infected with bacteria at MOI 10→ 60,000 x 10 = 600,000 bacteria needed.
- Calculate number of bacteria/ml using the OD measurements and know relationship between OD and bacteria concentration in the suspension. Measured OD of bacteria is 0.5 in the suspension → 1 x 109 bacteria/ml x 0.5 = 5 x 108 L. monocytogenes/ml.
- Calculate volume of suspension needed to add bacteria to cells. To add 600,000 bacteria to cells, calculated in (a) from a suspension of 5 x 108 L. monocytogenes/ml, calculated in (b) →600,000 ÷ 5 x 108=0.0012 ml=1.2 µl.
- Perform live cell imaging, starting at 2-3 h post infection, or fix cells for 15 min in 4% paraformaldehyde using a pre-warmed imaging chamber.
- Acquire live-cell high-resoultion images at 37 °C using a 60x oil immersion objective. Image fixed cells at room temperature, using a 60x oil immersion objective. Time-lapse imaging of infected, live MCF-7 cells is performed at 1 image/5 s for brief periods of time (e.g., 2 min), taking care to limit photobleaching.
- For highest resolution, deconvolve images acquired using the DeltaVision microscope using Softworx.
- Analyze single time-point images and time-series image sets using ImageJ/FIJI software, which provide numerous plugins for image presentation and quantifying phenotypes. For example, score the number of BODIPYTM FL C12 puncta (EVs) per cell using the “Cell Counter” plugin. Score subcellular EV dynamics by tracking the localization of BODIPYTM FL C12 puncta in time series using the “Manuel Tracking” plugin.
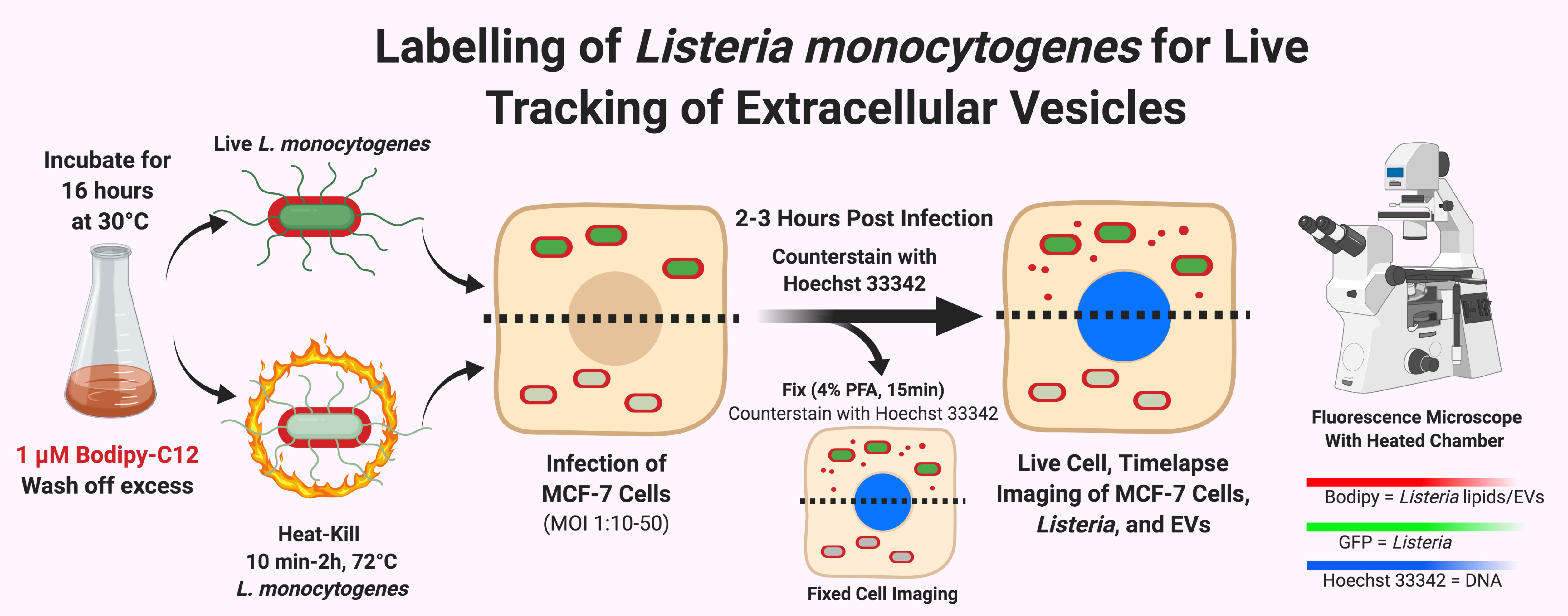
Figure 4. Diagram of BODIPYTM FL C12 labeling for live-imaging of EV secretion during L. monocytogenes infection
Recipes
- RBC Lysis buffer
Dissolve BSA into pH 5.5 PBS for a final concentration of 0.1% BSA (w/v)
Acknowledgments
A.C. was supported by National Institutes of Health (NIH) awards 5R01HL059842, 5R01AI033774, 5R37AI033142, and 5R01AI052733. Schematics in Figures 1-4 were created with BioRender.com.
