- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Protocols for Processing and Interpreting cryoEM Data Using Bsoft: A Case Study of the Retinal Adhesion Protein, Retinoschisin
Published: Vol 10, Iss 2, Jan 20, 2020 DOI: 10.21769/BioProtoc.3491 Views: 4816
Reviewed by: Edgar Soria-GomezBeatrice LiAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Mar 2019

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

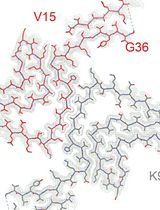
Abstract
The goal of cryoEM is to determine the structures of biomolecules from electron micrographs. In many cases the processing is straightforward and can be handled with routine protocols. In other cases, the properties and behavior of the specimen require adaptions to properly interpret the data. Here I describe the protocols for examining the higher order assemblies of the retinal adhesion protein, retinoschisin (RS1), using the Bsoft package. The protocols for micrograph preprocessing, 2D classification and 3D alignment and reconstruction follow the usual patterns for the majority of cryoEM specimens. The interpretation of the results is specific to the branched network of RS1 filaments. The 2D class averages are used to determine the relative positions of the RS1 molecules, thus defining the interacting interfaces in the network. The major interface of the linear filament is then further examined by reconstructing the “unit cell” and fitting the molecular models.
Keywords: Electron microscopyBackground
In cryoEM, the aim is to determine the structures of biomolecules to obtain biologically relevant information. The usual operation is to align, classify and average the 2D particle images obtained from electron micrographs, as well as align and reconstruct these images in 3D. There are several different approaches and many software packages that accomplish these tasks (https://en.wikibooks.org/wiki/Software_Tools_For_Molecular_Microscopy). The one I developed and used here is Bsoft (Heymann, 2001; Heymann and Belnap 2007; Heymann et al., 2008; Heymann, 2018a and 2018c).
In addition to the normal processing protocols, I describe how to approach a specific example, retinoschisin (RS1) (Tolun et al., 2016; Heymann et al., 2019), that exhibits behavior that requires specialized treatment for interpretation (Figure 1A). RS1 forms a network of filaments that adhere to the air-water interface (Figures 1B-1D). Biomolecules typically locate to the air-water interface (Noble et al., 2018; Noble et al., 2019), and often present a preferred orientation that complicates 3D reconstruction. The RS1 molecules in the filaments mainly present only one view. However, these still show many interactions between the individual molecules. Therefore, the strategy is to extract images large enough to contain several RS1 molecules, perform 2D classification and examine the prevalent interaction types (Figure 1A) (Heymann et al., 2019). To deal with the preferred orientation, a partial solution is to process micrographs of 30° tilted specimens to obtain a 3D map of the most common interaction. The 3D reference used here is a “unit cell” containing two RS1 molecules of the linear filament. Two molecules of RS1 are then fitted into the map to examine the interacting residues at their interface.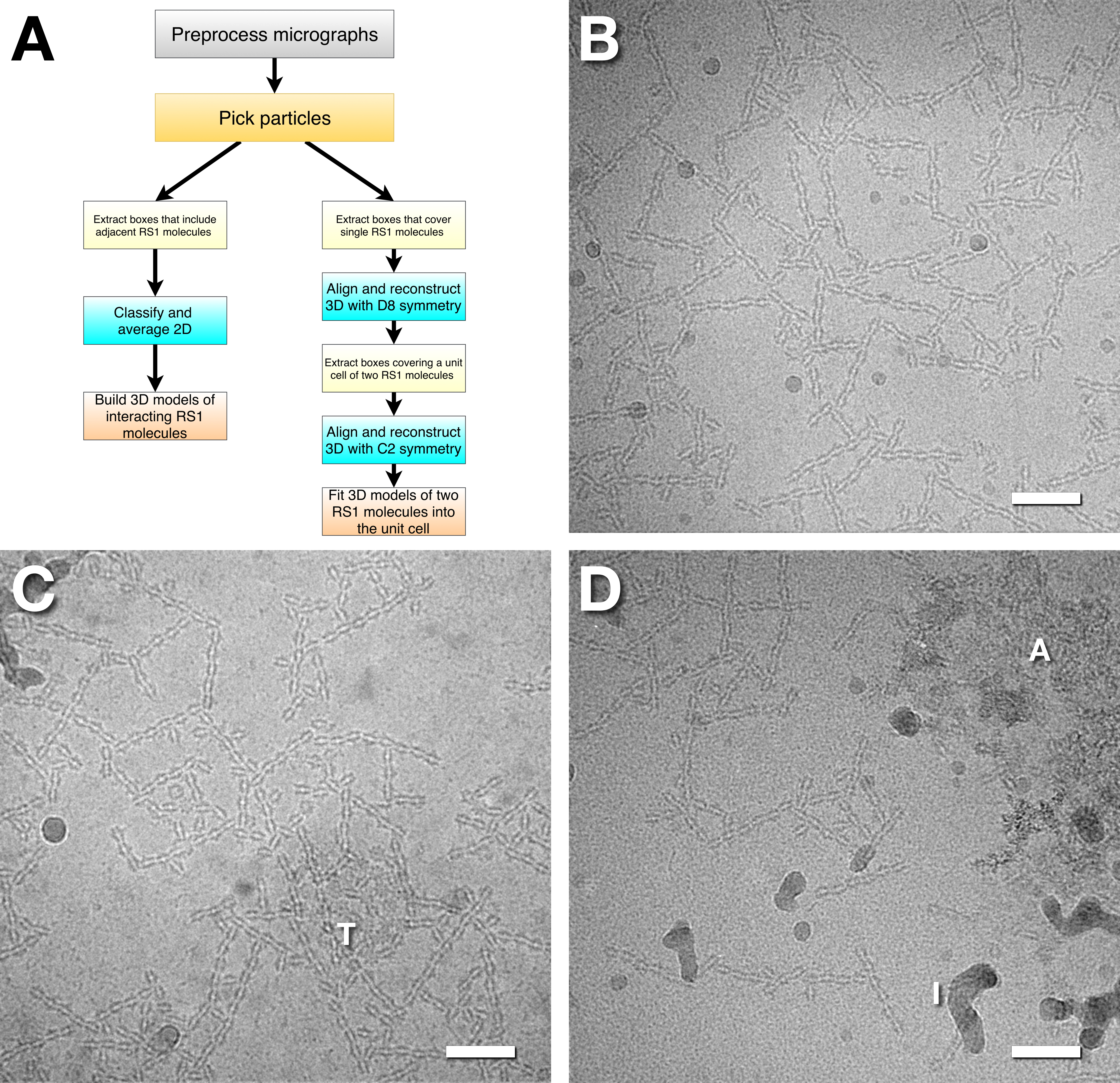
Figure 1. Protocol scheme and examples of micrographs of RS1. A. The protocol scheme starts with two processing sections common to cryoEM, followed by either 2D classification and averaging or 3D alignment and reconstruction. The results are then interpreted by modeling the interactions between the RS1 molecules. B. A good micrograph, showing a gradient of intensity from the top left to the bottom right. The intensity indicates the specimen thickness, where the lighter parts are thinner and more suitable for particle picking. C. A reasonable micrograph with some ice crystals. The “T” shows a cluster of top views of RS1 located in a somewhat thicker region. D. A micrograph with crystalline ice (“I”) and aggregated RS1 (“A”). Scale bars = 500 Å.
Equipment
- Computational setup
The computational work I describe can be done on any UNIX-based platform, such as Linux and MacOSX. All programs in the Bsoft package (Heymann, 2018a) can run on a single machine with multiple CPU cores. However, single particle analysis is computationally intensive and typically requires a cluster to run in a reasonable amount of time. The computational power needed scales with the number and size of particle images, as well as the choice of reference and processing parameters. In this particular case, I used approximately 900 CPU cores (Linux and MacOSX) in our cluster for the following tasks:- Micrograph preprocessing: ~70 h for 1,354 micrographs, each with 50-54 frames.
- 2D alignment and classification of 39,536 particles of size 330 x 330: ~12 h over five iterations.
- 3D alignment of 26,702 particles of size 168 x 168: ~10 h over six iterations.
- 3D alignment of 26,702 particles of size 224 x 224: ~4 h over four iterations.
Software
- Bsoft basics
All of the processing I describe here can be done with the current version of Bsoft (version 2.0.6) (Heymann, 2018a).- brun
Bsoft contains a large number of command-line programs. The program brun can be used to launch these command line programs in a more interactive way. Typing any program name on the command line (without options) provides usage information, including all the options. The options for these programs can be abbreviated, as long as they are unique. All the programs have a “-verbose” option that sets the level of information generated. Without this option, no information will be generated. With quick tasks the verbosity level is typically set to 7, which will give image statistics and parameters for the operation being executed. With extensive tasks (such as aligning particle images or performing reconstructions), the verbosity is usually set to 1 to limit the information to the most important results. - bshow
The main program for interactive work is bshow. It presents the user with an image processing environment where many different operations can be performed. For some programs there are options to plot the output data in Postscript files. These are text files with the data encoded as tables that can be easily imported into other plotting programs for better visualization. The conventions in Bsoft adhere to those we specified in 2005 (Heymann et al., 2005). Because one can use operations other than Fourier transformation to calculate frequency terms, I use the term “frequency space” rather than “Fourier space”. Many of the programs in Bsoft have been parallelized using Grand Central Dispatch on Mac OSX and OpenMP on Linux and will automatically use as many cores as available. In some cases the user can specify the number of threads (i.e., cores) to use (e.g., breconstruct). The Bsoft package (Heymann, 2018a) is freely available at http://bsoft.ws.
- brun
- Distributed processing
The command line programs in Bsoft are executable in any Unix-like environment and from any commonly used script language (such as bash, perl and python). Because each particle is aligned on its own, the philosophy is to separate a set of particles into smaller subsets and run those on different machines. This means that one can use a heterogenous mixture of computers to distribute the tasks. We use the Peach distributed processing system (Leong et al., 2005), but the same can be done on any cluster with a proper queueing manager. - Validation as an integral part of the processing
The success and validity of all micrograph processing depends on understanding the influence of noise and how to avoid bias imposed by user decisions. Most of the principles are laid out in Heymann (Heymann, 2018a). The best practices involve three recommendations:- Process at least two independent sets of particle images–I typically split the set of micrographs into manageable subsets before processing.
- Use conservative resolution limits during particle alignment.
- Test the final reconstruction for coherency: i.e., whether the averages or reconstructions from particle images are better than from noise images.
Procedure
Outline
The following sections cover the processing done for the paper on RS1 (Heymann et al., 2019) with the Bsoft package (Heymann, 2018a) (https://lsbr.niams.nih.gov/bsoft/), following the scheme in Figure 1A and using micrographs such as those in Figures 1B-1D. The micrograph processing, 2D averaging and 3D reconstruction are general operations that can be done for any type of specimen. The interpretation of the 2D class averages and 3D map of the filament “unit cell” are more specifically targeted towards understanding the interactions between RS1 molecules. With suitable modification, these can be applied to other cases. The first two sections (micrograph preprocessing and particle picking) deal with general processing of micrographs suitable for almost any cryoEM data set with the appropriate choices of parameters. The next two sections detail 2D classification and 3D reconstruction that can be adapted for other projects, taking into account the differences and peculiarities for each case. The final sections explain how I fitted the RS1 molecules to the 2D averages and 3D map.
The conceptual aim of single particle processing is:
•To find (pick) relevant particle images in the micrographs and extract them.
•To align these particle images and orient them in a common 2D or 3D coordinate frame.
•To generate 2D class averages and/or 3D reconstructions from the particles.
