- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Unbiased and Tailored CRISPR/Cas gRNA Libraries by Synthesizing Covalently-closed-circular (3Cs) DNA
(*contributed equally to this work) Published: Vol 10, Iss 1, Jan 5, 2020 DOI: 10.21769/BioProtoc.3472 Views: 9049
Reviewed by: David PaulAli Asghar KermaniAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Mar 2019
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Simplicity, efficiency and versatility of the CRISPR/Cas system greatly contributed to its rapid use in a broad range of fields. Applications of unbiased CRISPR/Cas screenings are increasing and thus there is a growing need for unbiased and tailored CRISPR/Cas gRNA libraries. Conventional methods for gRNA library generation apply PCR and cloning techniques, thus coupling library diversity with distribution. Here, we provide additional technical expertise to apply our covalently-closed-circular synthesized (3Cs) gRNA library generation technology for the generation of high-quality CRISPR/Cas gRNA libraries. F1-origin of replication-containing plasmid DNA is transformed into CJ236 bacteria for single colony outgrow followed by M13KO7 bacteriophage superinfection for the production and preparation of circular dU-containing ssDNA. dU-ssDNA is annealed with homology- and gRNA-encoding DNA oligonucleotides for their T7 DNA polymerase-mediated extension to form hetero-duplexed CCC-dsDNA (3Cs-dsDNA). 3Cs-dsDNA is electroporated for the selected amplification of the newly synthesized, gRNA-containing strand. To remove wild-type plasmid remnants, the purified plasmid DNA is digested with restriction enzymes targeting the gRNA-placeholder sequence in the template DNA. Undigested plasmid is electroporated for the extraction of the final 3Cs gRNA library. Due to the absence of PCR amplification and conventional cloning steps, the 3Cs technology uncouples sequence diversity from sequence distribution, thereby generating gRNA libraries with near-uniform distribution in diversities being only limited by electroporation efficiencies.
Keywords: CRISPR/CasBackground
CRISPR/Cas gene editing has developed to be the gold standard for tailored and unbiased high-throughput screening (Doench, 2018). Unbiased pooled screening enables the simultaneous testing of thousands of individual genetic perturbations in a single experiment. However, a prerequisite is the presence of a gRNA library, in which each individual construct defines a specific genomic target site (gRNA sequence), at which the gene perturbation is desired, with a library’s gRNA distribution and diversity dictating the experimental scale (Sanson et al., 2018). Classically, protocols to generate CRISPR/Cas gRNA libraries for arrayed or pooled applications contain T4 ligase or recombination-based ligations steps, requiring DNA polymerase-dependent amplification of the gRNA-encoding oligonucleotides coupled with restriction enzyme-digested plasmid DNA for subsequent gRNA cloning (Koike-Yusa et al., 2014; Shalem et al., 2014; Wang et al., 2014; Schmidt et al., 2015; Vidigal and Ventura, 2015; Arakawa, 2016; Ong et al., 2017). Due to these technical limitations, the resulting gRNA libraries contain undesired PCR and cloning-dependent sequence biases in their gRNA sequence distribution that directly affects the experimental scale needed for robust hit calling (Shalem et al., 2014; Wang et al., 2014). Unbiased and tailored CRISPR/Cas gRNA screening is becoming more ubiquitous, highlighting the need for novel protocols to generate gRNA libraries that can readily be established in non-expert laboratories.
Our recently described protocol to generate covalently-closed-circular synthesized (3Cs) gRNAs avoids DNA polymerase-dependent amplification steps of gRNA-encoding oligonucleotides, as well as enzyme-digested plasmid DNA (Figure 1, Wegner et al., 2019), thereby circumventing the occurrence of PCR-dependent gRNA sequence bias, as well as cloning artifacts within the final gRNA library, resulting in the decoupling of sequence distribution from sequence diversity and leading to nearly uniformly distributed gRNA libraries. As a consequence, the quality and distribution of the gRNA sequences within the original gRNA-encoding oligonucleotide pool is very important and particular caution should be paid to only use high-quality oligonucleotide pools.