We thank previous members of Casadevall laboratory for optimizing the protocol through the years. The labelling protocol was devised and optimized by Anne Hamacher-Brady and Nathan R. Brady.
Competing interests
We declare no competing interests.
References
- Brown, L., Kessler, A., Cabezas-Sanchez, P., Luque-Garcia, J. L. and Casadevall, A. (2014). Extracellular vesicles produced by the Gram-positive bacterium Bacillus subtilis are disrupted by the lipopeptide surfactin. Mol Microbiol 93(1): 183-198.
- Brown, L., Wolf, J. M., Prados-Rosales, R. and Casadevall, A. (2015). Through the wall: extracellular vesicles in Gram-positive bacteria, mycobacteria and fungi. Nat Rev Microbiol 13(10): 620-630.
- Burnum-Johnson, K. E., Kyle, J. E., Eisfeld, A. J., Casey, C. P., Stratton, K. G., Gonzalez, J. F., Habyarimana, F., Negretti, N. M., Sims, A. C., Chauhan, S., Thackray, L. B., Halfmann, P. J., Walters, K. B., Kim, Y. M., Zink, E. M., Nicora, C. D., Weitz, K. K., Webb-Robertson, B. M., Nakayasu, E. S., Ahmer, B., Konkel, M. E., Motin, V., Baric, R. S., Diamond, M. S., Kawaoka, Y., Waters, K. M., Smith, R. D. and Metz, T. O. (2017). MPLEx: a method for simultaneous pathogen inactivation and extraction of samples for multi-omics profiling. Analyst 142(3): 442-448.
- Camacho, E., Vij, R., Chrissian, C., Prados-Rosales, R., Gil, D., O'Meally, R. N., Cordero, R. J. B., Cole, R. N., McCaffery, J. M., Stark, R. E. and Casadevall, A. (2019). The structural unit of melanin in the cell wall of the fungal pathogen Cryptococcus neoformans. J Biol Chem 294(27): 10471-10489.
- Coelho, C., Brown, L., Maryam, M., Vij, R., Smith, D. F. Q., Burnet, M. C., Kyle, J. E., Heyman, H. M., Ramirez, J., Prados-Rosales, R., Lauvau, G., Nakayasu, E. S., Brady, N. R., Hamacher-Brady, A., Coppens, I. and Casadevall, A. (2019). Listeria monocytogenes virulence factors, including listeriolysin O, are secreted in biologically active extracellular vesicles. J Biol Chem 294(4): 1202-1217.
- Deatherage, B. L. and Cookson, B. T. (2012). Membrane vesicle release in bacteria, eukaryotes, and archaea: a conserved yet underappreciated aspect of microbial life. Infect Immun 80(6): 1948-1957.
- Dehghani, M., Gulvin, S. M., Flax, J., Gaborski, T. R. (2019). Exosome labeling by lipophilic dye PKH26 results in significant increase in vesicle size. bioRxiv 532028.
