- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Automated Analysis of Cell Surface Ruffling: Ruffle Quantification Macro
.jpeg)


Published: Vol 10, Iss 2, Jan 20, 2020 DOI: 10.21769/BioProtoc.3494 Views: 5203
Reviewed by: Alka MehraYang FuAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Nov 2018

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Cell surface protrusions include F-actin rich, wave-like ruffles that are erected transiently in response to stimuli and during cell migration. Macrophages are innate immune cells that ruffle constitutively and more dramatically in cells activated by pathogens. Dorsal ruffles and their resulting macropinosomes are key sites for environmental sampling, pathogen detection and immune signaling. Quantitative assessment of ruffling is important for assessing pathogen responses in macrophages and for analysis of growth factor responses in other cell types but automated and quantitative methods are lacking, and rely on manual and qualitative assessments. Here we present an automated ImageJ macro for quantifying dorsal cell surface protrusions from 3D microscope images. The assay presented here is suitable for high-throughput screening applications to detect drug, pathogen, or growth factor induced changes in cell ruffling by measuring ruffle area and intensity and providing normalized values in an easy to read combined spreadsheet.
Keywords: RufflingBackground
Cells develop ruffles or erect veils of membrane on their surfaces during migration and in responses to growth factors or other mediators. As innate immune cells, macrophages ruffle both constitutively and upon stimulation by growth factors, pathogens or other activating stimuli. Ruffles can take on different forms. Dorsal ruffles on the upper surface of adherent cells are typically large erect. F-actin-rich veils which can be circular (hence the alternative name ‘circular dorsal ruffles’) usually rise up transiently and then collapse back onto the surface. Ruffles often form macropinosomes as described in Condon et al., 2018. Macropinosomes internalise the ruffle membrane, simultaneously ingesting fluid and soluble or small particulate cargo (Kerr and Teasdale, 2009; Commisso et al., 2013). Dorsal ruffles or circular dorsal ruffles have been described in depth in macrophages, epithelial cells and fibroblasts where they have gained recognition as sites for immune cell activation (Luo et al., 2014; Wall et al., 2017), growth factor signaling (Buccione et al., 2004) and activated receptor clustering, and internalisation that play pivotal roles in cancer cell signaling (Buccione et al., 2004; Orth and McNiven, 2006). Dorsal ruffles are also induced by a number of pathogens, including Salmonella entericia serovar typhimurium at sites of pathogen entry (Ly and Casanova, 2007).
The large, circular dorsal ruffles that have been observed on different cell types are dependent on the transient production of F-actin, and inhibitors of F-actin polymerisation such as cytochalasin D abolish their formation (Hedberg et al., 1993; Buccione et al., 2004). As dorsal ruffles are highly enriched in F-actin and contribute a much greater area than other F-actin rich projections, such as filopodia, on the dorsal surface, fluorescent markers of F-actin are relatively enriched in dorsal ruffles. Many studies utilize the presence of dorsal ruffles as a qualitative read-out of cellular responses (Gurung et al., 2003; Gandhi et al., 2004; Singla et al., 2019). The ability to quantify ruffling or F-actin cell surface protrusions, would provide useful analysis for a wide range of biological research ranging from stimulation assays to high-throughput screening. Ruffling has been assessed in images by taking whole-cell, whole field of view F-actin measurements (Schlam et al., 2016) or by performing manual selection and user-interactive regions of interest (Venter et al., 2015).
Here, we have developed a robust and automated assay for dorsal ruffle quantification that requires no user input for cell selection or segmentation of both basal and dorsal regions, defined by the mid-point of the nucleus. Measurements for ruffles are also made on Sum-slices images, meaning depth information is taken into account (i.e., taller ruffles contribute to higher intensity counts). This assay enables batch processing of multiple images to analyse large cell numbers and provide tabulated results for easy statistical analysis using third party software. This robust, automated assay is well suited for screening cell populations to detect changes in dorsal ruffling induced by extracellular stimuli such as pathogens, growth factors, hormones or cytokines and/or large scale screening of pharmacological inhibitors, natural products, or nutritional conditions.