•Assess the resolution and validity of the averages and reconstructions.
•Interpret the results to derive biologically relevant information.
The protocols I present here cover only a fraction of the processing attempted. In a typical project, I try various strategies and tweak parameters to obtain the best possible result. I would therefore encourage anyone to experiment with the protocols and not view it as a simple linear path.
Preliminary information and imaging configuration
The details of the micrographs recorded on the microscope camera may vary based on the type and configuration of the camera, imaging parameters and software used. Important parameters are:
- The acceleration voltage used.
- The spherical aberration (Cs) of the microscope.
- The camera mode used (integrative, counting, superresolution).
- Whether the images were gain-corrected, or otherwise where to obtain the gain reference.
- Whether the images were compressed and how.
- The calibrated magnification and pixel size.
- The number of frames per image and dose fractionation.
The pixel size is important because there are certain inviolable limits that must be considered. The hard limit in all image processing is the Nyquist frequency, which corresponds to two times the pixel size. Due to interpolation and sampling issues, it is unlikely to reliably reconstruct a map with information to Nyquist frequency. A rule of thumb is the maximum that can be achieved is three to four times the pixel size. Also, at a smaller pixel size, there are fewer particles in a field-of-view and more micrographs are required to have enough particles. Another issue is that the particle alignment is a fourth order function of the box size (Heymann, 2018a). The pixel size should be as large as possible, but still small enough to reach a desired resolution. In concrete numbers, for a pixel size of 0.5 Å, information can be recovered up to a resolution of 1.5 Å. However, the resolution is often limited by the conformational flexibility of the particle and pixel sizes of 1 Å or larger is more efficient.
The signal-to-noise ratio (SNR) increases with dose making higher dose desirable. However, the electron beam is highly damaging, eventually destroying the structural information of interest. In addition, it induces specimen movement that further degrades the information. With movies on direct detection cameras, specimen movement can be compensated by taking fast frames and subsequently aligning them. To decrease the effect of radiation damage, the frames can be weighed based on the accumulated dose (Grant and Grigorieff, 2015). A typical dose fraction scheme involves taking 3-10 frames per second at a low dose per frame of ~1-3 e-/Å2, accumulating a total dose of 50-100 e-/Å2. There is a tradeoff between the keeping the dose per frame low, but high enough to align.
In the RS1 case, the samples were imaged with a 300 kV Krios microscope (Thermo Fisher Scientific) fitted with a Cs corrector and a K2 camera (Gatan). The images were acquired in superresolution mode with a dose of ~0.25 e-/Å2 per superresolution pixel (see Heymann et al., 2019 for more details). The images were recorded without gain-correction, compressed with the LZW algorithm and written to storage as TIFF files. Bsoft programs can decompress these files on reading. The images also need to be binned by two before alignment to have sufficient signal for cross-correlation (binned dose ~1 e-/Å2). Figure 2A shows a histogram of the frames of a representative micrograph taken in superresolution mode. Most pixels are zero, with only about a quarter showing one electron hit, and few multiple hits. It follows a Poisson distribution (dots), where the probability that k electrons hit a pixel is given by: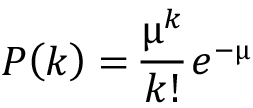
The average, µ, is the overall electron dose per pixel. When all 54 frames are added, the curve shifts to a higher average (approximately 54 times higher) (Figure 2B). Because the field of view contains objects that are correlated and the signal is therefore not completely random, the data does not follow the Poisson curve exactly.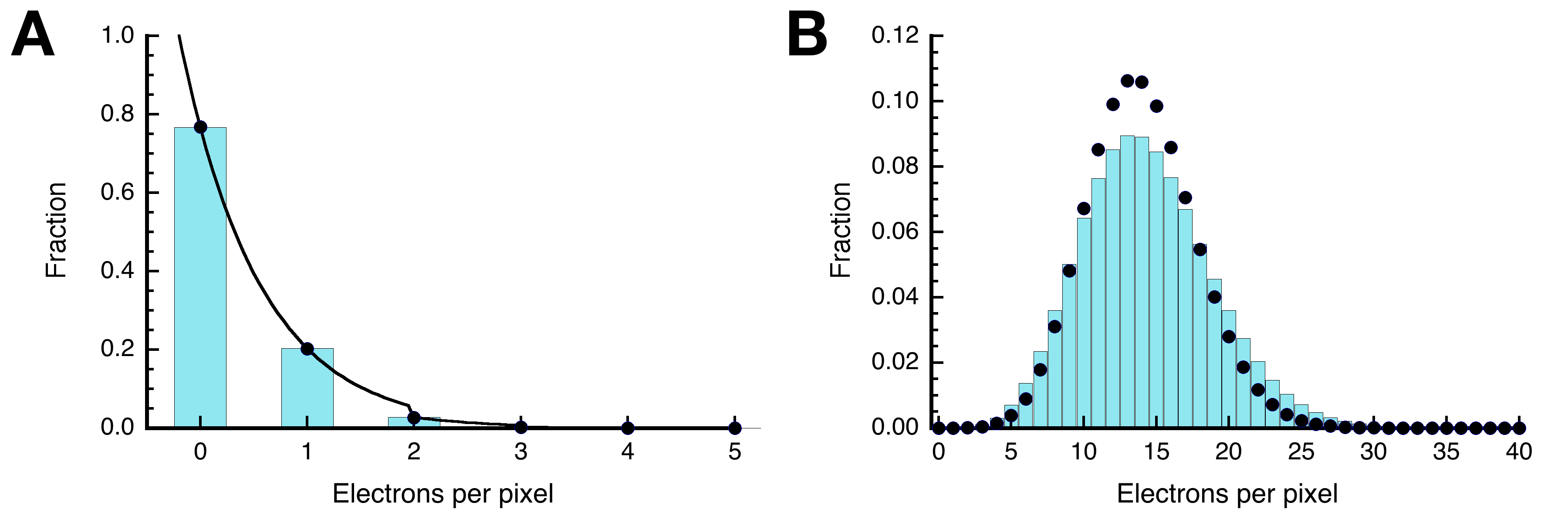
Figure 2. Electron counts (bars) in an electron micrograph follows a Poisson distribution (dots). A. Raw counts from the frames of a representative micrograph (average = 0.26 e-/pixel). B. Counts after summing the 54 frames (average = 14 e-/pixel).
Setup of a processing environment
The current high-end electron microscopes produce large numbers of big files. It is therefore important to organize the files in subdirectories for easier access and processing. However, this means that to find these files, the paths to the files must be considered. The parameter files of Bsoft (usually STAR files) embed the paths to the micrographs and other images. Once the parameter files have been created, it is not advisable to move them to other directories, unless care is taken to fix the paths. The paths can be specified in two forms: an absolute path (such as “/mydisk/joe/project1/mg”) or a relative path (such as “../mg”). The latter allows the user to move the whole project without compromising the paths in the parameter files.
A typical approach is to create a directory for the project, with a subdirectory called “mg” for the micrographs and files generated during preprocessing. The particle images are extracted into another subdirectory called “part”, and alignment iterations performed in subdirectories called “run1”, “run2”, etc. The relative paths in parameter files are then “../mg” for the micrographs and “../part” for the particle images.
Micrograph preprocessing and contrast transfer function fitting
The first task is to set up a parameter file for each micrograph (program bmg). This is then followed by gain-correcting (if necessary), aligning the frames and generating a frame-summed micrograph (program bseries). Finally, a power spectrum is calculated and the contrast transfer function (CTF) fitted (program bctf). All of these steps are typically done in Bsoft using the script (Appendix): mgprep.pl (executed in the micrograph subdirectory).
- Preparing for gain-correction by checking the orientation of the gain-reference:
- First sum the frames of one micrograph to obtain a high-dose image that would show detector defects:
bimg -verb 7 -sum Sep02_14.30.09.tif Sep02_14.30.09_sum.mrc - Open both the gain reference and summed micrograph:
bshow Sep02_14.30.09_sum.mrc
bshow gain_20160902.dm4
Locate corresponding defects and determine if and how the gain reference should be transformed. This could be a 90° or 180° rotation, or a flip in x or y. - If needed, transform the gain reference using the “-reslice” option:
bimg -verb 7 -reslice y-xz gain_20160902.dm4 gain_20160902_resliced.mrc
The argument to the “-reslice” option means that the x and y axes are switched to get a 90° rotation. To flip in y, use “x-yz”.
- First sum the frames of one micrograph to obtain a high-dose image that would show detector defects:
- The parameters for all micrographs can be encoded in one parameter file. However, the large size of typical multi-frame micrograph images makes it more practical to preprocess each micrograph separately and combine the records later. This is also suitable for distributing tasks over a cluster of computers. The script, mgprep.pl, processes each micrograph as follows:
- Create the first parameter file (usually a STAR format file):
bmg -verb 7 -extract frame -Pixel 1.04 -Volt 300 -Amp 0.07 -Cs 0.01 -out Sep02_14.30.09.star Sep02_14.30.09.mrc
The “-extract frame” option indicates that these are movie frames. I set the pixel size in case it was not properly embedded in the original micrographs. The other options set the acceleration voltage (in kV), the amplitude fraction (0.07 is typical for cryo-electron micrographs, or 1.0 for phase plate images) and the spherical aberration, Cs (in mm). The Cs is specific to a microscope and usually ranges from 1-4 mm. We acquired this particular data set on a microscope with a Cs corrector, reflected in the 0.01 mm value specified (Heymann et al., 2019). - Align the movie frames with optional gain correction and binning:
bseries -verb 1 -frames -dose 1.0 -Gainreference ../mg/gain_20160902.dm4 -align 10 -resol 20,1000 -shift 100 -bin 2 -exposure 20 -write sum -out Sep02_14.30.09_aln.star Sep02_14.30.09.star
The “-frames” option indicates that the micrograph frames will be aligned (instead of a series of micrographs). The “-dose” option invokes dose-weighting of the frames with the specified dose per frame in e-/Å2 (Grant and Grigorieff, 2015). The “-Gainreference” option specifies the gain reference image to use. The “-align” option activates alignment using the indicated frame as initial reference. The “-resolution” option sets limits for cross correlation that are typically on the low side because the images have not been CTF-corrected. The “-shift” option limits translation (in pixels), avoiding spurious results for very noisy images. The “-bin” option downsizes the images internally to speed up processing. The “-exposure” option indicates the total acquisition time of all frames (in seconds) to estimate the frame drift rate. The “-write sum” option activates writing a sum of the aligned frames to disk. As the program runs, it reports the shifts per frame that can be used to eliminate micrographs with too much drift. Whole frames are aligned, producing a single shift vector for each frame and the corresponding correlation coefficient. - Fit the CTF:
bctf -verb 1 -action prepfit -tile 512,512,1 -resolution 5,20 -Range 0.5,4,0.1 -out Sep02_14.30.09_ctf.star Sep02_14.30.09_aln_avg.star
Use the option “-action prepfit” to calculate a power spectrum and fit the CTF automatically. The “-tile” option indicates that the movie frames will be tiled (size 512 x 512 x 1), Fourier transformed and the individual power spectra averaged to generate a noise-reduced power spectrum. The noise in the final power spectrum decreases with the number of tiles. However, if the tiles are too small, the oscillations of the CTF cannot be adequately sampled at high frequencies. As explained in Heymann (2018a), the optimal tile size is: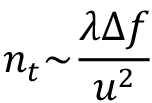
where λ is the electron wavelength, ∆f is the defocus and u is the pixel size. For example, with λ ~ 0.02 Å (at 300 kV), u = 1 Å/pixel, and ∆f ~ 2 µm = 20,000 Å, the tile size should be larger than 400 x 400 pixels. The “-resolution” option sets the limits for fitting the CTF. The low resolution limit (20 Å) is intended to eliminate the very strong inelastic scattering at low frequencies. The high resolution limit (5 Å) cuts out high frequency noise that may affect the fit. The “-Range” option limits the search for defocus values by specifying a minimum, maximum and step size (in µm). If this option is omitted, the default range may be too wide and lead to incorrect defocus estimates. If the specimen was tilted, the tilt axis angle and tilt angles must be specified to calculate a tilt-adjusted power spectrum. Other parameters for the CTF fitting are already set in the parameter file (see Step 3).