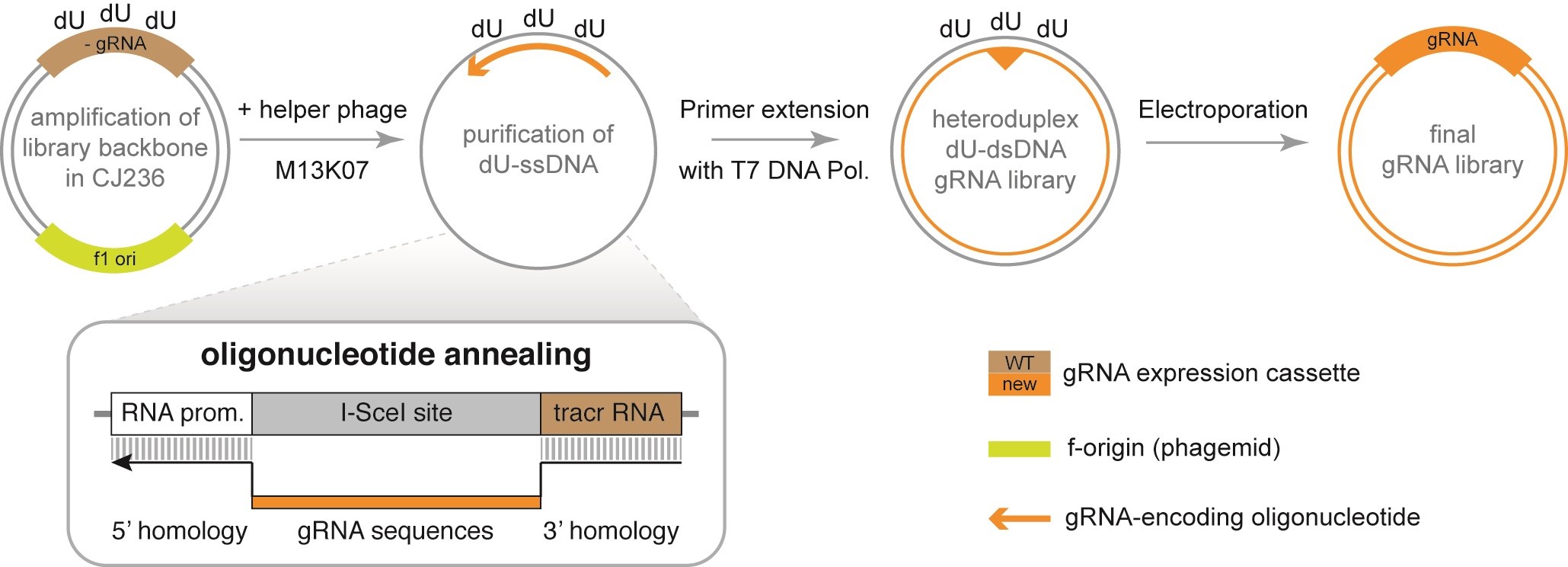
Figure 1. Covalently-closed-circular (3Cs) synthesized gRNA reagents. Amplification of f1-ori-containing plasmids in M13KO7-infected dut-/ung- E. coli CJ236 bacteria and subsequent purification yields uracilated single-stranded DNA (dU-ssDNA). Annealing of gRNA-encoding oligonucleotides, extension with T7 polymerase, and ligation with T4 ligase produces a heteroduplex dU-dsDNA plasmid library. Electroporation into dut-/ung- E. coli yields the final library. The gRNA-encoding oligonucleotide is designed to anneal 5′ and 3′ relative to the I-SceI placeholder site in the library backbone and acts as a primer for T7 polymerase in the synthesis step of the 3Cs protocol.
Materials and Reagents
- 2 mm electroporation cuvette (BTX, catalog number: 45-0125)
- Conical centrifuge tube, 50 ml (Falcon, catalog number: 352070)
- Safe-lock tubes, 1.5 ml (Eppendorf, catalog number: 0030120086)
- Bacteria (E. coli) CJ236 (TaKaRa, catalog number: 9053 or Lucigen, catalog number: 60701), storage -80 °C
- Bacteria (E. coli) 10-beta, electrocompetent (New England Biolabs, catalog number: C3020K), storage -80 °C
- Bacteria (E. coli) SS320 (MC1061 F’), electrocompetent (Lucigen, catalog number: 60512)
- dNTP mix (Carl Roth, catalog number: 0178), storage -20 °C
- T4 polynucleotide kinase (PNK) (New England Biolabs, catalog number: M0201), storage -20 °C
- T4 DNA ligase(New England Biolabs, catalog number: M0202), storage -20 °C
- T7 DNA polymerase (unmodified) (New England Biolabs, catalog number: M0274), storage -20 °C
- Restriction enzyme I-SceI (New England Biolabs, catalog number: R0694), storage -80 °C
- Restriction enzyme EcoRV-HF (New England Biolabs, catalog number: R3195), storage -20 °C
- Helper phage M13KO7 (New England Biolabs, catalog number: N0315)
- LB broth with agar (Lennox) (Sigma-Aldrich, catalog number: L2897)
- LB medium (Lennox) (Carl Roth, catalog number: X964)
- 2x YT medium (Carl Roth, catalog number: 6676)
- S.O.C. medium (Thermo Fisher, catalog number: 15544034)
- Polyethylene glycol 8000 (PEG 8000) (Carl Roth, catalog number: 263)
- Sodium chloride (NaCl) (Sigma-Aldrich, catalog number: 31434-M)
- Dulbecco’s phosphate-buffered saline (PBS) (Sigma-Aldrich, catalog number: D8662)
- E.Z.N.A. M13 DNA Mini Kit (Omega Bio-Tek, catalog number: D69001-01)
- GeneJET Gel Extraction Kit (Thermo Fisher, catalog number: K0692)
- GeneJET Plasmid Miniprep Kit (Thermo Fisher, catalog number: K0503)
- DNA Clean and Concentrator-5 Kit (Zymo Research, catalog number: D4013)
- Plasmid Midi Kit (Qiagen, catalog number: 12143)
- Plasmid Maxi Kit (Qiagen, catalog number: 12163 or Promega, catalog number: 12163)
- Ampicillin sodium salt (Amp) (Carl Roth, catalog number: K0292)
- Chloramphenicol (Cmp) (Carl Roth, catalog number: 3886)
- Kanamycin sulphate (Kan) (Carl Roth, catalog number: T832)
- Adenosine 5'-triphosphate (ATP) (New England Biolabs, catalog number: P0756), storage -20 °C
- Dithiothreitol (DTT) (Cell Signaling Technology, catalog number: 7016), storage -20 °C
- Agarose, standard (Applichem, catalog number: 732-2789)
- SYBR Safe DNA gel stain (Thermo Fisher, catalog number: S33102)
- KCl (Carl Roth, catalog number: P017)
- CaCl2 (Carl Roth, catalog number: CN93)
- MgCl2 (Carl Roth, catalog number: KK36)
- Tris base (Carl Roth, catalog number: A411)
- Tris-HCl (Carl Roth, catalog number: 9090)
- EDTA (Applichem, catalog number: A5097,0500)
- Acetic acid (Sigma-Aldrich, catalog number: 33209)
- KCM buffer (see Recipes)