Materials and Reagents
- Polystyrene Petri Dish for cell passaging (Thermo Fisher Scientific, catalog number: LBSPD1002X, or equivalent)
- 24-well tissue culture plate (Thermo Fisher Scientific, catalog number: 142475, or equivalent)
- 12 mm round #1 coverslip (Thermo Fisher Scientific, catalog number: MENCSC121GP, or equivalent)
- Microscope slide 26 x 76 mm (VWR, catalog number: VWRI631-1558, or equivalent)
- RAW264.7 macrophage-like cell line (ATCC, catalog number: TIB071)
- Clear Nail Polish (Ultra3, catalog number: 14111130, or equivalent)
- RPMI 1640 media (Lonza Australia, catalog number: BE12-702F)
- Fetal Bovine Serum (Thermo Fisher Scientific, catalog number: 16000044)
- L-glutamine 100x (Thermo Fisher Scientific, catalog number: 25030-081)
- 4% Paraformaldehyde (ProSciTech, catalog number: EMS15735-100, or equivalent)
- Triton X-100 (Sigma-Aldrich, catalog number: X100-500ML)
- Phosphate buffered saline (PBS) (Thermo Fisher Scientific, catalog number: 10010023, or equivalent)
- AlexaFluor 488-Phalloidin (Invitrogen, catalog number: A12379, see Notes)
- DAPI nuclei stain (Roche, catalog number: 10236276001, or equivalent, see Notes)
- ProLong Diamond Antifade reagent (Thermo Fisher Scientific, catalog number: P10144, or equivalent)
- Poly-D-Lysine (Thermo Fisher Scientific, catalog number: A3890401, or equivalent)
- Cell culture medium (see Recipes)
General
- Cell passaging supplies
- 15 ml conical tubes
- Pipette tips (10, 50, 100, 1,000 μl)
- Kimwipes
- Lens wipes
- Parafilm
- Paper towel
- Immersion oil
- dH2O
- 70% Ethanol (EtOH)
Equipment
- Automated serological pipette (Thermo Fisher Scientific, S1 Pipette Filler, catalog number: 14-387-163, or equivalent)
- Graduated pipettes (P2, P20, P100, P1000) (John Moris, Gilson #115209, or equivalent)
- Precision forceps (Dumont, style 7, catalog number: T07-912, or equivalent)
- Temperature and CO2 controlled incubator (VWR, catalog number: 390-2012, or equivalent)
- Tissue culture inverted phase contrast microscope
- Hemocytometer
- Light-proof staining chamber
Note: An aluminum foil coated shallow chamber is sufficient. - Zeiss AxioImager Upright fluorescence Apotome Microscope (see Notes, or equivalent)
Software
- Zeiss Zen Blue 2.3 (Carl Zeiss, Germany, or equivalent acquisition software)
- FIJI Image Analysis program (Schindelin et al., 2012)
- Ruffle Quantification Macro (https://github.com/NickCondon/RuffleQuantification)
- Microsoft Excel (or equivalent)
- GraphPad Prism 8 (or equivalent)
Procedure
- Cell culture
- Create a suspension of your cell line choice.
- Determine cell concentration using hemocytometer and dilute to a concentration 0.1 x 106 cells/ml in tissue culture medium (Recipe 1).
- Place one coverslip into each well of 24-well tissue plate.
- Add 500 μl of diluted cell suspension (0.5 x 105 cells) to each well.
Note: Desired confluency the next day = 60%. - Press coverslip into base of well using forceps to remove any air bubbles that have formed under the coverslip.
- Allow cells to adhere and spread out overnight (at least 16 h) prior to experiments.
Note: For longer treatment or transient transfection, lowering the initial cell seeding density may be required (60% confluency on day of fixation is desirable). - Perform stimulation/pharmacological inhibition experiment as required.
Note: We routinely use 10-100 ng/μl lippopolysaccharide for 30 min to induce ruffle formation. - Aspirate media and wash with ice cold PBS.
- Fix cells for at least 30 min with 4% Paraformaldehyde.
- Thrice wash cells with room temperature PBS.
- Staining coverslips
- Place a wet paper towel into the base of the staining chamber.
- Lay out parafilm on top of wet paper towel.
- Remove coverslips from 24-well plate and place cell-side up onto parafilm.
- Add a drop of PBS onto each coverslip.
Note: Coverslips should be kept ‘wet’ at all times to ensure cell integrity and to prevent dehydration. - Permeabilise cells by placing a drop of 0.1% Triton X-100 onto each coverslip for no more than 5 min.
- Wash coverslips with PBS three times by aspiration.
- Prepare AlexaFluor 488-Phalloidin and DAPI (2 mg/ml) labelling mix by diluting stock solutions 1:500, and 1:1,000 respectively into ice cold PBS.
- Place a 50 μl drop of labelling mix onto parafilm adjacent to each coverslip.
- Aspirate PBS from coverslip and invert the coverslip onto the drop of labelling mixture (cell side down).
- Incubate for at least 30 min at room temperature (RT, 25 °C), protected from the light.
- Invert coverslips and wash with PBS three times.
- Wash coverslips with dH2O once to prevent PBS crystals forming.
- Place a small drop of mounting medium (we use ProLong Diamond) onto microscope slides.
Note: For best results microscope slides should be wiped with 70% EtOH prior to use to remove any dust and lint. - Lift coverslip from parafilm using forceps and remove any excess water by gently placing the edge of the coverslip against a clean Kimwipe tissue.
- Place the coverslip cell side-down onto the drop of mounting medium.
- Gently press the forceps against the top of the coverslip and aspirate any excess mounting medium.
- Seal the coverslip to the microscope slide using clear nail polish.