- Create the first parameter file (usually a STAR format file):
- Combine all the parameter files and check the CTF fits:
- Concatenate all the parameter files:
bmg -verb 7 -out rg4_all.star Sep*.star - Open the parameter file:
bshow rg4_all.star - Select a power spectrum image in the dialog box (it is indicated as “:ps:”).
- The power spectrum image has a high dynamic range, which means that it needs to be rescaled to see the Thon rings. Move the “Max” slider to the left and then click to the right to increase it by increments. If the increments are too small or large, change it in the “Range step” field and press “return”.
- To see the CTF fit, select the “Micrograph/Fit CTF” menu item (Figure 3).
- If the power spectrum has a big broad peak around 3.8 Å, it indicates the specimen was too thick and the micrograph should be deselected (using the select button at the bottom of Figure 3).
- Make sure the acceleration voltage, spherical aberration (Cs) and amplitude contrast are the correct values.
- Adjust the scale to enlarge the graph: typical values for the first is 4-8 and for the second is 100-5,000.
- Adjust the defocus so that the minima in the red curve (CTF) fit the minima in the blue curve (radial power spectrum or RPS). The CTF curve should agree with the RPS curve to high resolution. The frequency or resolution at which it stops agreeing is called the “information limit”. If this indicates too low a resolution, the micrograph should not be used in any further processing.
- Improving the fits can be done with the “Fit” buttons. The “Quick” button does an initial fit and is typically used if the automated fit failed. The “Baseline”, “Envelope”, “Defocus” and “Astigmatism” buttons refine each aspect of the fit and can be used in any order.
- There are 6 baseline types. Choose the one that gives the nicest baseline where the minima of the blue curve (RPS) are close the x axis with the “Subtract” button selected. The last three baseline types attempt to fit the peak at 3.8 Å if it is present (arrow in Figure 3).
- There are 4 envelope types. They increase in complexity with number 4 the default. Choose other types if they seem to fit better to the maxima of the blue curve (RPS).
- Click on the “Next micrograph” button to advance to the next one.
- Periodically save the modifications using the “Micrograph/Write parameters” menu item.
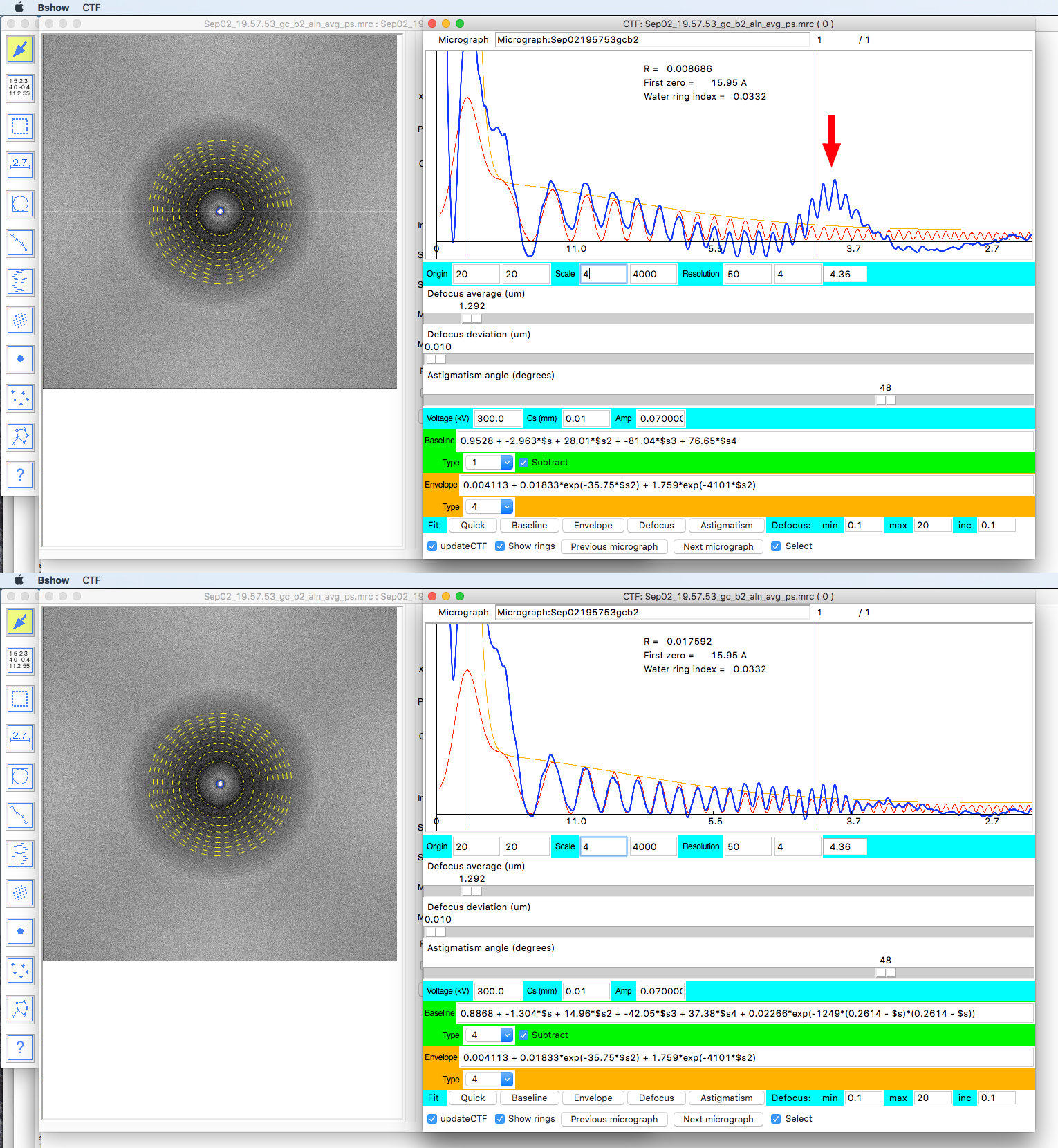
Figure 3. Checking the CTF fit in bshow. The top panel shows the fit with baseline 1 and the prominent peak at 3.8 Å (red arrow). The bottom panel shows the fit with baseline 4, compensating for the peak at 3.8 Å. The main aim is to align the minima in the radial power spectrum (RPS, blue curve) to the minima in the CTF (red curve). If the parameter file contains records for multiple micrographs, step through the micrographs using the “Previous micrograph” and “Next micrograph” buttons.
- Concatenate all the parameter files:
Particle picking
Particles may be picked by hand or automatically using a template. The typical approach is to generate a template from a representative set of particles and use it to automatically pick the rest. The results must be checked to eliminate bad particles (such as blobs of ice, strong carbon edges and other undesirable objects) and add more desirable particles. The box size must be larger than the particle, but how much larger? The use of defocus to increase contrast in cryo-EM means that the signal is delocalized to some extent (Downing and Glaeser, 2008). To properly correct the images for the CTF, enough of a border around the particle must be included. A good rule of thumb is to pick a box size about three times the diameter of the particle. The specific box size also determines the speed of processing. The most computationally intensive operation is the Fourier transform. To choose a box size for a reasonable processing speed, run the bfft program with the range of desired box sizes: bfft -test 2,100,200. The results rank the speed of each box size.
In the RS1 case (Heymann et al., 2019), the box was set to a small size (168 x 168) to pick individual RS1 molecules. Because the RS1 molecules arranged into larger assembles, larger boxes (330 x 330) were extracted to cover three RS1 molecules in a string for 2D classification. For the 3D reconstruction of RS1, the smaller box was used for initial alignment with high symmetry, and then re-extracted with a box size suitable for two molecules in the string (224 x 224) as the “unit cell”. This was then processed with C2 symmetry.
Pick particles:
- Open a parameter file (with one or more micrograph records):
bshow rg4_ctf.star - Select the one micrograph from the dialog box (it is indicated as “:mg:”).
- Select the boxing tool (
 ):
):
It opens the dialog box for picking and examining particles. This dialog box can also be open using the “Micrograph/Particles” menu item, but the boxing tool must be selected to add more particles. - Change the box size. Only the first two values need to be changed for 2D images. For the RS1 example, I chose a box size of 168 x 168 to comfortably cover a single molecule of the double ring structure. This can be changed later when extracting the particles into images (see Step 12).
- Set a temporary low pass filter to make the particles more visible (it will not change the micrographs themselves):
- Select the menu item “Image/Filter image”
- Make sure the type is “Averaging”
- Set the kernel size to 11 (or a value that gives a nice presentation of the micrograph)
- Click on “Set preference”. This sets it so micrographs are automatically filtered when opened. It can be turned off in the “Help/Preferences” dialog box.
- Click on “Filter”.
- If the result is not acceptable, re-read the image by using the “File/Revert” menu item and go back to c.
- Assess whether the micrograph is acceptable. If not, click on the “Select” button in the “Particles” dialog box to deselect it. The de-selected micrographs can be removed from a parameter file after saving it from bshow as follows:
bmg -verb 7 -remove -out rg4_sel.star rg4_ctf.star - Pick a representative set of particles (about 10-20).
- From the Particles dialog box, open the template picker dialog box using the “Boxes/Template picker” menu item. It shows an average of the currently picked particles (see Figure 4).
- Set the high and low resolution limits in the “Template Picker” dialog box. Because the images have not been corrected for the CTF, it is best to set the high resolution limit to around first zero, typically about 20 Å. The exclusion limit sets the closest distance allowed between picked particles. Set it somewhat smaller than the box size (usually about 80%). Set the bin option to 2 to speed up the automatic picking. Then click the “Pick” button to do automatic picking on the current micrograph.
- Eliminate all the bad picks (not particles) and add any that should be included. In the “Template Picker” dialog box, click the “Update” to generate a new template. If this template is adequate, save it using the “File/Save…” menu item. If not, repeat Step 8.