- Elluri, S., Enow, C., Vdovikova, S., Rompikuntal, P. K., Dongre, M., Carlsson, S., Pal, A., Uhlin, B. E. and Wai, S. N. (2014). Outer membrane vesicles mediate transport of biologically active Vibrio cholerae cytolysin (VCC) from V. cholerae strains. PLoS One 9(9): e106731.
- Horstman, A. L. and Kuehn, M. J. (2000). Enterotoxigenic Escherichia coli secretes active heat-labile enterotoxin via outer membrane vesicles. J Biol Chem 275(17): 12489-12496.
- Morales-Kastresana, A., Telford, B., Musich, T. A., McKinnon, K., Clayborne, C., Braig, Z., Rosner, A., Demberg, T., Watson, D. C., Karpova, T. S., Freeman, G. J., DeKruyff, R. H., Pavlakis, G. N., Terabe, M., Robert-Guroff, M., Berzofsky, J. A. and Jones, J. C. (2017). Labeling extracellular vesicles for nanoscale flow cytometry. Sci Rep 7(1): 1878.
- Nakayasu, E. S., Nicora, C. D., Sims, A. C., Burnum-Johnson, K. E., Kim, Y. M., Kyle, J. E., Matzke, M. M., Shukla, A. K., Chu, R. K., Schepmoes, A. A., Jacobs, J. M., Baric, R. S., Webb-Robertson, B. J., Smith, R. D. and Metz, T. O. (2016). MPLEx: a robust and universal protocol for single-sample integrative proteomic, metabolomic, and lipidomic analyses. mSystems 1(3).
- Nicora, C. D., Burnum-Johnson, K. E., Nakayasu, E. S., Casey, C. P., White, R. A., 3rd, Roy Chowdhury, T., Kyle, J. E., Kim, Y. M., Smith, R. D., Metz, T. O., Jansson, J. K. and Baker, E. S. (2018). The MPLEx protocol for multi-omic analyses of soil samples. J Vis Exp (135). doi: 10.3791/57343.
- Rodrigues, M. L., Nakayasu, E. S., Oliveira, D. L., Nimrichter, L., Nosanchuk, J. D., Almeida, I. C. and Casadevall, A. (2008). Extracellular vesicles produced by Cryptococcus neoformans contain protein components associated with virulence. Eukaryot Cell 7(1): 58-67.
- Théry, C., Witwer, K. W., Aikawa, E., Alcaraz, M. J., Anderson, J. D., Andriantsitohaina, R., Antoniou, A., Arab, T., Archer, F., Atkin-Smith, G. K., Ayre, D. C., Bach, J. M., Bachurski, D., Baharvand, H., Balaj, L., Baldacchino, S., Bauer, N. N., Baxter, A. A., Bebawy, M., Beckham, C., Bedina Zavec, A., Benmoussa, A., Berardi, A. C., Bergese, P., Bielska, E., Blenkiron, C., Bobis-Wozowicz, S., Boilard, E., Boireau, W., Bongiovanni, A., Borràs, F. E., Bosch, S., Boulanger, C. M., Breakefield, X., Breglio, A. M., Brennan, M. Á., Brigstock, D. R., Brisson, A., Broekman, M. L., Bromberg, J. F., Bryl-Gorecka, P., Buch, S., Buck, A. H., Burger, D., Busatto, S., Buschmann, D., Bussolati, B., Buzás, E. I., Byrd, J. B., Camussi, G., Carter, D. R., Caruso, S., Chamley, L. W., Chang, Y. T., Chen, C., Chen, S., Cheng, L., Chin, A. R., Clayton, A., Clerici, S. P., Cocks, A., Cocucci, E., Coffey, R. J., Cordeiro-da-Silva, A., Couch, Y., Coumans, F. A., Coyle, B., Crescitelli, R., Criado, M. F., D'Souza-Schorey, C., Das, S., Datta Chaudhuri, A., de Candia, P., De Santana, E. F., De