- 20% PEG/NaCl (see Recipes)
- 10x TM buffer (see Recipes)
- 50x TAE buffer (see Recipes)
Equipment
- Bacterial shaker (Eppendorf, model: New Brunswick Innova 44/44R, catalog number: M1282-0012; or similar)
- Gene Pulser electroporation system (Bio-Rad, catalog number: 164-2076, or similar system for electroporation of bacteria)
- Mini-sub cell GT horizontal electrophoresis system, 7 x 10 cm (Bio-Rad, catalog number: 1704467, or similar)
- Centrifuge Avanti J-E (Beckman, catalog number: 369003, or similar)
- Rotor JA-12 (fixed-angle aluminum rotor- 12 x 50 ml, 12,000 rpm, 23,200 x g, Beckman, catalog number: 360993)
- NanoDrop (Thermo Fisher, catalog number: ND-1000, or similar)
Software
- bCl2fastq (Illumina, https://support.illumina.com/)
- cutadapt (https://cutadapt.readthedocs.io/en/stable/; Martin, 2011)
- Bowtie2 (http://bowtie-bio.sourceforge.net/bowtie2/index.shtml; Langmead and Salzberg, 2012)
- PinAPL-Py (http://pinapl-py.ucsd.edu/; Spahn et al., 2017)
Procedure
- Preparation of plasmid single-stranded DNA (ssDNA)
- Transform 100-200 ng of plasmid DNA into 25-50 μl competent E. coli CJ236 bacteria. Use either heat-shock (1 min, 42 °C, heat-block) or KCM buffer (use the same volume of bacteria and a mix of 5x KCM buffer diluted 1:5 with water, ice/15 min, room temperature/10 min) for transformation. Add 50-200 μl S.O.C. medium and plate on LB plates containing 25 μg/ml Cmp and additional antibiotic matching plasmid resistance (usually 100 μg/ml Amp*, depending on plasmid), leave overnight at 37 °C.
*Note: In theory, any plasmid containing f1-origin of replication can be used, such as lentiCRISPR v2 (Addgene 52961). The plasmid should have an antibiotic resistance gene other than Amp, Cmp and Kan. - Next day, pick several single colonies of E. coli CJ236 with a sterile pipette tip, resuspend each into 1 ml of 2x YT medium supplemented with Amp* (100 μg/ml), Cmp (25 μg/ml) and 1 μl helper phage. Shake for 2 h (37 °C, 200 rpm); add 100 μg/ml Kan and continue shaking for 6-10 h (37 °C, 200 rpm).
- Transfer 1 ml of initial culture into 30 ml of 2x YT medium containing Amp* (100 μg/ml) and Kan (100 μg/ml) and shake 16-20 h (37 °C, 200 rpm).
- Transfer 30 ml bacterial culture into a 50 ml Falcon tube; centrifuge (10 min, 12,000 x g, 4 °C).
- Transfer phage-containing supernatant (Figure 2A) into 6 ml of sterile-filtered PEG/NaCl buffer, invert 4-6 times, incubate for 30-60 min at room temperature; centrifuge (10 min at 12,000 x g, 4 °C).
- Decant supernatant and aspirate the remaining supernatant.
- Resuspend phage pellet (Figure 2B) in 1 ml 1x PBS and transfer to a new 1.5 ml reaction tube.

Figure 2. Preparation of ssDNA from bacterial culture. E. coli CJ236 bacterial culture before (A) and after (B) centrifugation (pellet contains bacteria, whereas supernatant contains phage). C. Phage pellet following PEG/NaCl precipitation and subsequent centrifugation. D. Phage resuspended in 1x PBS buffer (transparent solution). E. After addition of ssDNA lysis buffer (turbid solution, turbidity reflects amount of phage). - Centrifuge for 5 min at 16,100 x g to remove cell debris, transfer the supernatant to a new 1.5 ml reaction tube and store at 4 °C.
- Purify ssDNA from phage supernatant according to the Omega Bio-Tek E.Z.N.A M13 DNA Mini Kit protocol.
- Determine ssDNA concentration using NanoDrop.
- Analyze ssDNA in a 0.8% TAE/agarose gel using 500 ng ssDNA and 250 ng of the respective dsDNA. The ssDNA should appear as a predominant single band (Figure 3A).