- Pick particles from the whole project from the command line:
bpick -verb 1 -box 168 -Template rs1_template.mrc -extension mrc -out rg4_part.star rg4_ctf.star
The “-box” option takes 1 to 3 size parameters (in pixels). Giving one parameter sets the others to the same value. Internally, the third size is set to 1 for 2D images. This command now saves all the coordinates for the particles in the parameter file. - Extract particle images from the micrographs (in a particle subdirectory):
bpick -verb 1 -extract -norm -back -box 330 -extension mrc -partpath ../part -out rg4_part.star ../mg/rg4_ctf.star
The “-extract -norm -back” options indicate extracting particle images from the micrograph images, normalizing them, and setting the background to the average. The background is defined as the area outside the circle fitting into the box frame (as shown in bshow when picking particles).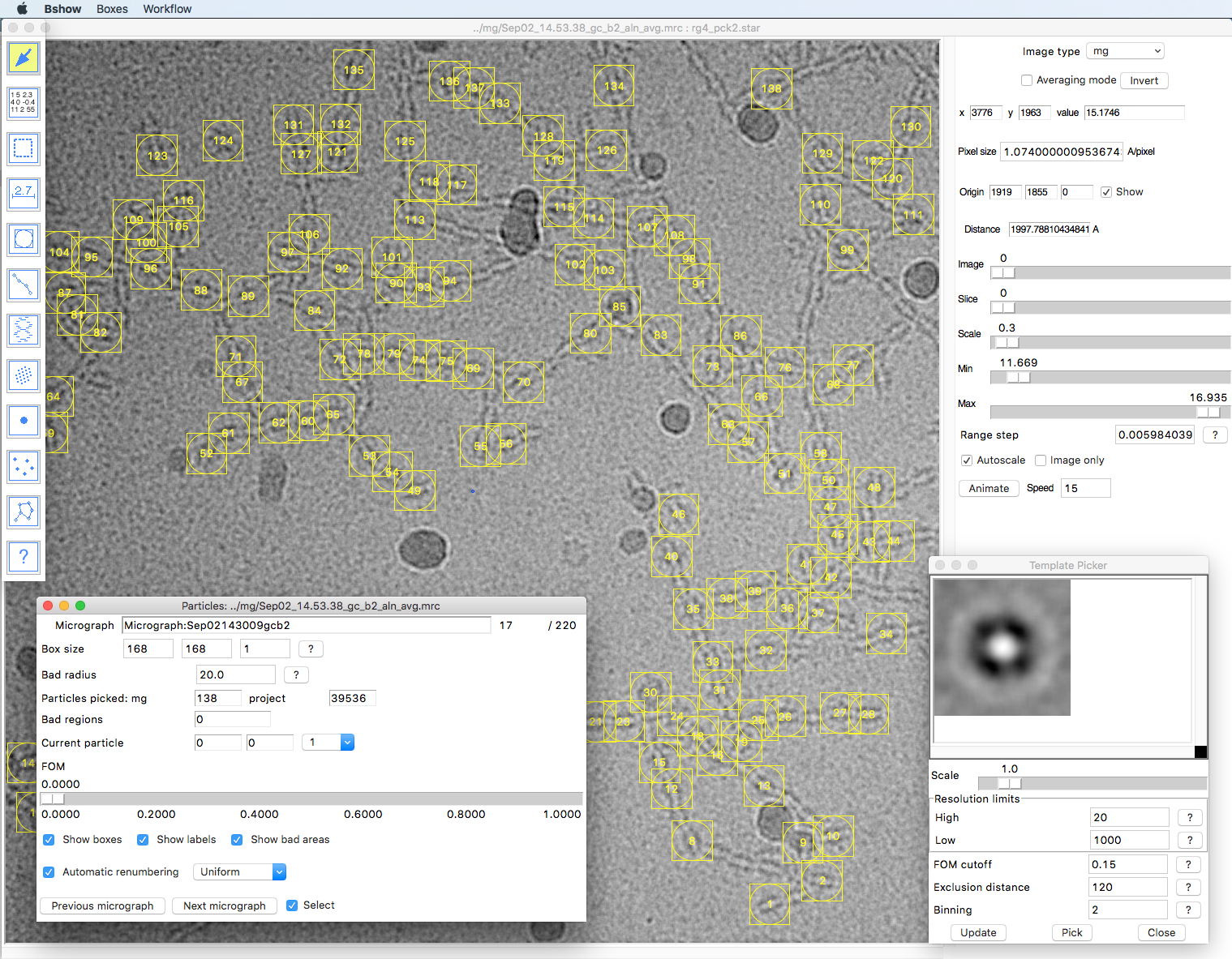
Figure 4. Particle picking in bshow. First, generate a template (lower right) from a small set of hand-picked particles. Then, picked a larger set automatically using the “Pick” button. Remove badly picked particles and then generate a new template with the “Update” button. Iterate this until it selects an acceptable set of particles. Save the template and use it to automatically pick particles from multiple micrographs.
Principles of particle alignment
The particle images extracted from micrographs are assumed to be different views of the same particle or at least very similar particles. The task is to align them to a common reference frame and average them, either in 2D or 3D (the latter is commonly referred to as “reconstruction”, although it is also an averaging process). The references are 2D images the same size and scaling as the particle images. These images can be any arbitrary set of 2D images considered to be related to the particle images, or projections from one or more 3D maps.
The typical processing for a particular set of micrographs iterates the alignment and averaging/reconstruction multiple times. The results are assessed at each iteration using measures such as the spectral signal-to-noise ratio (SSNR) (Unser et al., 1987) or Fourier shell correlation (FSC) (Harauz and van Heel, 1986). It is considered done when the results of the processing fail to improve.
The program borient, operates on any set of 2D reference images, including projections generated from a 3D map. It aligns each particle image to each reference projection using a combined polar Fourier transform and polar image transform algorithm (Figure 5). It then selects the best match and sets the corresponding orientation parameters (shift and rotation). If the aim is to perform 2D class averaging, the alignment parameters are used to calculate a new set of 2D class averages as references for the next iteration. This is effectively a k-means clustering approach, with the opportunity to modify the set of reference images at each iteration. If the aim is to reconstruct 3D maps, program aligns particle images against the projections from a reference map using a global search grid within the asymmetric unit. This is then followed by 3D reconstruction with the program breconstruct, generating a new reference map for the next iteration.
The program brefine uses previously determined particle translations and orientations (e.g., determined with borient or a previous run of brefine) and refine them locally together with magnification and defocus:
- Translation or shift in x and y, encoded as the origin in each particle image in pixels
- Rotation, encoded as a view (an alternative to three Euler angles)
- Magnification, encoded as a fraction
- Defocus, encoded as the average defocus per particle
The program searches around the initial view either in a grid pattern (“-grid” option) or a random search (“-monte” option). The two arguments for the grid pattern indicate the initial and final search angular step sizes. It searches in a 3 x 3 x 3 angular grid, calculating a correlation coefficient at each orientation. It then shifts the search to the best one and repeat it. If the best one is the central on of the grid, it contracts the grid to half the size (divides the angular step size by 2). This is repeated until the step size drops below the final angular step size. The other parameters are also refined at the same time by searching linearly around their initial values. As for borient, the output is used to reconstruct a map or maps with breconstruct. The current implementation of brefine only addresses 3D alignment and not 2D alignment.
All comparisons by cross-correlation in Bsoft require resolution limits, aimed at excluding high frequency noise as well as high amplitude low frequency components that don’t contain much orientation information. For the initial alignment where the reference images or map may not contain much orientation information, the appropriate high resolution limit is typically 20-60 Å. This depends on the size of the box and whether there are features in the reference map apparent at this resolution. The choice of high resolution limit also determines what the best angular step is to use in a global search (with borient). For a resolution of r and a box size D (both in Å), a proper angular step size should be (in degrees):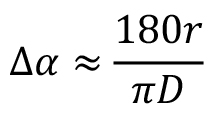
The low resolution limit is often not critical, but depends on the case. Typical values are 100 or 200 Å to avoid strong amplitudes at low frequencies.
The initial reference map for 3D alignment can be generated in a number of ways. If no previous information is available, there are many approaches to produce a low resolution map that show some of the particle features. If a previous map exists, it can be used with the appropriate resizing. A map can also be generated from an atomic model of a biomolecule that is close enough in structure to the particle of interest using program bsf. In case nothing suitable exists, a synthetic map can be constructed using a program such as beditimg.
Figure 5. Illustration of the algorithmic concepts used in borient. A. Top view projection. B. Side view projection. C. Top view power spectrum (calculated as the logarithm here for display). D. Side view power spectrum. E. Polar transform of the top view (the angle is on the horizontal axis from 0 to 360° and the annulus is on the vertical axis). F. Polar transform of the side view. G. Polar transform of the top view power spectrum. H. Polar transform of the side view power spectrum. The algorithm in the program borient aligns each particle image to 2D reference images, including projections from a 3D map such as the top (A) and side (B) views of RS1. The algorithm first calculates the power spectra (C, D) corresponding to (A) and (B). The next step transforms the power spectra to polar coordinates (G, H). The polar power spectra of the particle and the reference projection are then compared by cross-correlating their annuli to determine an initial rotation angle. The program then rotates the reference projection image to this angle and determine an initial shift by 2D cross-correlation. The program transforms the real space particle images and reference projections to their polar forms (E, F), using the previously determined origins for the particle images. The final step refines the rotation angle by cross-correlating the annuli of the polar images. The final figure-of-merit is the correlation coefficient from the 2D cross-correlation of the particle image with the reference projection rotated through the best angle. The white and gray circles in (B) are set by the “-annuli” option to limit the annuli used in the calculations. The annuli inside the white circle don’t have useful orientation information, while the annuli outside the gray circle only contain background.
2D alignment and classification
Classification of the 2D particle images is initiated by selecting a defined number of the particles as initial reference images, or any other set of reference images. This number needs to be large enough to cover most of the variability in the particle images, but not so large that there are only a few particles per class. For a data set of 105 particle images, a number of classes between 200 and 1,000 is appropriate. The reference images can be prepared in any number of ways other than that described in the protocol below.
2D classification with borient:
- Do the first alignment to a subset of particle images as references (in its own subdirectory):
borient -verb 1 -mode ccc -prepare 420,100 -CTF -resol 25,100 -ann 10,150 -ppx -TwoD rg4_run1.mrc -out rg4_run1.star ../part/rg4_part.star
The “-prepare” option selects the first number of particle images (420 in this case) as initial references for classification, starting from particle 100. If a different set of 2D images have been prepared, it can be specified with the “-reference” option. The “-CTF” option imposes the CTF on the reference images before comparison with the particle images. The “-resolution” option sets the frequency limits for alignment. For the initial run, the high resolution limit should be relatively low, around the first zero in the CTF. The “-annuli” option specifies the particle annuli to consider in rotational alignment (see Figure 5B). The “-ppx” option saves intermediate results in a “ppx” subdirectory. If the program crashes, just rerun the original command line and it will skip those particles that have already been completed. For a rerun of borient with different parameters, the “ppx” subdirectory should first be removed. The “-TwoD” option specifies the filename for the output class averages. With the “-CTF” option these are CTF corrected during averaging. - Assess the output:
bpartsel -verb 2 -FOM rg4_run1_fom.ps rg4_run1.star
This lists the number of particles incorporated into each class average and the FOM, plotted on the third page of the Postscript file specified by the “-FOM” option. In this case the FOM is the resolution calculated at an FSC cutoff of 0.3 using the spectral signal-to-noise method of Unser et al. (Unser et al., 1987), converted to FSC. An example plot is shown in Figure 6. The Postscript file can be opened in a suitable reader (such as Preview on Mac OSX or Ghostscript on Linux). The data in the Postscript file is arranged as a table and can be imported in other programs such as Excel or Kaleidagraph.
Figure 6. Resolution of the RS1 2D class averages calculated for an SSNR cutoff of 0.5. The linearity of the plot indicates that a better resolution can be achieved with more particles than was used here. Above some number (~105) the gain will level off and adding more particles will not improve the resolution. - Visualize the class averages as a montage (Figure 7):

Figure 7. The 70 final class averages. The most prevalent class is 37, composed from more than half of the particle images. The white numbers indicating the contributing particle images.- Open the parameter file in bshow:
bshow rg4_run1.star - Select the last line in the dialog box with the type “Class”
- Open the montage tool with the “Image/Montage” menu item.
- Enter numbers for the column and row and click the “Montage” button.
The numbers in the lower left of each cell is the class average image number, and the number in the lower right is the number of particles incorporated into each class average. The montage can be saved in its own image using the “Save” button in the montage dialog box, but without the overlay with the numbers. To retain the overlay representation, take a screen shot.