- Imaging and Data Analysis
- Capture 3D z-stacks using Nyquist sampling with a fluorescent microscope capable of serial optical sections, i.e., Apotome, Confocal or widefield system (Figures 1A-1C) to capture the full 3-dimensional image of the cells and ruffles.
- Acquire multiple fields of view for each condition (collecting at least 100 cells per treatment).
Notes:- Images here were captured using a Zeiss Axioscan fluorescence upright microscope using a 40x 1.3 NA Plan Neofluar objective with an Axiocam 506 CCD camera. Samples were illuminated with HXP 120W whitelight source and filtered with Zeiss DAPI 49/FITC 44 filter cubes. Z-stacks were captured at 0.2 μm intervals beyond both the visible top and bottom of the cells with 5 apotome phase shifts for optical sectioning to identify the midpoint of the nucleus and ruffles on the dorsal surface (Figures 1D-1F).
- Nuclei Mid-point Offset determination. In its native form the script determines the mid-point of the nucleus by finding the slice where the cross-sectional area is greatest (Figure 1G). In macrophages, this suitably allows for the selection and segmentation of dorsal ruffles which are only present above this mid-point, however for alternate use cases, or different cell lines it may be required to use a different selection cut-off point. Figure 1H shows where peripheral ruffles may not be originally selected by using the nuclei mid-point, instead by applying an offset (of a few slices) the additional peripheral ruffle is selected (Figure 1H inset, grey arrow). The nuclei mid-point offset value allows for greater flexibility and broadens the use of the script to further analysis applications.
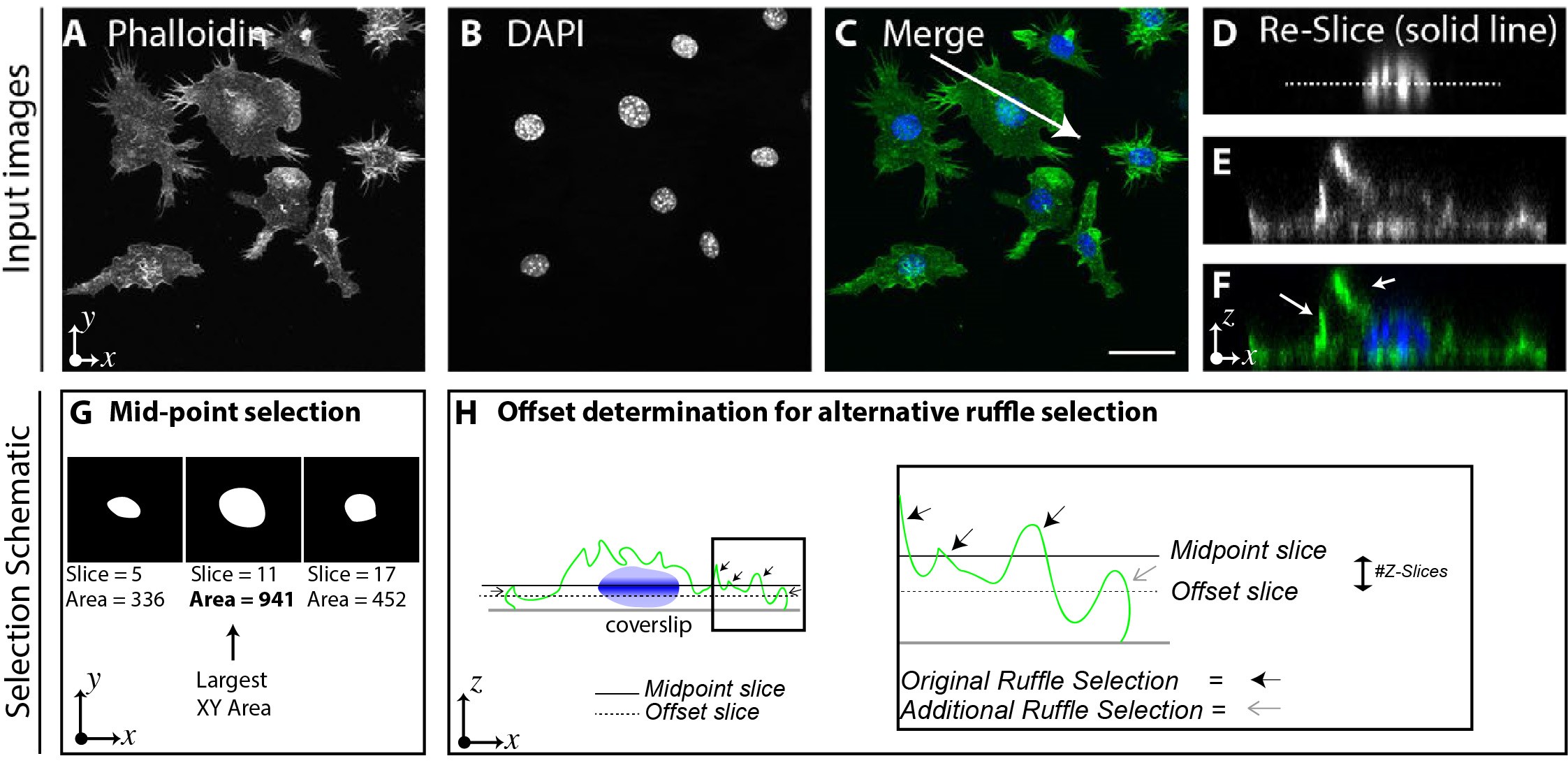
Figure 1. Processing steps of the Ruffle Quantification Macro. 3-Dimensional, 2 channel images are required for the macro captured here using a 40x 1.3 NA objective. Images A-C represent Z-projected (for graphical display purposes only) example images. A. Phalloidin stained RAW 264.7 Macrophages (channel 1). B. Nuclei labelled with DAPI (channel 2). C. Merged two channel image, solid arrow represents location of reslice. D-F. Reslices of C (arrow), showing X-Z axis (cut vertically along the arrow in panel C) of 3D images. D. Dotted line represents mid-point of the nucleus, as selected by the macro E-F. F-actin rich ruffles with merged DAPI highlighted by white arrows (F). Scale bar = 20 μm. G. Example cross-sectional images (X-Y axis) from three different Z-planes of the nuclei showing differences in the measured area, with the largest cross-sectional area determined to be the nuclei ‘mid-point.’ H. Example resliced (X-Z axis) of cell showing a solid line where the determined mid-point of the nucleus is, as well as where the offset line would be if used (example, -3 offset), small open arrows (grey) highlight additional ruffle that would be selected if offset is used, larger black arrows show ruffles selected by the existing mid-point slice with the differential between slices being the offset. - Images here were captured using a Zeiss Axioscan fluorescence upright microscope using a 40x 1.3 NA Plan Neofluar objective with an Axiocam 506 CCD camera. Samples were illuminated with HXP 120W whitelight source and filtered with Zeiss DAPI 49/FITC 44 filter cubes. Z-stacks were captured at 0.2 μm intervals beyond both the visible top and bottom of the cells with 5 apotome phase shifts for optical sectioning to identify the midpoint of the nucleus and ruffles on the dorsal surface (Figures 1D-1F).
- Utilising FIJI, install the macro file: ‘Ruffle Analysis Macro’ and select Run.
Note: To load the macro into FIJI either open the .ijm file, or copy the text from the Github website (see Software section above) and paste this into a new script window (File>New>Script). For continued use, the file Ruffle_Analysis_Macro_.ijm can be copied into the “scripts” folder of the FIJI application, where it will appear under the plugins menu from the next time you open FIJI. - Read the Acknowledgement screen for assay description and instructions (Figure 2A).
- Navigate to the directory location for your files for processing (Figure 2B).
- Confirm the file extension in use (e.g., “.czi”) in the dialog box and enter a Nuclei Mid-Point Offset if required and select “OK” (Figure 2C).
Note: The script will run through the entire directory only opening images that contain the file extension chosen.
Upon completion of the script a dialog will appear. - Confirm success running of the macro with the batch completion dialog box (Figure 2D).
- Navigate to the output directory location (within the chosen working directory location) and confirm thresholding selections are accurately selecting your regions of interest using the output threshold images.
Note: Due to the streamlined nature of the macro, the time required to re-run the macro with new or modified parameters is minimal and it is encouraged that output data is checked and re-run if necessary.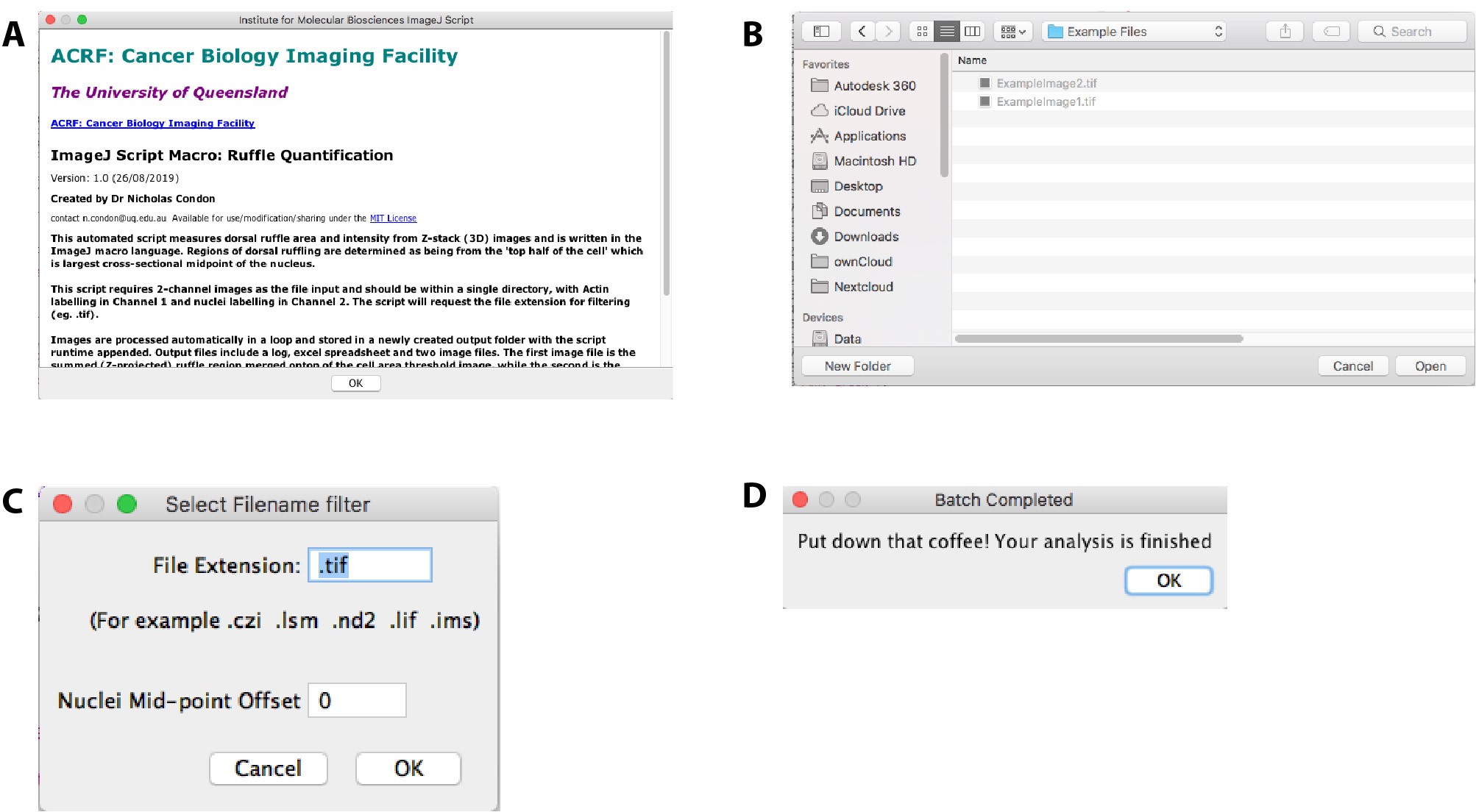
Figure 2. User-interactive macro windows. Dialog boxes requiring user-input during the running of the Ruffle Quantification Macro. A. Acknowledgement and splash screen describing the macro, contact details of the creator, input requirements, and output files. B. OS-dependent file navigator window to select the directory containing images for processing and analysis. C. File extension filter dialog window allows for the user to enter the extension for file filtering and mid-point offset value. D. Macro completion window to notify the user the successful completion of the macro.