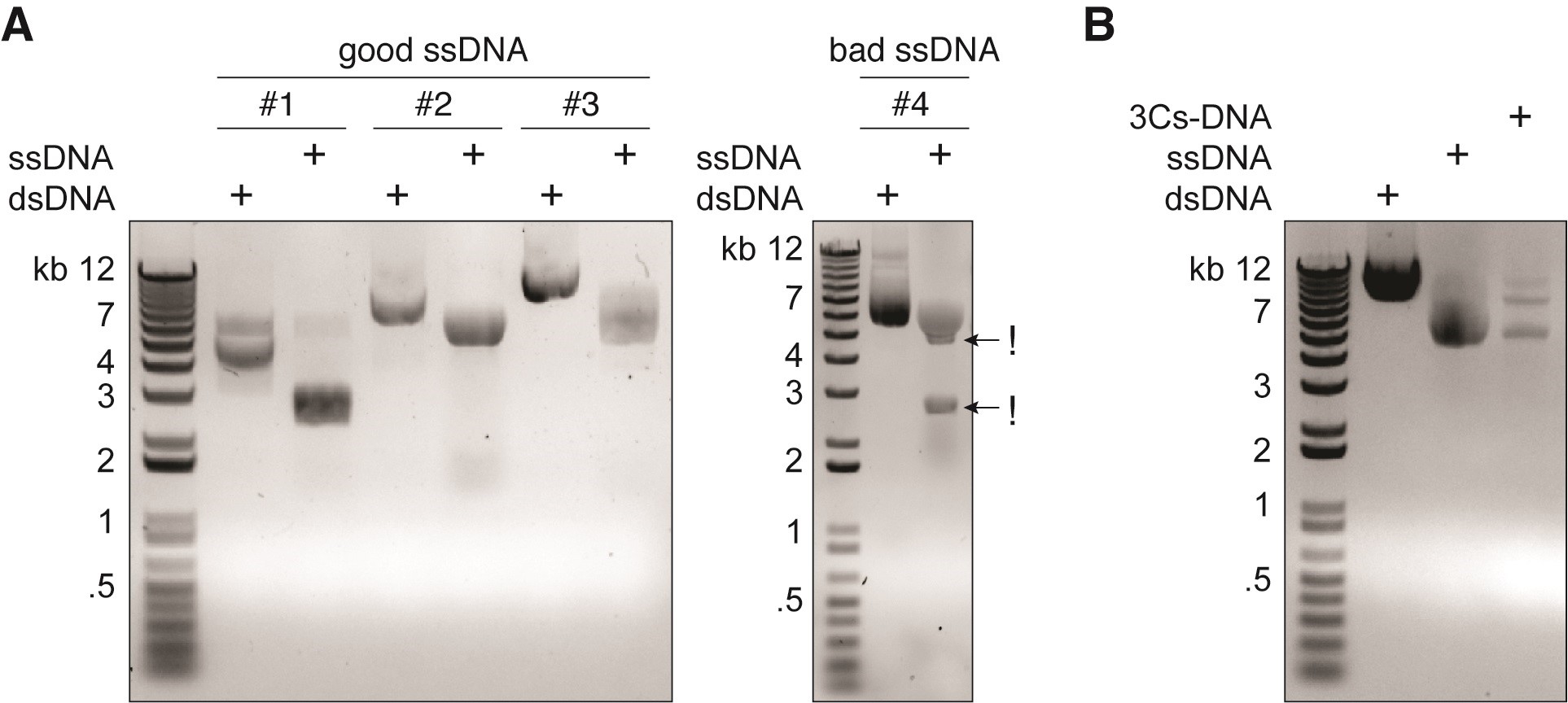
Figure 3. Quality control of ssDNA and 3Cs reaction products. A. ssDNA appears as a single band on an agarose gel and usually exhibits higher electromobility than its dsDNA plasmid counterpart. #1, #2, and #3 refer to example plasmids of different sizes. Low quality ssDNA exhibits multiple bands indicating phage-derived recombination products that will lead to poor performance during 3Cs (!). B. The 3Cs reaction product typically exhibits three bands on an agarose gel. All three electrophoreses were run on 0.8% 1x TAE agarose for 30 min at 110 V.
- Transform 100-200 ng of plasmid DNA into 25-50 μl competent E. coli CJ236 bacteria. Use either heat-shock (1 min, 42 °C, heat-block) or KCM buffer (use the same volume of bacteria and a mix of 5x KCM buffer diluted 1:5 with water, ice/15 min, room temperature/10 min) for transformation. Add 50-200 μl S.O.C. medium and plate on LB plates containing 25 μg/ml Cmp and additional antibiotic matching plasmid resistance (usually 100 μg/ml Amp*, depending on plasmid), leave overnight at 37 °C.
- Oligonucleotide phosphorylation with T4 polynucleotide kinase
In a 1.5 ml reaction tube, combine 600 ng oligonucleotide pool with 2 μl 10x TM buffer, 1 mM ATP, 5 mM DTT and 20 units T4 polynucleotide kinase in a total volume of 20 μl; incubate for 2 h at 37 °C.
Note: Oligonucleotide pools can be ordered from various commercial sources. Hand-pooled oligonucleotides generally lead to libraries with very variable gRNA distribution and should therefore be avoided.
- Annealing of the oligonucleotides to the template
Mix 20 μg freshly prepared ssDNA with 25 μl 10x TM buffer (final concentration 1x TM) and 20 μl phosphorylated oligonucleotides in a total volume of 250 μl; incubate in a thermoblock as follows: 90 °C (3 min), 50 °C (5 min), room temperature (5 min).
Note: Molar ratio between oligonucleotides and vector ssDNA should be determined experimentally and might be different for a particular plasmid. Usually, ratio ranges of 3:1, 5:1 and 7:1 are optimal for 3Cs reactions.
- Enzymatic synthesis of 3Cs-dsDNA
Add the following components to the previous mixture: 10 μl of 10 mM ATP, 10 μl of 100 mM dNTPs, 15 μl of 100 mM DTT, 3 μl of T4 DNA ligase (stock: 400,000 units/ml) and 5 μl of T7 DNA polymerase (stock: 10,000 units/ml); mix carefully by pipetting up and down or flicking the tube; incubate for 16 h at 22 °C. - Purification of 3Cs-dsDNA with DNA Clean and Concentrator-5 Kit
- Mix the 3Cs reaction with 2x volume of DNA binding buffer and incubate for 10 min at room temperature.
- Divide the 3Cs reaction on 2 columns.
- Spin down (1 min, 2,300 x g, room temperature); re-spin each flow through twice.
- Wash column twice each with 200 μl wash buffer (1 min, 2,300 x g, room temperature).
- Discard flowthrough and dry the column (1 min, 7,400 x g, room temperature).