- Open the parameter file in bshow:
- Eliminate poorly represented classes:
- In the montage, click in a cell to toggle selection. Selected cells have red frames while non-selected cells have yellow frames.
- Select all the bad class averages (usually those that are noisy indicating few contributing particles).
- Delete these by clicking the “Delete selected” button in the montage dialog box.
- Save the image under a new name using the “File/Save as…” menu item (I used “rg4_run1_sel.mrc”). This can now be used as reference for further 2D classification runs.
- Refine the classes:
borient -verb 1 -mode ccc -ref ../run1/rg4_run1_sel.mrc -CTF -resol 20,100 -ann 20,150 -ppx -TwoD rg4_run2.mrc -out rg4_run2.star ../run1/rg4_run1.star
The “-reference” option specifies the reference images to use for alignment, in this case the class averages from the previous run. The class refinement can be performed several times, optionally changing the number of classes by either eliminating bad reference images or adding more reference images (Steps 3-5). Stop refinement when the results don’t change much between iterations.
The 2D classification described above produces class averages in arbitrary orientations. To compare them, they have to be oriented to a common coordinate frame. In the RS1 case, the side view is common to all the class averages and can be used as a common reference. The class averages from the previous classification are now treated as particles and aligned to the reference.
- Prepare a common reference to align the class averages:
- Select one class average for the reference:
bimg -verb 7 -select 105 rg4_run2_sel.mrc rg4_run2_105.mrc
This image has three RS1 molecules almost vertically. - Rotate it to be as close to vertical as possible:
bint -verb 7 -rotate 0,0,1,2 rg4_run2_105.mrc rg4_run2_105_rot.mrc - Generate a mirror image by flipping the y axis:
bimg -verb 7 -reslice x-yz -trans 0,1,0 rg4_run2_105.mrc rg4_run2_105_flipy.mrc - Cross correlate to check the rotation:
bcc -verb 7 -resol 10,1000 -Cross rg4_run2_105.mrc rg4_run2_105_flipy.mrc
The correlation coefficient shows how well the top and bottom molecules superimpose. - Redo steps b-d with different angles to find the best correlation coefficient. Then do step b with this rotation angle for the final reference.
- Select one class average for the reference:
- Align the class averages to obtain a common orientation:
- Set up a parameter file for the class averages:
bmg -verb 7 -extract part -out rg4_run2_sel_part.star rg4_run2_sel.mrc - Align the images to the common reference:
borient -verb 1 -resol 20,100 -ann 20,150 -ref rg4_run2_105_rot.mrc -out rg4_run2_sel_part_out.star rg4_run2_sel_part.star - Tag the images as separate entities:
bpartsel -verb 7 -sets 1 -out rg4_run2_sel_part_sets.star rg4_run2_sel_part_out.star
The “-sets” option assigns selection numbers to the images. In this case it sets each one to a different number to indicate they represent different classes. - Write new images correctly oriented:
borient -verb 1 -TwoD rg4_run2_sel_aln.mrc -oriented -out rg4_run2_sel_aln.star rg4_run2_sel_part_sets.star
This output multi-image file is now suitable to use as reference in classification steps 3-5 to refine the class averages in a common orientation and before comparing them in the next step.
- Set up a parameter file for the class averages:
- Compare the class averages:
- Calculate correlation coefficients between each pair of images and cluster them:
bmapdist -verb 1 -TwoD -resolution 10,200 -matrix rg4_run5_cc.dat -preference 0.7 -out rg4_run5_cc.star rg4_run5.star
The option “-matrix” generates a matrix with pairwise correlation coefficients between the images and classifies them using affinity propagation (Frey and Dueck 2007) with the “-preference” option. The output multi-image file now contains the new smaller set of class averages that can be visualized as in step 3b (Figure 8). - Convert the matrix to an image and color it:
bmatrix -verb 7 -color -scale 5 -outimage rg4_run5_cc.png rg4_run5_cc.dat
The “-color” option converts the data to an 8-bit byte color presentation suitable for display. The “-scale” option enlarges the output image (Figure 8). (This map is just for display and not used further.)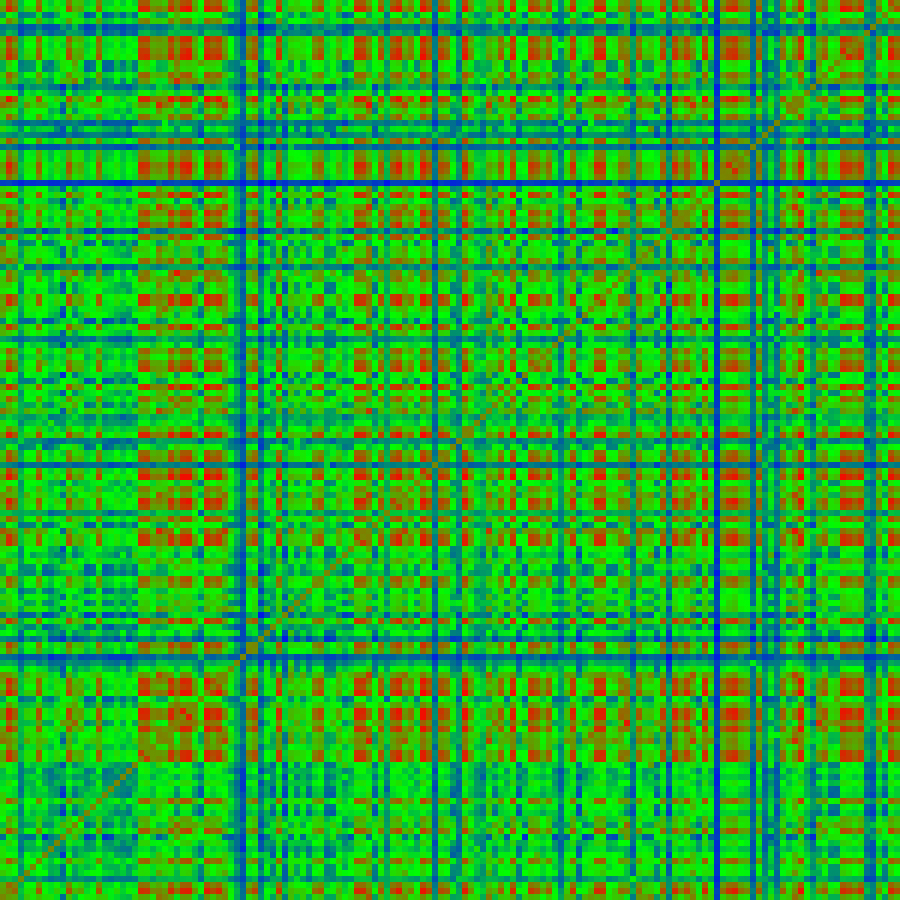
Figure 8. A “heat map” of correlation coefficients between pairs of class averages calculated as in step 3b where the red blocks show high correlations. As indicated in step 3a, the program bmapdist clusters the images based on these correlation coeficients to form new aggregate classes using affinity propagation (Frey and Dueck, 2007).
- Calculate correlation coefficients between each pair of images and cluster them:
3D alignment and reconstruction
For the RS1 case, the strategy is to first align the particle images with a smaller box size (168 x 168) against a previously determined 3D map with D8 symmetry. One iteration is performed using borient for global alignment, followed by several iterations of refinement with brefine. Once the best resolution is achieved, the box is enlarged to the “unit cell” to include neighboring densities (224 x 224) and the particles images re-extracted into a new subdirectory. These are then aligned against a “unit cell” reference, first globally with borient, and then locally with brefine.
The following protocols described how to calculate the initial reference maps for the RS1 case. It is followed by a protocol to calculate a corresponding mask. The mask will be used to eliminate some of the background noise in the reference maps. This improves cross-correlation and may be key to successfully align the particle images.
Initial D8 reference map
Generating a map of RS1 with D8 symmetry from an existing atomic model of the RS1 subunit (PDB: 3JD6; http://www.rcsb.org/pdb/home/home.do):
- Apply D8 symmetry to the RS1 subunit:
bmolsym -verb 7 -apply D8 -rename A 3jd6.pdb 3jd6_D8.pdb - Rotate the model to the orientation in the micrographs:
bmol -verb 7 -rotate 0,0,1,22.5 3jd6_D8.pdb 3jd6_D8_r22.5.pdb - Calculate a map from the model at the correct size and sampling (pixel size):
bsf -verb 7 -coor 3jd6_D8_r22.5.pdb -real -size 168,168,168 -ori c -sam 1.07516 -resol 2 3jd6_D8_r22.5.map
Initial “unit cell” C2 reference map
Generating an initial reference map of the RS1 type 1 “unit cell” by positioning three transformed versions of the single-molecule map (EMDB: 6425; https://www.ebi.ac.uk/pdbe/emdb/index.html/) and combining them (Figure 9):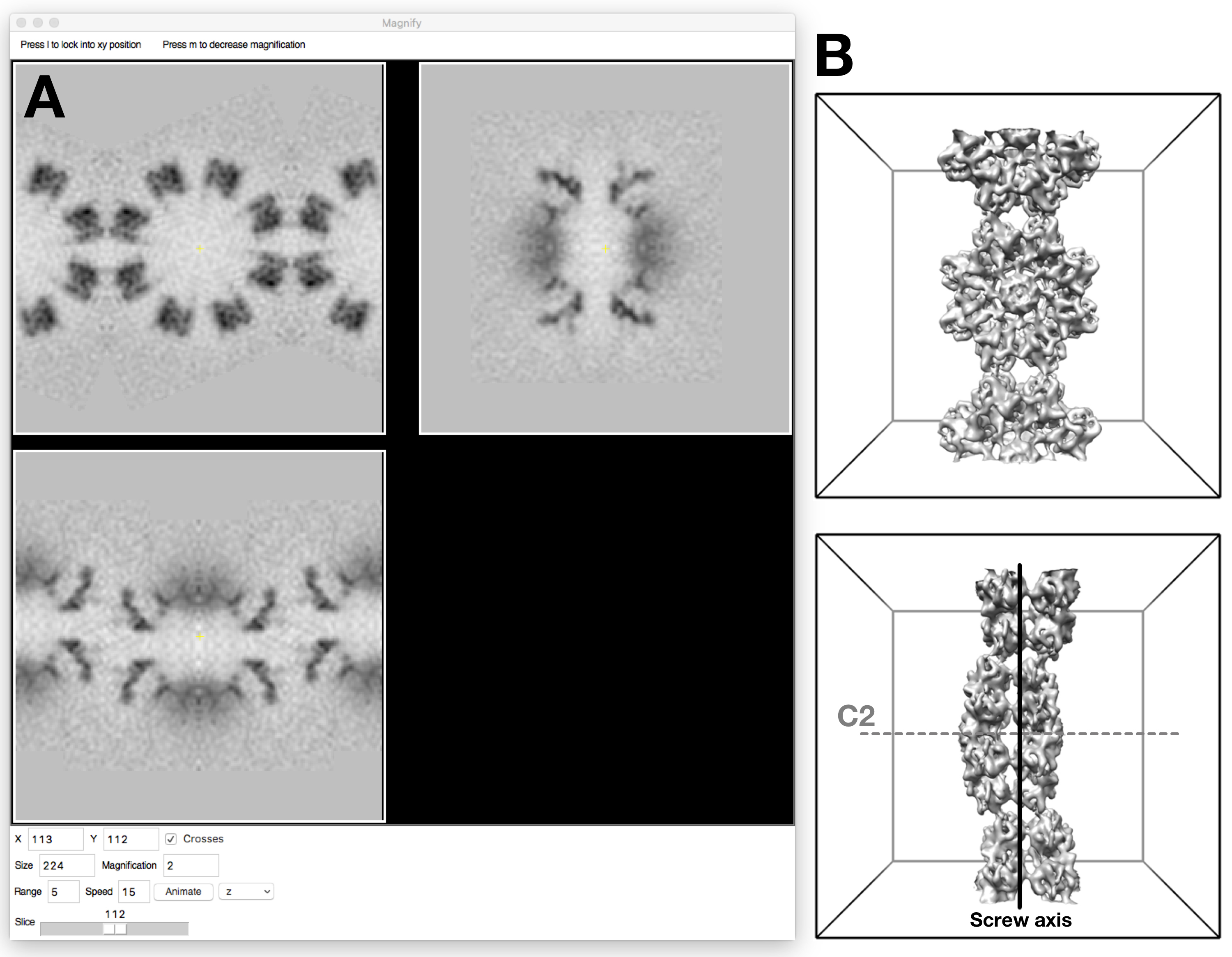
Figure 9. Reference map for the main string interactions (Type I) of the RS1 molecules. A. Orthogonal slices of the map shown in bshow using the “Magnify” window. B. The map shown in the unit cell frame (top and side views). The type I filament can be described as a one-dimensional crystal consistent with the space group P2221, with a 2-fold axis passing through the center of one molecule (gray dashed line labelled C2), and a two-fold screw axis (vertical black line) relating the two molecules.