Wever, O., Del Portillo, H. A., Demaret, T., Deville, S., Devitt, A., Dhondt, B., Di Vizio, D., Dieterich, L. C., Dolo, V., Dominguez Rubio, A. P., Dominici, M., Dourado, M. R., Driedonks, T. A., Duarte, F. V., Duncan, H. M., Eichenberger, R. M., Ekström, K., El Andaloussi, S., Elie-Caille, C., Erdbrügger, U., Falcón-Pérez, J. M., Fatima, F., Fish, J. E., Flores-Bellver, M., Försönits, A., Frelet-Barrand, A., Fricke, F., Fuhrmann, G., Gabrielsson, S., Gámez -Valero, A., Gardiner, C., Gärtner, K., Gaudin, R., Gho, Y. S., Giebel, B., Gilbert, C., Gimona, M., Giusti, I., Goberdhan, D. C., Görgens, A., Gorski, S. M., Greening, D. W., Gross, J. C., Gualerzi, A., Gupta, G. N., Gustafson, D., Handberg, A., Haraszti, R. A., Harrison, P., Hegyesi, H., Hendrix, A., Hill, A. F., Hochberg, F. H., Hoffmann, K. F., Holder, B., Holthofer, H., Hosseinkhani, B., Hu, G., Huang, Y., Huber, V., Hunt, S., Ibrahim, A. G., Ikezu, T., Inal, J. M., Isin, M., Ivanova, A., Jackson, H. K., Jacobsen, S., Jay, S. M., Jayachandran, M., Jenster, G., Jiang, L., Johnson, S. M., Jones, J. C., Jong, A., Jovanovic-Talisman, T., Jung, S., Kalluri, R., Kano, S. I., Kaur, S., Kawamura, Y., Keller, E. T., Khamari, D., Khomyakova, E., Khvorova, A., Kierulf, P., Kim, K. P., Kislinger, T., Klingeborn, M., Klinke, D. J., 2nd, Kornek, M., Kosanović, M. M., Kovács, Á. F., Krämer-Albers, E. M., Krasemann, S., Krause, M., Kurochkin, I. V., Kusuma, G. D., Kuypers, S., Laitinen, S., Langevin, S. M., Languino, L. R., Lannigan, J., Lässer, C., Laurent, L. C., Lavieu, G., Lázaro-Ibáñez, E., Le Lay, S., Lee, M. S., Lee, Y. X. F., Lemos, D. S., Lenassi, M., Leszczynska, A., Li, I. T., Liao, K., Libregts, S. F., Ligeti, E., Lim, R., Lim, S. K., Linē, A., Linnemannstöns, K., Llorente, A., Lombard, C. A., Lorenowicz, M. J., Lörincz, Á. M., Lötvall, J., Lovett, J., Lowry, M. C., Loyer, X., Lu, Q., Lukomska, B., Lunavat, T. R., Maas, S. L., Malhi, H., Marcilla, A., Mariani, J., Mariscal, J., Martens-Uzunova, E. S., Martin-Jaular, L., Martinez, M. C., Martins, V. R., Mathieu, M., Mathivanan, S., Maugeri, M., McGinnis, L. K., McVey, M. J., Meckes, D. G., Jr., Meehan, K. L., Mertens, I., Minciacchi, V. R., Möller, A., Møller Jørgensen, M., Morales-Kastresana, A., Morhayim, J., Mullier, F., Muraca, M., Musante, L., Mussack, V., Muth, D. C., Myburgh, K. H., Najrana, T., Nawaz, M., Nazarenko, I., Nejsum, P., Neri, C., Neri, T., Nieuwland, R., Nimrichter, L., Nolan, J. P., Nolte-'t Hoen, E. N., Noren Hooten, N., O'Driscoll, L., O'Grady, T., O'Loghlen, A., Ochiya, T., Olivier, M., Ortiz, A., Ortiz, L. A., Osteikoetxea, X., Østergaard, O., Ostrowski, M., Park, J., Pegtel, D. M., Peinado, H., Perut, F., Pfaffl, M. W., Phinney, D. G., Pieters, B. C., Pink, R. C., Pisetsky, D. S., Pogge von