- Incubate column with preheated 10 μl of elution buffer (10 min, 50 °C), spin down at maximum speed (2 min), re-spin each flow through twice.
- Repeat elution with 20 μl flow through (final volume will be around 40 μl).
- Run a gel to control the quality of dsDNA alongside to ssDNA (Figure 3B).
- Determine concentration by Nanodrop.
- Electroporation of electrocompetent E. coli bacteria with 3Cs dsDNA
- Prepare 20 ml pre-warmed S.O.C. medium at 37 °C in a small Erlenmeyer flask.
- Incubate the entire purified 3Cs dsDNA and a 2 mm electroporation cuvette on ice.
- Thaw 400 μl of electrocompetent E. coli strain SS320 or NEB 10-beta bacteria on ice (20 min).
- Add the cells to the pre-chilled and purified 3Cs DNA and mix slowly by pipetting, avoid generating bubbles.
- For electroporation, use the following settings: 2.5 kV field strength, 25 µF capacitance, 200 ohms resistance (optimal duration: 3.8-4.2 ms).
- Place the dried cuvette into the electroporator holder.
- Transfer the mixture gently (without bubbles) into the cuvette and tap the cuvette (avoid air bubbles that lead to arcing!).
- Tightly close the holder, put the lid on the cuvette.
- Electroporation: press both buttons on the power supply at once, release once beeping starts (usually 3.8-4.2 ms).
- Immediately rescue the electroporated cells by rinsing in 2x 1 ml pre-warmed S.O.C. medium. Any delay will significantly decrease the electroporation efficiency.
- Incubate Erlenmeyer flask (with 20 ml S.O.C. medium and electroporated bacteria) for 30 min (37 °C, 140 rpm).
- Remove 40 μl of bacterial culture, keep for preparing serial dilutions.
Note: In a 96-well plate prepare 3 columns (i.e., 3 x 8 wells) with 90 μl 1x PBS per library. Add 10 μl of bacterial culture into the first 90 μl 1x PBS (prepare in triplicate), use it for subsequent serial dilutions (1:10). Plate 5 μl of diluted bacteria on Amp* LB plate, incubate at 37 °C overnight. Calculate library diversity as follows: count colonies in a dilution where separate colonies are nicely visible and calculate the mean number of colonies per 1 μl diluted bacterial culture. Knowing the final volume of the bacterial culture (20.4 ml) and dilution from which colonies were counted (for example, lane 3 dilution in a 96-well plate corresponds to 103 dilution), calculate the library diversity: Diversity = (number of bacterial colonies in 1 μl diluted bacterial culture) x 10(dilution) x bacterial culture volume (μl). - Transfer the remaining content of the small Erlenmeyer flask into 180 ml of pre-warmed 2x YT medium (with 100 μg/ml Amp*), shake overnight (37 °C, 140 rpm).
- Next day prepare a mini-prep from 5 ml of the overnight culture and use it to check library quality (see Steps G1-G3).
- Spin the remaining culture down (4 °C, 6,000 x g, 20 min) for subsequent midi-prep.
- Analysis of the pre-library by restriction enzymes
- Digest 0.5 μg pre-library dsDNA with the appropriate restriction enzymes; always use original vector as cleavage control as well.
- Load undigested and digested pre-library and original vector on 0.5-0.8% 1x TAE agarose gel.
- Check the quality of the pre-library and compare it with the original vector (Figure 4).
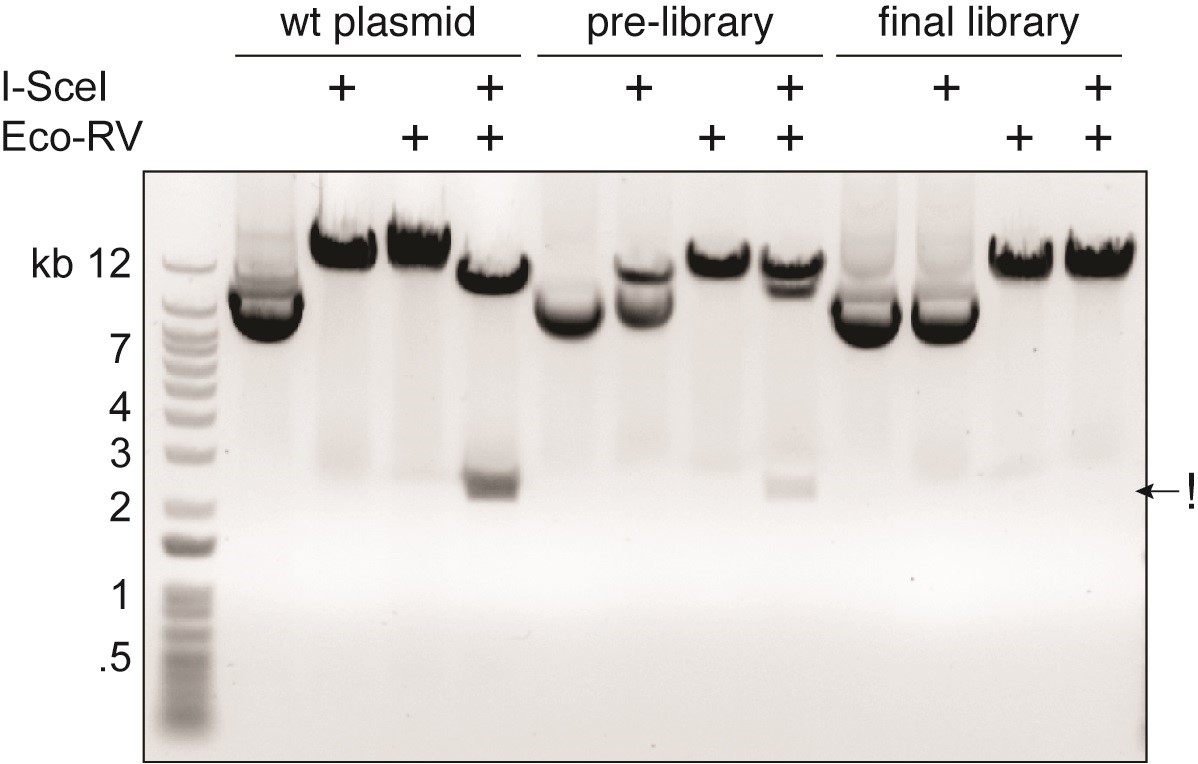