- Using a previously existing map of a related structure, rescale it to correspond to the particle size and pixel size of the current particle images:
bint -verb 7 -invert -rescale 0,1 -scale 0.958,0.958,0.958 -size 224,224,224 -translate 32,32,25.9 -rotate 0,0,1,22.5 -newori 112,112,112 EMD-6425.map rs1_uc1.mrc
The “-invert” option changes the map so that density is black, the same as the micrographs. The “-rescale” option modifies the density to an average of 0 and a standard deviation of 1 (a nice distribution to work with). The “-scale” option argument is calculated as the ratio of the original pixel size to the desired pixel size. The “-size” option sets the output image size. The “-translate” option moves the origin of the original image to a new location. Because this also sets the origin in the new image, the “-newori” is used to set it to the desired value. The “-rotate” option here rotates the map around the z-axis (0,0,1) by 22.5° to orient it with the correct 2-fold axis on the x-axis. The final result is that the map is translated and rotated to set the C2 axis in the middle on the z-axis, and the screw axis on the x-axis. - Generate a second molecule, displaced in the positive x-direction:
bint -verb 7 -rotate 1,0,0,180 -translate 112,0,0 -newori 112,112,112 rs1_uc1.mrc rs1_uc_tp.mrc
The “-rotate” option here rotates the map 180° around the x-axis (1,0,0) which is also the screw axis of the unit cell. The “-translate” option moves the map 112 pixels on the x-axis to position it appropriately with respect to the density in Step 1. - Generate a third molecule, displaced in the negative x-direction:
bint -verb 7 -rotate 1,0,0,180 -translate -112,0,0 -newori 112,112,112 rs1_uc1.mrc rs1_uc_tn.mrc
This map is translated in a negative direction compared to the density in Step 2. - Add the three maps together:
badd -verb 7 -out rs1_uc.mrc rs1_uc1.mrc rs1_uc_tn.mrc rs1_uc_tp.mrc - Low-pass filter and rescale the map, suitable for an initial reference:
bfilter -verb 7 -band 20,200 -rescale 0,1 rs1_uc.mrc rs1_uc_r20.mrc
Real space mask
The main aim of a real space mask is to remove as much background noise as possible. However, care must be taken to not introduce high frequencies that may compromise alignment. The most important part of the protocol below is the hard low-pass filter in Step 3 that erases all high frequencies (Figure 10). The approach to making the mask here is to exploit the high variance of the particle in the map compared to the low variance of the background, generating a threshold for binarization.
Figure 10. Mask construction. A. The initial binary mask. B. Power spectrum of (A), showing extensive high frequency components. C. Smoothed and low-pass filtered mask. D. Power spectrum of (C), showing that all frequencies beyond 20 Å (arrow) have been erased.
- Generate a variance image and convert to a binary mask:
bmask -verb 7 -variance 21 -dilate 2 rs1_uc.mrc rs1_uc_varmask.mrc
The “-variance” option sets each pixel to the variance calculate within the indicated kernel size around the pixel. The histogram of the variance image is separated into two clusters that defines a threshold between foreground and background in a resultant binary mask. The “-dilate” option enlarges the mask to fully cover the original map density. - Smooth the mask with an averaging filter:
bfilter -verb 7 -average 15 -datatype float rs1_uc_varmask.mrc rs1_uc_varmaskf.mrc
The “-average” option sets each pixel to the average within the kernel around it. The “-datatype” option changes the data type from “byte” to “float” to accommodate non-integer values generated by bfilter. - Low-pass filter with a hard cutoff:
bfilter -verb 7 -band 20 rs1_uc_varmaskf.mrc rs1_uc_varmaskf_r20.mrc
The “-band” option erases all values in frequency space beyond 20 Å. - Check the mask by applying it to the original map:
bop -verb 7 -mult 1,0 rs1_uc.mrc rs1_uc_varmaskf_r20.mrc rs1_uc_masked.mrc
Open the resultant map in bshow and check if the mask follows the density but does not cut into it.
Alignment and reconstruction
Each iteration is a sequence of similar actions: an alignment run with borient or brefine, selecting particles for reconstruction, reconstruction, resolution assessment and preparing the new reconstruction as reference for the next iteration. The following protocol is for one iteration of particle alignment and reconstruction that is repeated until the resolution stops improving. The first iteration should always be a global alignment with borient, followed by either borient or brefine in subsequent iterations. The reuse of borient is to reset the processing, hopefully to recover some previously excluded particle images with a better reference map. Figure 11A shows the progression of the processing, indicating the high resolution limit used and the resolution estimates for the reconstructions. The data set is split into two separate processing streams to be able to validate the results. This can be done in various ways, but the easiest is to just split the micrographs into two sets and process them independently. All operations are done separately for the two sets, except where the reconstructions of the two sets are compared by Fourier shell correlation (FSC).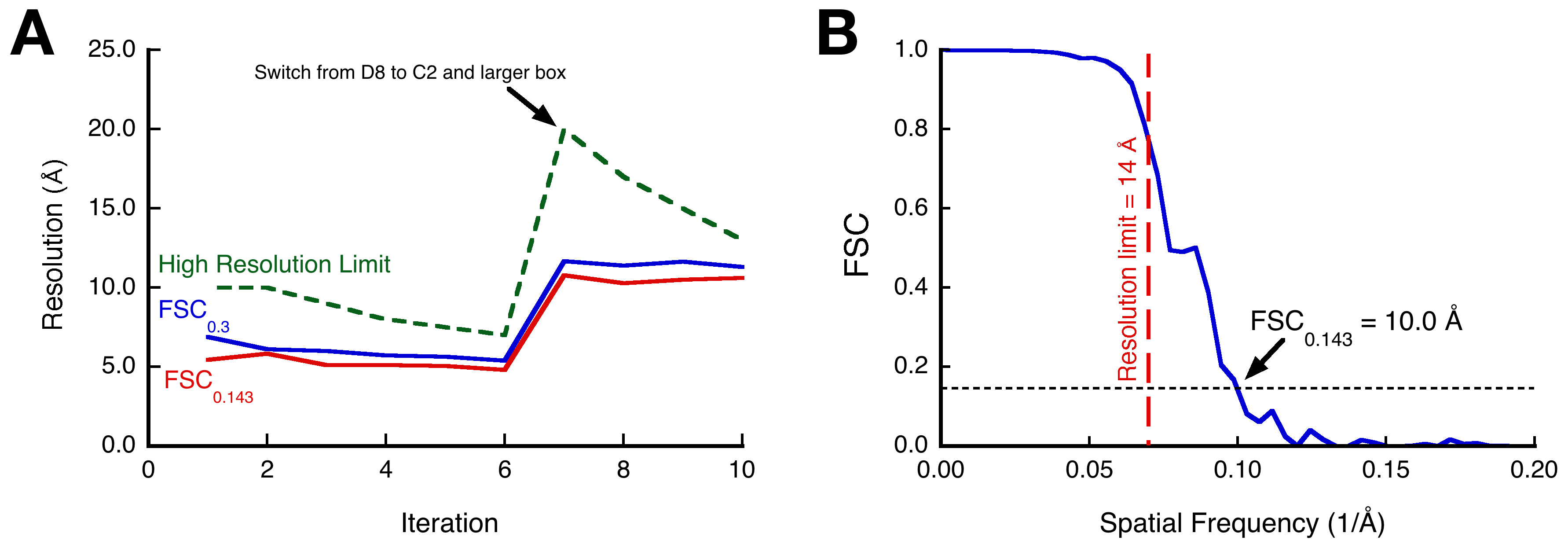
Figure 11. Assessment of processing progress by Fourier shell correlation. A. Particle alignment and reconstruction iterations from micrographs of the 30° tilted specimen of RS1. The first iteration was done with borient, using D8 symmetry and a box size of 168 x 168, followed by 5 iterations of refinement with brefine. The box was then enlarged to the “unit cell” size (224 x 224), the newly extracted particles aligned with borient and C2 symmetry, followed by 3 iterations of refinement with brefine. B. The final resolution estimate by FSC for the 3D reconstruction of the RS1 “unit cell” (iteration 10) is ~10 Å at the 0.143 cutoff, showing that information is recovered beyond the 14 Å high resolution limit used for the last alignment.
- Align a set of particles:
Choose option a or b:- Do a global alignment when no previous orientations have been determined or to try and recover formally excluded particle images.
borient -verb 1 -ppx -mode ccc -sym C2 -ang 5 -resol 20,200 -ann 30,100 -CTF -ref rs1_uc_r20.mrc -out rg4_uc_run1.star ../part_uc/rg4_part_uc.star
The “-mode” option selects different search strategies. The “ccc” mode is the slowest but most accurate. The “-symmetry” option limits the search to an asymmetric unit. The “-angles” option sets the angular step size for the search. - Do a local refinement when previous orientations are encoded in the parameter file
brefine -verb 1 -ppx -grid 5,0.5 -step 5,0.5 -res 17,100 -defocus 0.0005 -mag 0.001 -ref ../uc_run1/rg4_uc_run1m.mrc -out rg4_uc_run2.star rg4_uc_run1r.star
The “-grid” option sets the initial and final angular step sizes (degrees). The “-step” option sets the initial and final translational step sizes (pixels). The “-magnification” and “-defocus” options set their respective step sizes (fraction and µm). - Options common to both programs:
The “-ppx” saves a file for each individual particle in a subdirectory called “ppx”. If the same command line is run again, the program will skip over the particles that have already been done (i.e., those in the “ppx” subdirectory). The “-resolution” options sets the high and low resolution limits for cross-correlation (in Å). The “-reference” option specifies the path and file name of the reference map. - The programs report the translational and rotational parameters determined for each particle, together with cross-correlation and cross-validation coefficients (FOM’s or figures-of-merit).
- Do a global alignment when no previous orientations have been determined or to try and recover formally excluded particle images.