- Overview of key steps for Ruffle Quantification Macro
- Nuclei images are extracted (channel 2) and thresholded using the Otsu algorithm. The mid-point is determined by progressively measuring nuclei area within each slice. The slice with the greatest area of the nuclei is stored. The number of nuclei is also counted and stored for output (Figure 3A).
Note: The Otsu thresholding method is used here to detect nuclei, the Otsu method finds the maximum point of inter-class variance between the foreground and the background and is suited where bi-modal distributions of intensity are expected. - The F-actin (phalloidin) channel (channel 1) is extracted from the image stack into two separate images stacks, (1) the bottom half of the cell is taken (slice 1 to nuclei midpoint slice (± offset); Figure 3B) and (2) the top, ruffle area is taken (midpoint slice slice (± offset) to the top most slice; Figure 3E).
- The cell area is determined by maximum Z-projecting the image slices (from slice 0 to the determined nuclei midpoint slice (± offset) and thresholding using the Triangle algorithm (to select total cell peripheries). and measured for output (Figure 3C). The thresholded flattened bottom image is then reduced using a distance map to separate close and/or touching cells. The distance map is then thresholded using the IJ_IsoData threshold algorithm and the number of ‘objects’ (cells) is estimated and stored for output (Figure 3D).
Note: The Triangle algorithm is used here to detect cells, the algorithm is best suited for finding objects that may only be slightly above background (e.g., the very edges of cells) and is therefore used to create accurate selections for measuring cell area. For cell counting, the distance mapped thresholded cells are subjected to a Moments IJ_IsoData threshold as it selects for high-value maximums (e.g., the very center or cells) and thus will not be influenced by touching cells, provided accurate cell number counts. - The ruffles are determined by sum Z-projecting the top F-actin channel (midpoint ± offset to total/top number of slices) and measuring the intensity of the resultant 32-bit image (multiplied by the detected area). The flattened top/ruffle image is then thresholded using the Moments algorithm and the area of the thresholded ruffles is measured (Figure 3F) and used to determine regions of interest for directly measuring summed ruffle intensity.