Figure 4. Removal of wild-type remnants from pre-library preparation and quality control of final library. Restriction enzyme digestion of the wild-type plasmid, the pre-library, and the final library indicates the ratio of wild-type and 3Cs plasmids at each step. As control, the wild-type plasmid (wt plasmid) is linearized with either I-SceI or EcoRV. A double digest with both enzymes removes a fragment of approximately 2,500 bp as both enzymes cut the plasmid (!). In the pre-library pool, the incomplete digestion with I-SceI and the faint band resulting from the double-digest indicate the majority of DNA being 3Cs products. The double digest reveals a band at ~2,500 bp with a significantly lower intensity than in the wild-type plasmid pool. In the final library, the I-SceI digest reveals no visible linearized fragment. The double digest linearizes the library and shows no visible band at ~2,500 bp, indicating the final library to be free of wild-type plasmid. Electrophoresis was run on 0.8% 1x TAE agarose for 30 min at 110 V.
- Digestion of pre-library library prior final electroporation
- Digest 10-20 μg of the pre-library with the appropriate enzyme and purify by Clean & Concentrator Kit.
- Transform and process 5-10 μg of the cleaved pre-library into 400 μl electrocompetent bacteria as described under Steps F1-F12.
- Grow 200-1,000 ml of 2x YT-Amp* (100 µg/ml) bacterial culture (volume depends on library size, future needs and amount of remaining oligonucleotides) at 37 °C overnight.
- Next day spin down bacteria (prepare multiple aliquots of bacterial pellets from either 50, 100 or 150 ml bacterial culture, depending on the maxi-prep kit that you are using and size of your library).
- Prepare a maxi-prep from one bacterial pellet, keep the remaining pellets at -20 °C until needed.
- Analysis of the final library by restriction enzymes
Follow Steps G1-G3 (Figure 4). - Sequence analysis of the final library by Sanger sequencing
Send appropriate amount (usually 700-1,200 ng) of the final library for Sanger sequencing. Choose a sequencing primer that is near your gRNA cassette, for example U6 primer. Representative data is presented in Figure 5.
Figure 5. SANGER sequencing of final library. As a first quality control step, SANGER sequencing of the final library with the U6-primer reveals randomization at the I-SceI placeholder side. In this example, digestion of the final optimized truly genome-wide library (oTGW) from Wegner et al. (2019) results in a randomized nucleotide pattern resembling the intended sequence composition of that library. - Sequence analysis of the final library guide distribution by next-generation sequencing (NGS)
For a detailed description of data processing and data analysis we refer to Wegner et al., 2019, a brief overview of the procedures is described below.
Data analysis
A detailed description of data processing and data analysis can be found in Wegner et al., 2019. To quality control 3Cs reagents, multiplexed Next Generation Sequencing (NGS) of several different plasmid libraries is possible when using barcoded Illumina adapter sequences. Raw NGS data can be demultiplexed using bCl2fastq from Illumina, preferably in the most recent version. To count individual reads of a library, use a designated tool like PinAPL-Py (Spahn et al., 2017) or design a customized pipeline that performs read trimming and sequence alignments according to your needs using, e.g., cutadapt and bowtie2.