- Check the figures-of-merit (FOM’s):
bpartsel -verb 7 -all rg4_uc_run1.star
This report statistics for two FOM’s in the order: minimum, maximum, average and standard deviation. FOM0 is the correlation coefficient between the resolution limits set for the alignment and is typically in the range 0.1 to 0.5. FOM1 is the crossvalidation coefficient calculated beyond the high resolution limit. Negative values of FOM1 usually indicate bad particles that should be deselected in the following step. - Select the best particles:
bpartsel -verb 7 -all -sym C2 -setasu C2 -fom 0.2,1 -cv 0.01,1 -Part rg4_uc_run1_views.ps -FOM rg4_uc_run1_fom.ps -out rg4_uc_run1s.star rg4_uc_run1.star
The “-sym” and “setasu” options ensure that the orientations fall within an asymmetric unit, ready for display of the views (orientations) and translations in the Postscript file specified by the -Particle” option. The “-fom” and “-cv” options specify cutoffs for the correlation coefficient (FOM0) and the cross-validation coefficient (FOM1), respectively. They also each have a flag to specify that the coefficients should be scaled according to micrograph defocus. The program reports the number and percentage of particles selected. Modify these values to make sure that a significant number of particles are selected and rerun the command line. If the particles look good and there is little expected structural variation, the percentage selected should be high (say ~80%). However, if there is significant conformational or compositional variation, a much lower percentage is appropriate and 3D classification should be considered (not covered here). Many other types of selection can be explored with bpartsel. - Reconstruct:
breconstruct -v 1 -sym C2 -rescale 0,1 -resol 3 -CTF baseflip -full -half -threads 20 -rec rg4_uc_run1.mrc -out rg4_uc_run1r.star rg4_uc_run1s.star
This reconstructs a map from all the selected particles, correcting for the CTF up to a resolution of 3 Å and imposing C2 symmetry. It produces a full map and two halfmaps and estimates the resolution by FSC between the halfmaps. The “-threads” option sets the number of computer threads to use and the amount of memory required. The program will indicate the memory required and whether it will exceed the available computer memory. If so, reduce the number of threads. - Compare the two maps by FSC (Figure 11B):
bresolve -verb 1 -resol 3 -fsccut 0.8,0.3,0.143 -Post rg4_uc_run1_res.ps -mask rs1_uc_varmaskf_r20.mrc -map rg4_uc_run1_01.mrc rg4_uc_run1_02.mrc
The “-resolution” option should match that used in the reconstruction. The “-mask” option indicates the real space mask to remove background noise (see the procedure on appropriate mask generation). The “-Postscript” option specifies the file name for the FSC graph. The “-fsccut” option sets the desired cutoffs to report. The 0.3 and 0.143 cutoffs indicate conservative and optimistic estimates, respectively, of the detail in the map. While the 0.143 cutoff is commonly reported in the literature, it should always be viewed in the context of the whole curve and how the map looks. Sometimes, spurious apparent correlations at high frequency may exceed this cutoff and lead to an exaggerated resolution estimate not supported by the visual detail in the map. It is better to trust the 0.3 cutoff and to follow its improvement as processing proceeds. The other cutoff at 0.8 represents a good choice for the high resolution limit for the next iteration of alignment using this map as reference. Any information recovered beyond this resolution after further alignment and reconstruction provides a measure of validation. - Mask the reference map for the next run:
bop -verb 7 -mult 1,0 rg4_uc_run1.mrc rs1_uc_varmaskf_r20.mrc ../rg4_uc_run1m.mrc
Make sure to use an appropriate mask as described in the section “Real space mask”.
Model building based on 2D averages
Model building is most often associated with fitting coordinates into 3D densities. However, in cases where only 2D class averages are available, valuable information can still be obtained if each has a defined orientation relative to a 3D map. Here, each of the 2D class averages show multiple copies of the RS1 molecules in two major orientations: a side view and a top view. To understand the geometry of the interactions between the molecules, their exact locations and orientations must be determined by matching the two projections from the 3D map to the different copies in each class average. A 3D map is then built from all the properly oriented copies of the single-molecule map. The assumption is that all the molecules showing side views are interacting with the air-water interface and are therefore in the same plane. Although this is a very specific example of such modeling, the details can be adapted to accomplish similar results for other cases.
Building maps based on 2D images:
- Generate two reference images, top and side view:
- Project the 3D map into top and side views:
bproject -verb 7 -angles 90 -symmetry D8 -kernel 8,2 rs1_temp_lrg_r22.5.mrc rs1_temp_lrg_r22.5_top_side.mrc
The “-symmetry” option limits the projections to the asymmetric unit. The “-angles” selects views at 0° (top) and 90° (side). The “-kernel” option specifies that the projection is performed in frequency space with the indicated interpolation parameters. The resultant file contains one top view and three side views (two of them not desired). - Extract the relevant top and side views:
bimg -verb 7 -delete 1,3 rs1_temp_lrg_r22.5_top_side.mrc rs1_temp_lrg_r22.5_top_side.mrc
The “-delete” option removes the indicated images (where the first image in the file is 0).
- Project the 3D map into top and side views:
- Find the RS1 molecules in the 2D images:
bmatch -verb 1 -resolution 20,100 -angle 1 -radius 70 -exclusion 70 -thresh 0.15 -reference rs1_temp_lrg_r22.5_top_side.mrc -out rg4_2d_r5_avg_cluster_p10_mod.star rg4_2d_r5_avg_cluster_p10.pif rg4_2d_r5_avg_cluster_p10_cc.grd
The “-resolution” option sets the high and low resolution limits for cross-correlation (in Å). The “-angle” option sets the angular search step (in degrees). The “-radius” option sets the mask radius to apply for cross-correlation. The “-exclusion” option imposes a minimum distance between hits. The “-threshold” option specifies the cutoff for identifying hits in the cross-correlation map (it is used in the initial step before masking, and is therefore relatively low compared to the final correlation coefficients obtained after masking). The “-reference” option gives the template file with one or more projections. - Build a composite 3D map:
bxb -verb 1 -build 330,330,330 -origin center -sampling 1.04-map rs1_temp_lrg_r22.5.mrc -fom 0.8 -cons rg4_2d_r5_avg_cluster_p10_bld.mrc -out rg4_2d_r5_avg_cluster_p10_bld.star rg4_2d_r5_avg_cluster_p10_mod.star
The “-build” option indicates that a new map will be built with the given size. The “-map” option specifies the template to be built into the new map. The “-fom” option selects only those hits with a better correlation coefficient. Its value is typically determined by trial and error. It is set high here because the images have little noise. - Examine the model in bshow:
bshow rg4_2d_r5_avg_cluster_p10_mod. star
This opens the “Model” panel in bshow (Figure 12). - Project it down the z axis for comparison with the original image:
bproject -verb 7 -axis z rg4_2d_r5_avg_cluster_p10_bld.mrc rg4_2d_r5_avg_cluster_p10_bld_pz.mrc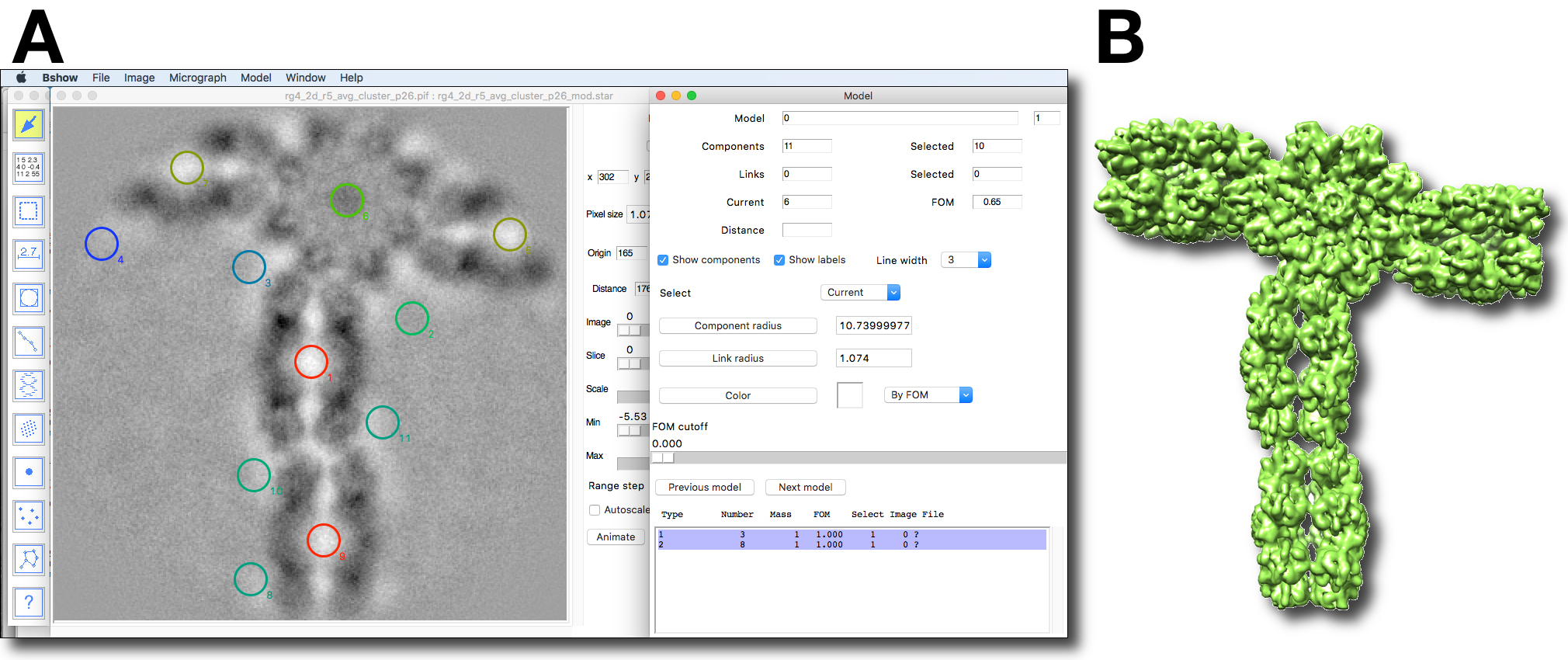
Figure 12. Fitting a map of RS1 to multiple molecules present in a 2D average. A. A screen shot of bshow with the model of the hits from cross-correlation of the class average of the type II interaction with the top and side views of the RS1 3D map using bmatch. To show the locations of the hits as color-coded with the correlation coefficient or FOM (red: high; blue: low), select the “By FOM” item from the drop-down menu next to the “Color” button, and click the “Color” button. Move the FOM cutoff slider to show only hits with higher FOM’s. B. The model with orientations determined in A were used to build a map for the type II RS1 interactions using bxb, but selecting only the components with high FOM’s.