Note: The Moments algorithm is used here to threshold the ruffles, the algorithm works on a series of averaging steps, and selects regions above background but of varying intensity. This facilitates an accurate selection of all ruffles from the 32-bit sum projected image and not just the brightest objects. - Calculations perform normalisations of ruffle area/cell area, and the intensity of the ruffles/cell area. These values are further normalized against the number of nuclei detected.
- Images at key points in the macro are saved into the output folder (nuclei detection, cell area (bottom) and ruffle area (top), and a merged (green/red) RGB image of the ruffle selection overlayed the cell area image) for verification purposes (Figures 3G-3H).
- All output values are written to the log window (Figure 3I), and the output .csv file into a subdirectory called Analysis_Results_
- Quantified data from the analysis is stored as a .csv file with ratios and normalization to cell number already calculated (Figure 3K). (Values and calculations explained in detail below).
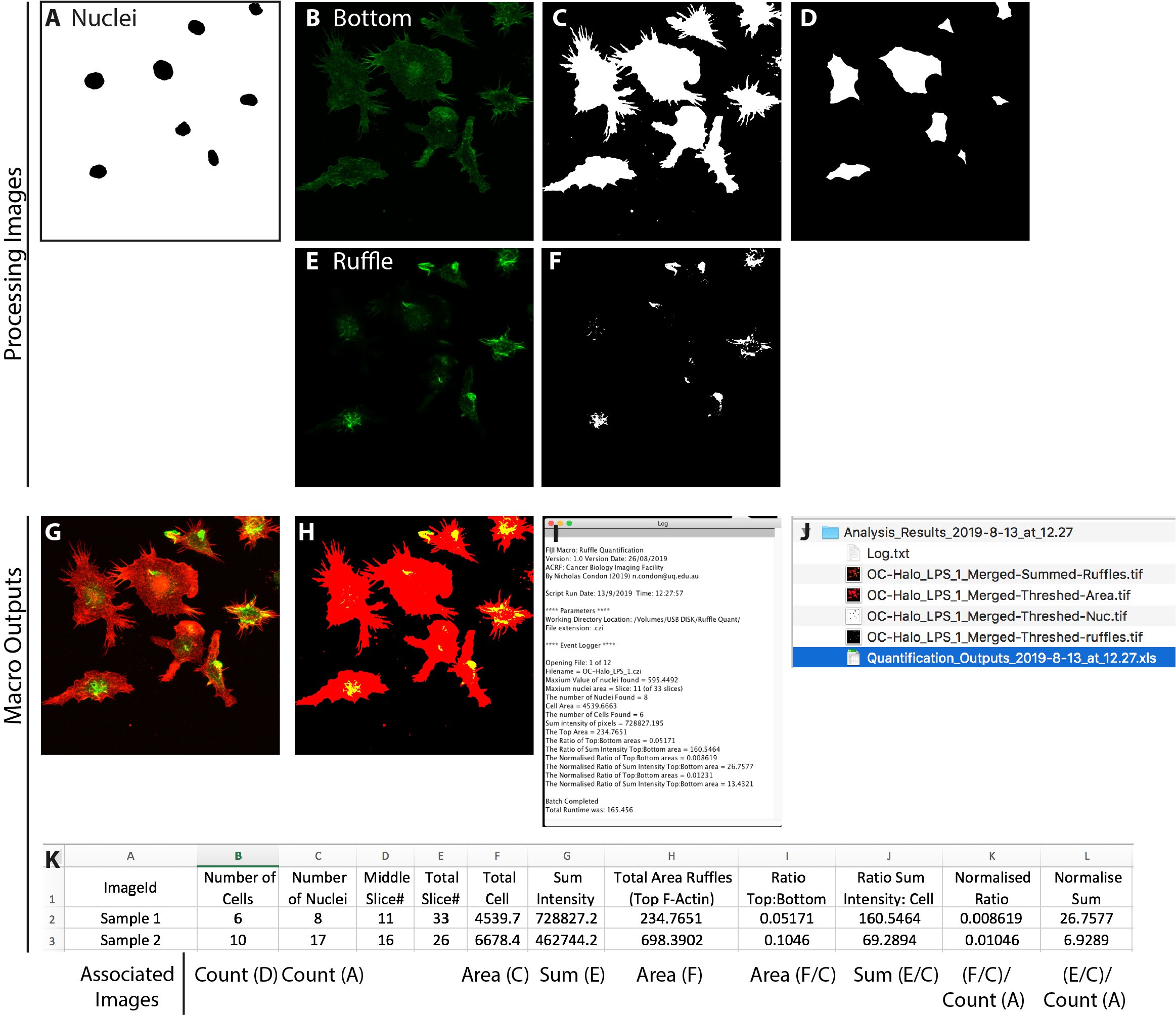
Figure 3. Processing steps of the Ruffle Quantification Macro. A. Thresholded nuclei mask as counted by the macro. B-C. Flattened F-actin base of the cell (bottom) and corresponding threshold for measuring cell area. D. Thresholded distance map showing individual cell segmentation for counting number of cells/field of view. E-F. Sum Z-projected top surface (ruffles) including thresholded image for measuring ruffle area. G. Example output image showing merged bottom (red) and top/ruffles (green). H. Example output image showing merged thresholded bottom (red) and top/ruffles (green) I. Screenshot of the Log window reporting status, macro run date, file names and results. J. Screenshot of the results directory containing output images, log file and a Results.csv file. K. Screenshot of the .csv output results file with explanation of source information below referring to images above where applicable. Scale bar = 20 μm.