Adapter trimming can be performed using cutadapt by trimming the read to the 20 gRNA-encoding nucleotides (Martin, 2011, https://cutadapt.readthedocs.io/en/stable/). Alignment of the trimmed reads against the library can then be performed with bowtie2 or custom scripts. The final library should contain all desired gRNA sequences. To assess the distribution of the library, generate Lorenz curves of gRNA representation and compute the area under the resulting curve according to (Wegner et al., 2019) (Figure 6A). This will allow you to compare different libraries. A histogram of the frequencies of the gRNA reads and calculating the ratio between the 90% and 10% percentiles reveals the skew of a library (Figure 6B). For the analyses of gene perturbation screens, we refer to the relevant literature as the experimental settings and read out determine the analyses techniques.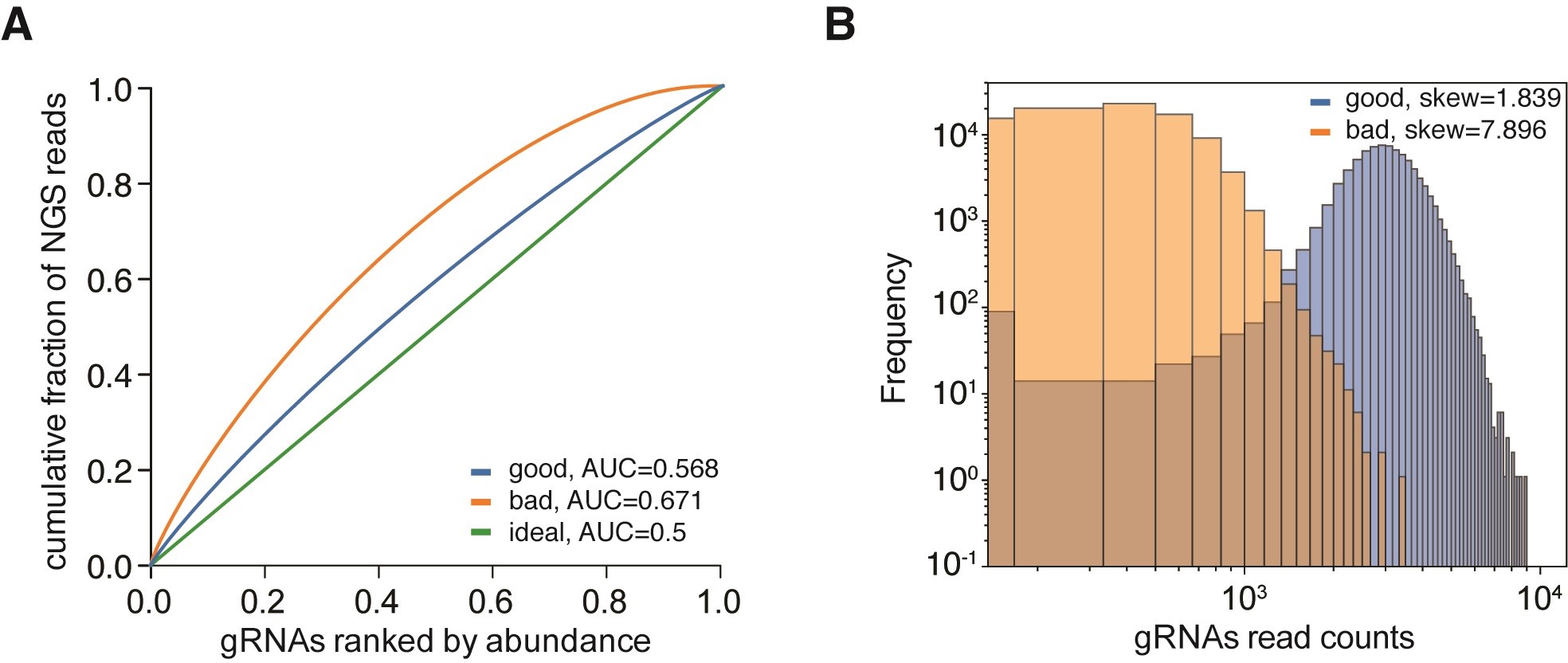
Figure 6. NGS analysis and visualization of expected sequence distributions. A. gRNA counts of three gRNA libraries are visualized as Lorenz plots, displaying the cumulative fraction of represented NGS reads against the gRNAs ranked by abundance. The ideal distribution would be uniform and resembles a diagonal line with an area under the curve (AUC) of 0.5 (green curve, ideal). A well distributed library contains every intended gRNA sequence and exhibits an AUC that differs only slightly from 0.5 (blue curve, good). An increased AUC value indicates an unevenly distributed library (orange curve, bad). Note that the slope of this curve decreases to 0 near x = 1.0, indicating that there are gRNA sequences missing in that library. B. Plotting the gRNA distributions from (A) as histograms visualizes the skew or symmetry of a library. A well distributed library exhibits a low skew near 1 (blue, good), indicating symmetry. A skewed library is asymmetrically distributed. The bad example is skewed towards higher gRNA read counts as revealed by the long right tail in the histogram (orange, bad).
Notes
- Oligonucleotides are usually 55-65 nucleotides in length, having a gRNA-encoding region in the middle, and plasmid sequence homology regions (3Cs homology) at its 5′ and 3′ ends. Homology of 18 nucleotides (on each side of the gRNA) and annealing temperature above 45 °C are optional for 3Cs reaction. The oligonucleotides should not have any modifications at either end as the efficiency of the ligation will decrease.
Important: The template plasmid must contain an f1-origin of replication. The E. coli CJ236 strain is Cmp-resistant and dut-/ung-. - Phage suspended in PBS can be stored for months at 4 °C.
- Once isolated, ssDNA is stable up to a few days or weeks. We recommend using freshly prepared ssDNA.
- To confirm the quality of your ssDNA, you can mix 1 μl of phage prep with KCM-competent bacteria and 200 μl S.O.C. medium. Incubate at 37 °C for 1 h and then dilute to 5 ml LB medium complemented with Amp* (100 μg/ml). Shake overnight (37 °C, 200 rpm), isolate dsDNA using a mini-prep kit and analyse dsDNA by using the same approach as in Procedure (Steps G1-G3). Use original plasmid dsDNA as positive control (compare size and restriction pattern in agarose gel).
- Use only freshly phosphorylated oligonucleotides for 3C reaction.
- High-quality electrocompetent E. coli strain SS320 or NEB 10-beta bacteria can also be prepared in the lab.
- The removal of dU-containing DNA in dut+/ung+ E. coli is not 100% efficient, therefore, it can be beneficial to introduce restriction sites (such as I-SceI) in the area of plasmid where the gRNAs will be introduced (gRNA placeholder sequence). Such an approach ensures that all the original sequences (still containing I-SceI site) will be depleted upon pre-library cleavage by I-SceI and subsequent electroporation. Importantly, make sure that the restriction recognition sequence does not resemble an active gRNA sequence in the genome of interest to avoid unintended editing events. A guide sequence containing an I-SceI restriction site is safe to use in the human genome. Also ensure that the restriction site is not a part of any of the gRNA sequences in the library to avoid unintended removal of a gRNA from the final pool.