Rigid model fitting into 3D density
The final map from 3D alignment and reconstruction is located in the “unit cell” as shown in Figure 9. It also only has C2 symmetry, while the screw axis symmetry was not imposed. The following protocol imposes the screw axis symmetry and places two almost intact molecules in the map to emphasize the interaction between them (this is similar to the protocol for generated a 3D reference map of the “unit cell”):
- Rotate the map around the screw axis and translates it in a negative direction:
bint -verb 7 -rotate 1,0,0,180 -translate -112,0,0 -newori 112,112,112 rs1_uc4.mrc rs1_uc4_tn.mrc - Rotate the map around the screw axis and translates it in a positive direction:
bint -verb 7 -rotate 1,0,0,180 -translate 112,0,0 -newori 112,112,112 rs1_uc4.mrc rs1_uc4_tp.mrc - Combine the three maps:
badd -verb 7 -out rs1_uc4_3.mrc rs1_uc4.mrc rs1_uc4_tn.mrc rs1_uc4_tp.mrc - Mask the map to remove background:
bop -verb 7 -multiply 1,0 rs1_uc4_3.mrc ../rs1_mask3df_uc_3tr10t.mrc rs1_uc4_3m.mrc - Weigh the map to have the same frequency space spectral distribution as the initial reference map used for 3D alignment:
bampweigh -verb 7 -Reference ../3jd6_D8_uc_3.mrc -resolution 10 -rescale 0,1 rs1_uc4_3m.mrc rs1_uc4_3m_awr10.mrc
The program Fourier transforms the map and the reference, then scales the map amplitudes to match those of the reference, and backtransforms it to real space. The “-resolution” option low-pass filters the result and the “-rescale” option sets the average to 0 and the standard deviation to 1. - Translate the map to position two molecules in the “unit cell”:
bimg -verb 7 -translate 56,0,0 -invert -rescale 0,1 -wrap rs1_uc4_3m_awr10.mrc rs1_uc4_3m_awr10_t.mrc
The “-wrap” option turns on wrap-around, i.e., the parts of the map that shifts off the one edge appears at the opposite edge to adhere to crystallographic periodicity. The map is now positioned so that the interface between the two molecules is in the middle and there is a two-fold axis along the y axis that relates the two molecules. (This map was deposited in the EMDB: 7907; https://www.ebi.ac.uk/pdbe/emdb/index.html/)
Generating a “unit cell” of two RS1 molecules:
The RS1 atomic model (PDB 3JD6; http://www.rcsb.org/pdb/home/home.do) is only one subunit of the 16-mer and must be transformed and copied to fit into the 3D density of the “unit cell” (Figure 13). The original model produced by Tolun et al., 2016 is oriented with the 16-mer twofold between apposing subunits in different rings. The RS1 molecules in the images here are mostly oriented to a view down the alternative twofold axis. Therefore, to get the molecule in the correct view, it must be rotated by 22.5°. In addition, the two molecules are offset from each other by about 12.2 Å, or offset from the central screw axis by 6.1 Å. Finally, to generate the second molecule from the first, it must be rotated by 180° and translated by 120.3 Å:
- Generate all symmetry-related subunits from the PDB file:
bmolsym -verb 7 -apply D8 -rename A 3jd6.pdb 3jd6_D8.pdb
The “-rename” option ensures that the chains have different labels, starting with “A”. - Rotate the model to get the correct view:
bmol -verb 7 -rotate 0,0,1,22.5 3jd6_D8.pdb 3jd6_D8_r22.5.pdb - Offset the molecule on the z axis:
bmol -verb 7 -trans 0,0, -6.1 3jd6_D8_r22.5.pdb 3jd6_D8_uc.pdb - Generate the second molecule along the screw axis:
bmol -verb 7 -rotate 1,0,0,180 -trans -120.3,0,0 3jd6_D8_uc.pdb 3jd6_D8_uc_tn.pdb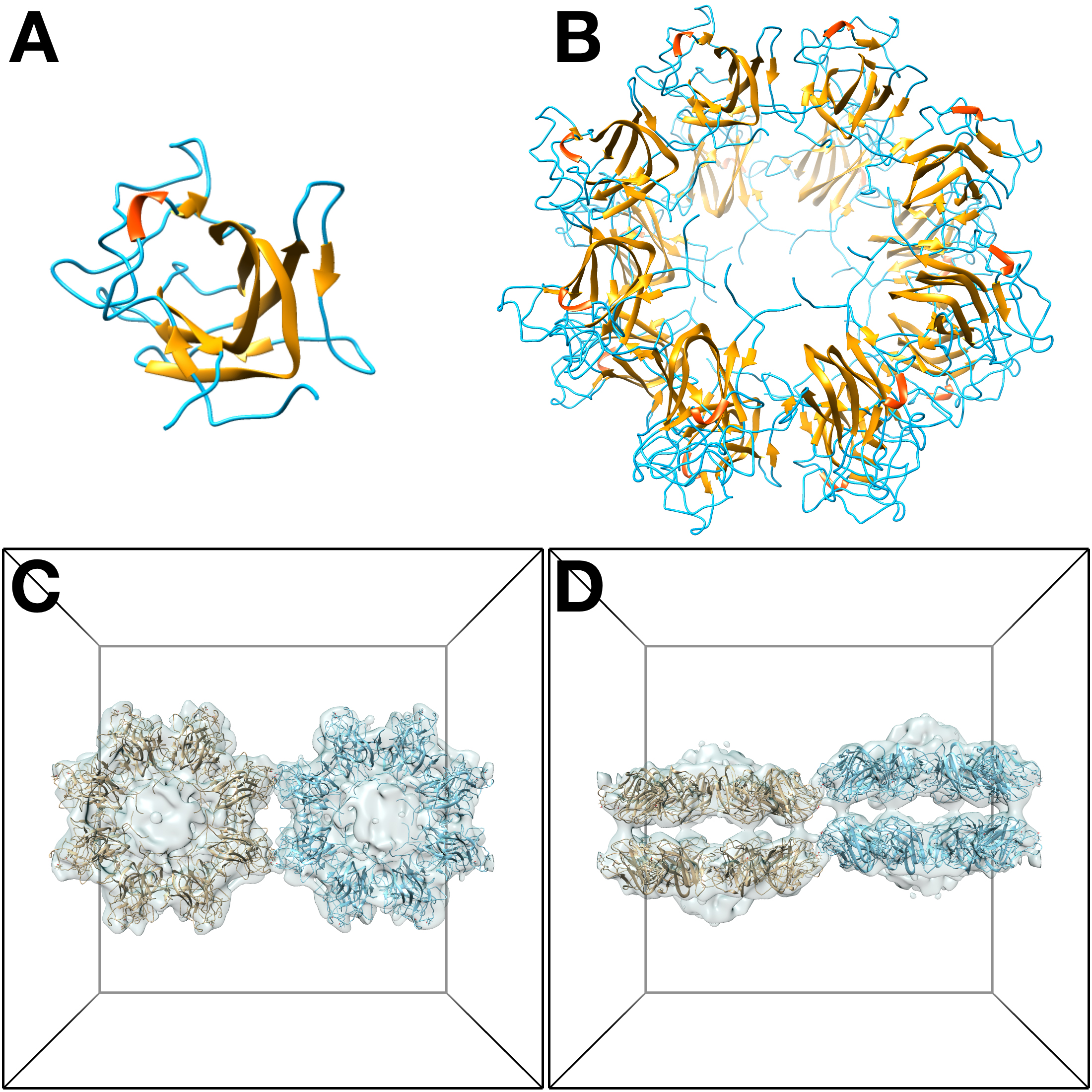
Figure 13. Model of the RS1 “unit cell” of the type I interaction. A. One subunit of RS1 (PDB: 3JD6). B. The 16-mer molecule of RS1 with D8 symmetry. C. Top view of the “unit cell”. D. Side view of the “unit cell”.
Acknowledgments
I am sincerely grateful for the useful comments and suggestions of Drs. Altaira Dearborn and Michael Buch. This work was supported by the Intramural Research Program of the National Institute for Arthritis, Musculoskeletal and Skin Diseases, NIH.
Competing interests
The author has no conflicts of interest to declare.
References
- Downing, K. H. and Glaeser, R. M. (2008). Restoration of weak phase-contrast images recorded with a high degree of defocus: the "twin image" problem associated with CTF correction. Ultramicroscopy 108(9): 921-928.
- Frey, B. J. and Dueck, D. (2007). Clustering by passing messages between data points. Science 315(5814): 972-976.
- Grant, T. and Grigorieff, N. (2015). Measuring the optimal exposure for single particle cryo-EM using a 2.6 A reconstruction of rotavirus VP6. Elife 4: e06980.
- Harauz, G. and van Heel, M. (1986). Exact filters for general geometry three dimensional reconstruction. Optik 73: 146-156.
- Heymann, J. B. (2001). Bsoft: image and molecular processing in electron microscopy. J Struct Biol 133(2-3): 156-169.
- Heymann, J. B. (2018a). Guidelines for using Bsoft for high resolution reconstruction and validation of biomolecular structures from electron micrographs. Protein Sci 27(1): 159-171.
- Heymann, J. B. (2018b). Map Challenge assessment: Fair comparison of single particle cryoEM reconstructions. J Struct Biol 204(2): 360-367.
- Heymann, J. B. (2018c). Single particle reconstruction and validation using Bsoft for the map challenge. J Struct Biol 204(1): 90-95.
- Heymann, J. B. and Belnap, D. M. (2007). Bsoft: Image processing and molecular modeling for electron microscopy. J Struct Biol 157(1): 3-18.
- Heymann, J. B., Cardone, G., Winkler, D. C. and Steven, A. C. (2008). Computational resources for cryo-electron tomography in Bsoft. J Struct Biol 161(3): 232-242.
- Heymann, J. B., Chagoyen, M. and Belnap, D. M. (2005). Common conventions for interchange and archiving of three-dimensional electron microscopy information in structural biology. J Struct Biol 151(2): 196-207 (Corrigendum, J Struct Biol 153, 312).
- Heymann, J. B., Vijayasarathy, C., Huang, R. K., Dearborn, A. D., Sieving, P. A. and Steven, A. C. (2019). Cryo-EM of retinoschisin branched networks suggests an intercellular adhesive scaffold in the retina. J Cell Biol 218(3): 1027-1038.
- Leong, P. A., Heymann, J. B. and Jensen, G. J. (2005). Peach: a simple Perl-based system for distributed computation and its application to cryo-EM data processing. Structure 13(4): 505-511.
- Noble, A. J., Dandey, V. P., Wei, H., Brasch, J., Chase, J., Acharya, P., Tan, Y. Z., Zhang, Z., Kim, L. Y., Scapin, G., Rapp, M., Eng, E. T., Rice, W. J., Cheng, A., Negro, C. J., Shapiro, L., Kwong, P. D., Jeruzalmi, D., des Georges, A., Potter, C. S. and Carragher, B. (2018). Routine single particle CryoEM sample and grid characterization by tomography. eLife 7: e34257.
- Noble, A. J., Dandey, V. P., Wei, H., Brasch, J., Chase, J., Acharya, P., Tan, Y. Z., Zhang, Z. N., Kim, L. Y., Scapin, G., Rapp, M., Eng, E. T., Rice, W. J., Cheng, A. C., Negro, C. J., Shapiro, L., Kwong, P. D., Jeruzalmi, D., des Georges, A., Potter, C. S. and Carragher, B. (2019). Cryoet of Single Particle CryoEM Grids Reveals Widespread, but Reducible, Particle Adsorption to the Air-Water Interface. Biophys J 116(3): 11a.
- Tolun, G., Vijayasarathy, C., Huang, R., Zeng, Y., Li, Y., Steven, A. C., Sieving, P. A. and Heymann, J. B. (2016). Paired octamer rings of retinoschisin suggest a junctional model for cell-cell adhesion in the retina. Proc Natl Acad Sci U S A 113(19): 5287-5292.
- Unser, M., Trus, B. L. and Steven, A. C. (1987). A new resolution criterion based on spectral signal-to-noise ratios. Ultramicroscopy 23(1): 39-51.
Article Information
Copyright
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Heymann, J. B. (2020). Protocols for Processing and Interpreting cryoEM Data Using Bsoft: A Case Study of the Retinal Adhesion Protein, Retinoschisin. Bio-protocol 10(2): e3491. DOI: 10.21769/BioProtoc.3491.
- Heymann, J. B., Vijayasarathy, C., Huang, R. K., Dearborn, A. D., Sieving, P. A. and Steven, A. C. (2019). Cryo-EM of retinoschisin branched networks suggests an intercellular adhesive scaffold in the retina. J Cell Biol 218(3): 1027-1038.
Category
Biochemistry > Protein > Imaging
Biophysics > Microscopy > Cryogenic microscopy
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.