- Nuclei images are extracted (channel 2) and thresholded using the Otsu algorithm. The mid-point is determined by progressively measuring nuclei area within each slice. The slice with the greatest area of the nuclei is stored. The number of nuclei is also counted and stored for output (Figure 3A).
- Overview data processing steps
- Output data (.csv file) from the macro is opened using Microsoft Excel (or equivalent).
- Data where the nucleus mid-point is incorrect (e.g., slice 33/33) suggests incorrect thresholding and detection these samples are excluded from the analysis, this can be further confirmed from the corresponding threshold output images.
- Data are copied and sorted into relevant groups (based upon image fields of view and/or treatment conditions) into GraphPad Prism 8 for statistical analysis.
- Depending on the data-sets. statistical analysis may include T-test, One- or Two-way ANOVAs. Data are compared using the in-built graphing tools.
- Output and Results of the Macro
The following details the source of values generated within the output .csv file:
Column A (Image ID) = Filename of source image for this row of data
Column B (Est. Number of cells) = Count of objects detected in Figure 3D
Column C (Number of Nuclei) = Count of objects detected in Figure 3A
Column D (Middle Slice#) = Image slice number of mid-point of nuclei
Column E (Total Slice#) = Number of slices in the source Z-stack
Column F (Total Cell Area) = Calculated area from Figure 3C
Column G (Sum Intensity) = Sum intensity from Figure 3E
Column H (Top Area Ruffles) = Calculated area from Figure 3F
Column I (Ratio Top:Bottom) = Ratio Area from Figure 3F/ Area Figure 3C
Column J (Ratio Sum Intensity:Cell Area) = Ratio Sum intensity Figure 3E/ Area Figure 3C
Column K (Normalised Ratio) = Result of Column I (Figure 3F/3C) divided by Column C (Number of Nuclei Figure 3A)
Column L (Normalised Sum) = Result of Column J (Figure 3E/3C) divided by Column C (Number of Nuclei Figure 3A)
Notes
- For these experiments the Zeiss AxioScan microscope with Apotome 2. was used to generate fluorescent images, however any fluorescence microscope with optical sectioning ability (e.g., confocal) can be substituted. Importantly, images must be 3-dimensional with the full range of the cells that are imaged, (from below the base, to above the top-most ruffle).
- Here, F-actin was labelled with Alexafluor 488-phalloidin, however any colour phalloidin can be substituted. Additionally, this macro works with genetically encoded F-actin labels such as GFP-LifeAct. Nuclei were stained with DAPI, however other nucleus/DNA markers would likely be suitable as well for determining the mid-point of the nuclei as part of the script.
- For these experiments RAW264.7 macrophage cells were utilized, however this assay has also been tested on primary bone-marrow derived macrophages (BMMs), MB231 cancer cells and BV2 microglial cells. The Ruffle Quantification Macro is therefore well suited to be used on multiple different cell types, whereby specific cell culture techniques may be modified.
Recipes
- Cell culture medium
RPMI 1640 Culture Medium
10% Feotal Bovine Serum
1% Poly-D-Lysine
Acknowledgments
Microscopy was performed at IMB Microscopy and the Australian Cancer Research Foundation (ACRF) funded Cancer Biology Imaging Facility at IMB. NDC received support from an Australian Post Graduate Award and the Yulgilbar Foundation, research support was from the National Health and Medical Research Council of Australia (1098710) and the Australian Research Council (DP180101910).
This assay was derived and modified from methods originally published in the Journal of cell Biology, Condon et al, 2018.
Competing interests
The authors declare no competing interests.
References
- Buccione, R., Orth, J. D. and McNiven, M. A. (2004). Foot and mouth: podosomes, invadopodia and circular dorsal ruffles. Nat Rev Mol Cell Biol 5(8): 647-657.
- Commisso, C., Davidson, S. M., Soydaner-Azeloglu, R. G., Parker, S. J., Kamphorst, J. J., Hackett, S., Grabocka, E., Nofal, M., Drebin, J. A., Thompson, C. B., Rabinowitz, J. D., Metallo, C. M., Vander Heiden, M. G. and Bar-Sagi, D. (2013). Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 497: 633.