- For pLentiGuide (Addgene 52963) and pLentiCRISPRv2 (Addgene 52961) plasmids, use a forward primer (5′-GGGCCTATTTCCCATGATTCCTTCATATTTGC-3′) that binds in the U6-promoter for Sanger sequencing.
Recipes
- KCM buffer
Note: KCM renders the dut-/ung- E. coli CJ236 cells competent.
0.5 M KCl
0.15 M CaCl2
0.25 M MgCl2 - 20% PEG/NaCl
Note: PEG/NaCl is used to precipitate phage particles containing dU-ssDNA.
20 % polyethylene glycol
2.5 M NaCl - 10x TM buffer
Note: Serves together with ATP and DTT as a reaction buffer for polynucleotide kinase T4 in the phosphorylation step of the 3Cs protocol.
0.1 M MgCl2
0.5 M Tris-HCl
pH 7.5 - 50x TAE buffer
2 M Tris base
0.5 M EDTA
1 M acetic acid
Acknowledgments
We thank Alkmini Kalousi for providing images related to bacteria and phage pellets. We further thank all members of the Frankfurt CRISPR/Cas Screening Center (FCSC) and the Kaulich laboratory who contributed to the original research paper from which this protocol is derived from (Wegner et al., 2019). This work was supported by the Hessian Ministry for Science and the Arts (HMWK, LOEWE-CGT, IIIL5-518/17.004), the German Research Foundation (DFG; CEF-MC - EXC115/2; ECCPS - EXC147/2) and in part by the LOEWE Center Frankfurt Cancer Institute (FCI) funded by the Hessen State Ministry for Higher Education, Research and the Arts (IIIL5-519/03/03.001-(0015)).
Competing interests
The Goethe University Frankfurt has filed a patent application related to this work on which Martin Wegner and Manuel Kaulich are inventors (WO2017EP84625). The Goethe University provides an exclusive license of the 3Cs technology to Vivlion GmbH for which Manuel Kaulich is co-founder, shareholder and chief scientific officer.
References
- Arakawa, H. (2016). A method to convert mRNA into a gRNA library for CRISPR/Cas9 editing of any organism. Sci Adv 2(8): e1600699.
- Doench, J. G. (2018). Am I ready for CRISPR? A user's guide to genetic screens. Nat Rev Genet 19(2): 67-80.
- Koike-Yusa, H., Li, Y., Tan, E. P., Velasco-Herrera Mdel, C. and Yusa, K. (2014). Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library. Nat Biotechnol 32(3): 267-273.
- Langmead, B. and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9(4): 357-359.
- Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 17(1): 10-12.
- Ong, S. H., Li, Y., Koike-Yusa, H. and Yusa, K. (2017). Optimised metrics for CRISPR-KO screens with second-generation gRNA libraries. Sci Rep 7(1): 7384.
- Sanson, K. R., Hanna, R. E., Hegde, M., Donovan, K. F., Strand, C., Sullender, M. E., Vaimberg, E. W., Goodale, A., Root, D. E., Piccioni, F. and Doench, J. G. (2018). Optimized libraries for CRISPR-Cas9 genetic screens with multiple modalities. Nat Commun 9(1): 5416.
- Schmidt, T., Schmid-Burgk, J. L. and Hornung, V. (2015). Synthesis of an arrayed sgRNA library targeting the human genome. Sci Rep 5: 14987.
- Shalem, O., Sanjana, N. E., Hartenian, E., Shi, X., Scott, D. A., Mikkelson, T., Heckl, D., Ebert, B. L., Root, D. E., Doench, J. G. and Zhang, F. (2014). Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343(6166): 84-87.
- Spahn, P. N., Bath, T., Weiss, R. J., Kim, J., Esko, J. D., Lewis, N. E. and Harismendy, O. (2017). PinAPL-Py: A comprehensive web-application for the analysis of CRISPR/Cas9 screens. Sci Rep 7(1): 15854.
- Vidigal, J. A. and Ventura, A. (2015). Rapid and efficient one-step generation of paired gRNA CRISPR-Cas9 libraries. Nat Commun 6: 8083.
- Wang, T., Wei, J. J., Sabatini, D. M. and Lander, E. S. (2014). Genetic screens in human cells using the CRISPR-Cas9 system. Science 343(6166): 80-84.
- Wegner, M., Diehl, V., Bittl, V., de Bruyn, R., Wiechmann, S., Matthess, Y., Hebel, M., Hayes, M. G., Schaubeck, S., Benner, C., Heinz, S., Bremm, A., Dikic, I., Ernst, A. and Kaulich, M. (2019). Circular synthesized CRISPR/Cas gRNAs for functional interrogations in the coding and noncoding genome. Elife 8: 42549.
Article Information
Copyright
![]() Wegner et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Wegner et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Wegner, M., Husnjak, K. and Kaulich, M. (2020). Unbiased and Tailored CRISPR/Cas gRNA Libraries by Synthesizing Covalently-closed-circular (3Cs) DNA. Bio-protocol 10(1): e3472. DOI: 10.21769/BioProtoc.3472.
- Wegner, M., Diehl, V., Bittl, V., de Bruyn, R., Wiechmann, S., Matthess, Y., Hebel, M., Hayes, M. G., Schaubeck, S., Benner, C., Heinz, S., Bremm, A., Dikic, I., Ernst, A. and Kaulich, M. (2019). Circular synthesized CRISPR/Cas gRNAs for functional interrogations in the coding and noncoding genome. Elife 8: 42549.
Category
Microbiology > Microbial genetics > CRISPR-Cas9
Molecular Biology > DNA > DNA synthesis
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.