Strandmann, E., Polakovicova, I., Poon, I. K., Powell, B. H., Prada, I., Pulliam, L., Quesenberry, P., Radeghieri, A., Raffai, R. L., Raimondo, S., Rak, J., Ramirez, M. I., Raposo, G., Rayyan, M. S., Regev-Rudzki, N., Ricklefs, F. L., Robbins, P. D., Roberts, D. D., Rodrigues, S. C., Rohde, E., Rome, S., Rouschop, K. M., Rughetti, A., Russell, A. E., Saa, P., Sahoo, S., Salas-Huenuleo, E., Sanchez, C., Saugstad, J. A., Saul, M. J., Schiffelers, R. M., Schneider, R., Schøyen, T. H., Scott, A., Shahaj, E., Sharma, S., Shatnyeva, O., Shekari, F., Shelke, G. V., Shetty, A. K., Shiba, K., Siljander, P. R., Silva, A. M., Skowronek, A., Snyder, O. L., 2nd, Soares, R. P., Sodar, B. W., Soekmadji, C., Sotillo, J., Stahl, P. D., Stoorvogel, W., Stott, S. L., Strasser, E. F., Swift, S., Tahara, H., Tewari, M., Timms, K., Tiwari, S., Tixeira, R., Tkach, M., Toh, W. S., Tomasini, R., Torrecilhas, A. C., Tosar, J. P., Toxavidis, V., Urbanelli, L., Vader, P., van Balkom, B. W., van der Grein, S. G., Van Deun, J., van Herwijnen, M. J., Van Keuren-Jensen, K., van Niel, G., van Royen, M. E., van Wijnen, A. J., Vasconcelos, M. H., Vechetti, I. J., Jr., Veit, T. D., Vella, L. J., Velot, E., Verweij, F. J., Vestad, B., Vinas, J. L., Visnovitz, T., Vukman, K. V., Wahlgren, J., Watson, D. C., Wauben, M. H., Weaver, A., Webber, J. P., Weber, V., Wehman, A. M., Weiss, D. J., Welsh, J. A., Wendt, S., Wheelock, A. M., Wiener, Z., Witte, L., Wolfram, J., Xagorari, A., Xander, P., Xu, J., Yan, X., Yáñez-Mó, M., Yin, H., Yuana, Y., Zappulli, V., Zarubova, J., Žėkas, V., Zhang, J. Y., Zhao, Z., Zheng, L., Zheutlin, A. R., Zickler, A. M., Zimmermann, P., Zivkovic, A. M., Zocco, D. and Zuba-Surma, E. K. (2018). Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles 7(1): 1535750.
- Vij, R., Cordero, R. J. B. and Casadevall, A. (2018). The buoyancy of Cryptococcus neoformans is affected by capsule size. mSphere 3: e00534-18.
Article Information
Copyright
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Coelho, C., Vij, R., Smith, D. Q., Brady, N. R., Hamacher-Brady, A. and Casadevall, A. (2020). Study of Microbial Extracellular Vesicles: Separation by Density Gradients, Protection Assays and Labelling for Live Tracking. Bio-protocol 10(2): e3502. DOI: 10.21769/BioProtoc.3502.
- Coelho, C., Brown, L., Maryam, M., Vij, R., Smith, D. F. Q., Burnet, M. C., Kyle, J. E., Heyman, H. M., Ramirez, J., Prados-Rosales, R., Lauvau, G., Nakayasu, E. S., Brady, N. R., Hamacher-Brady, A., Coppens, I. and Casadevall, A. (2019). Listeria monocytogenes virulence factors, including listeriolysin O, are secreted in biologically active extracellular vesicles. J Biol Chem 294(4): 1202-1217.
Category
Microbiology > Microbe-host interactions > Bacterium
Microbiology > Microbial cell biology > Cell imaging
Cell Biology > Organelle isolation > Extracellular vesicle
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.