- Condon, N. D., Heddleston, J. M., Chew, T. L., Luo, L., McPherson, P. S., Ioannou, M. S., Hodgson, L., Stow, J. L. and Wall, A. A. (2018). Macropinosome formation by tent pole ruffling in macrophages. J Cell Biol 217(11): 3873.
- Gandhi, P. N., Gibson, R. M., Tong, X., Miyoshi, J., Takai, Y., Konieczkowski, M., Sedor, J. R. and Wilson-Delfosse, A. L. (2004). An activating mutant of Rac1 that fails to interact with Rho GDP-dissociation inhibitor stimulates membrane ruffling in mammalian cells. Biochem J 378(2): 409-419.
- Gurung, R., Tan, A., Ooms, L. M., McGrath, M. J., Huysmans, R. D., Munday, A. D., Prescott, M., Whisstock, J. C. and Mitchell, C. A. (2003). Identification of a Novel Domain in Two Mammalian Inositol-polyphosphate 5-Phosphatases That Mediates Membrane Ruffle Localization: the inositol 5-phosphatase skip localizes to the endoplasmic reticulum and translocates to membrane ruffles following epidermal growth factor stimulation. J Biol Chem 278(13): 11376-11385.
- Hedberg, K. M., Bengtsson, T., Safiejko-Mroczka, B., Bell, P. B. and Lindroth, M. (1993). PDGF and neomycin induce similar changes in the actin cytoskeleton in human fibroblasts. Cell Motility 24(2): 139-149.
- Kerr, M. C. and Teasdale, R. D. (2009). Defining Macropinocytosis. Traffic 10(4): 364-371.
- Luo, L., Wall, A. A., Yeo, J. C., Condon, N. D., Norwood, S. J., Schoenwaelder, S., Chen, K. W., Jackson, S., Jenkins, B. J., Hartland, E. L., Schroder, K., Collins, B. M., Sweet, M. J. and Stow, J. L. (2014). Rab8a interacts directly with PI3Kγ to modulate TLR4-driven PI3K and mTOR signalling. Nat Commun 5(1): 4407.
- Ly, K. T. and Casanova, J. E. (2007). Mechanisms of Salmonella entry into host cells. Cell Microbiol 9(9): 2103-2111.
- Orth, J. D. and McNiven, M. A. (2006). Get Off My Back! Rapid Receptor Internalization through Circular Dorsal Ruffles. Cancer Res 66(23): 11094-11096.
- Schindelin, J., Arganda-Carreras, I., Frise, E., Kaynig, V., Longair, M., Pietzsch, T., Preibisch, S., Rueden, C., Saalfeld, S., Schmid, B., Tinevez, J. Y., White, D. J., Hartenstein, V., Eliceiri, K., Tomancak, P. and Cardona, A. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9(7): 676-682.
- Schlam, D., Canton, J., Carreño, M., Kopinski, H., Freeman, S. A., Grinstein, S. and Fairn, G. D. (2016). Gliotoxin suppresses macrophage immune function by subverting phosphatidylinositol 3,4,5-trisphosphate homeostasis. mBio 7(2): e02242-02215.
- Singla, B., Lin, H.-P., Ghoshal, P., Cherian-Shaw, M. and Csányi, G. (2019). PKCδ stimulates macropinocytosis via activation of SSH1-cofilin pathway. Cell Signal 53: 111-121.
- Venter, G., Polling, S., Pluk, H., Venselaar, H., Wijers, M., Willemse, M., Fransen, J. A. M. and Wieringa, B. (2015). Submembranous recruitment of creatine kinase B supports formation of dynamic actin-based protrusions of macrophages and relies on its C-terminal flexible loop. Eur J of Cell Biol 94(2): 114-127.
- Wall, A. A., Luo, L., Hung, Y., Tong, S. J., Condon, N. D., Blumenthal, A., Sweet, M. J. and Stow, J. L. (2017). Small GTPase Rab8a-recruited Phosphatidylinositol 3-Kinase γ Regulates Signaling and Cytokine Outputs from Endosomal Toll-like Receptors. J Biol Chem 292(11): 4411-4422.
Article Information
Copyright
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Condon, N. D., Stow, J. L. and Wall, A. A. (2020). Automated Analysis of Cell Surface Ruffling: Ruffle Quantification Macro. Bio-protocol 10(2): e3494. DOI: 10.21769/BioProtoc.3494.
- Condon, N. D., Heddleston, J. M., Chew, T. L., Luo, L., McPherson, P. S., Ioannou, M. S., Hodgson, L., Stow, J. L. and Wall, A. A. (2018). Macropinosome formation by tent pole ruffling in macrophages. J Cell Biol 217(11): 3873.
Category
Immunology > Immune cell imaging > High content microscopy
Cancer Biology > General technique > Cell biology assays > Cellular transformation
Cell Biology > Cell structure > Cell surface
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.