- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Protocol for Ribosome Profiling in Bacteria
Published: Vol 9, Iss 24, Dec 20, 2019 DOI: 10.21769/BioProtoc.3468 Views: 10322
Reviewed by: Miao HeYueqiang LengAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Feb 2019
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Ribosome profiling provides information on the position of ribosomes on mRNA on a genomic scale. Although this information is often used to detect changes in gene expression under different conditions, it also has great potential for yielding insight into the mechanism and regulation of protein synthesis itself. First developed in yeast, ribosome profiling involves the isolation and sequencing of ribosome-protected mRNA fragments generated by nuclease treatment. Since the application of ribosome profiling in bacteria has been problematic, we report here a systematically optimized protocol for E. coli that we have used with success for other bacteria as well. Cells are harvested by flash-freezing cultures directly in liquid nitrogen. After lysis, translation is arrested by high magnesium concentration without the use of antibiotics. These improvements eliminate artifacts induced by harvesting cells by centrifugation or filtration and by use of chloramphenicol to arrest translation. These improvements are especially appropriate for studies where the exact position of the ribosome is critical, and not merely the number of ribosomes per message, such as studies aimed at monitoring differences in local elongation rates.
Keywords: Ribosome profilingBackground
mRNAs are translated at varying efficiencies depending on properties inherent to the mRNA such as initiation rates, mRNA structure, and codon usage. In addition, physiological changes in the cell can alter how efficiently mRNAs are translated. Ribosome profiling was developed to monitor gene expression at the level of protein synthesis, making it possible to ask global questions about which regions of the genome are translated, when they are translated, and how translation is regulated. First developed by Ingolia and Weissman in yeast (Ingolia et al., 2009), ribosome profiling utilizes nuclease digestion to degrade regions of mRNA that are not occupied by ribosomes, generating ribosome-protected mRNA fragments that are isolated, cloned, and sequenced. When the reads are mapped back to the genome, they reveal the position of ribosomes throughout the transcriptome, providing a powerful tool to monitor translation and its regulation in vivo. Our focus here is the application of ribosome profiling to bacteria, first reported by Weissman and Bukau (Oh et al., 2011; Li et al., 2012; Becker et al., 2013). Our protocol is derived from these pioneering studies and incorporates recent improvements described by Ingolia for yeast and mammalian cells (McGlincy and Ingolia, 2017).
Ribosome profiling has been used in bacteria to identify which regions of the genome are translated, revealing novel small proteins and internal initiation sites in annotated ORFs (Shell et al., 2015; Meydan et al., 2019; Weaver et al., 2019). Many studies report changes in gene expression that occur upon a change in the environment, the imposition of an external stress, or the depletion of a regulatory factor (Liu et al., 2013; Subramaniam et al., 2013; Guo et al., 2014; Haft et al., 2014; Zhang et al., 2018). These studies are powerful because in exponentially growing bacteria the level of translation correlates strongly with steady-state protein levels (Li et al., 2014). When supplemented with sequencing of total RNA by conventional RNA-seq, ribosome profiling data provide estimates of translational efficiency (the number of ribosomes per message). These estimates have yielded important conclusions about how the translation of proteins that form a complex occurs in the correct stoichiometry, even from a single mRNA (Li et al., 2014; Lalanne et al., 2018), and identified mRNA structure as one of the primary determinates of translational efficiency (Burkhardt et al., 2017).
In addition to these studies of gene expression, ribosome profiling can also be used to probe features of the translational machinery and the distinct phases of translation. Mechanisms of action of elongation inhibitors can be studied in vivo at codon resolution (Kannan et al., 2014; Marks et al., 2016). Factors involved in regulating translation can also be studied in detail using profiling (Balakrishnan et al., 2014; Elgamal et al., 2014; Woolstenhulme et al., 2015; Baggett et al., 2017). Another successful approach has been immunoprecipitation of factors of interest that associate with ribosomes, followed by ribosome profiling to determine where on mRNAs these factors are engaged (Oh et al., 2011). This approach has led to insights into protein folding and complex-formation during translation.
We are primarily interested in using ribosome profiling to measure local elongation rates which vary as a result of ribosome pausing at mRNA structures, rare codons, or troublesome nascent peptide sequences. Sites where ribosomes pause are expected to yield a higher number of reads than sites where elongation is fast, leading to enriched ribosome density. For these studies which rely on high resolution, it is important that the procedure to capture ribosome footprints truly reflects ribosomes in their native environment.
In our efforts to optimize the protocol for bacteria, where no standard protocol has emerged, we identified artifacts that interfere with capturing the native distribution of ribosome footprints (Mohammad et al., 2019). First, ribosome footprints in bacteria vary widely in size from as short as 15 nt to as long as 40 nt. It is essential to capture all of these footprints to have an accurate view of translation. Second, the way that cells are harvested can alter translation and induce artifacts: addition of chloramphenicol before centrifugation induces strong pauses and leads to pile-ups of ribosomes at the 5’-end of ORFs. Harvesting by filtration also induces pauses at specific codons due to rapid depletion of aminoacyl-tRNAs. Third, translation continues after cell lysis, even in the presence of chloramphenicol. To address these problems, we developed a method of directly freezing the culture and immediately inhibiting translation with a high concentration of MgCl2. These improvements capture ribosomes as they were under growth conditions and will enable studies to more accurately detect ribosome position in vivo. For studies primarily concerned with the number of ribosomes per message (and not their exact position) we also provide the earlier filtration protocol which is simpler and useful for preparation of samples for RNA-seq.
Materials and Reagents
- Whatman cellulose nitrate 0.45 μm filters (VWR, catalog number: 7184-009)
- Corning® Costar® Spin-X® centrifuge tube filters (Sigma-Aldrich, catalog number: CLS8163-100EA)
- Corning® Costar Cell Lifter (plastic spatula) (Sigma-Aldrich, catalog number: CLS3008)
- 3M Overhead Sheets (PP2500)
- Razor blades
- Saran wrap
- 50 ml conical tube
- Ti 70 polycarbonate tube
- 18 G needle
- P1000 tips
- Aluminum foil
- CircLigaseTM ssDNA Ligase (Epicentre, catalog number: CL4115K)
- SuperScript® III Reverse Transcriptase (Invitrogen, catalog number: 18080-044)
- T4 RNA Ligase 2, truncated (NEB, catalog number: M0242L)
- T4 Polynucleotide Kinase (PNK) (NEB, catalog number: M0201L)
- Ambion SUPERase·InTM (Invitrogen Life Technologies, catalog number: AM2696)
- Nuclease S7 Micrococcal nuclease (MNase) (Roche, catalog number: 10107921001)
- RNase Free DNase I (Roche, catalog number: 04716728001)
- Phusion® High-Fidelity DNA Polymerase (NEB, catalog number: M0530L)
- O’RangeRuler 10 bp Ladder (ThermoFisher, catalog number: SM1313)
- Primers
- Universal miRNA cloning linker (New England BioLabs, catalog number: S1315S)
- Oligonucleotides from IDT:
Size control 15 mer (100 nmol) (5’-AUGUACACGGAGUCG)
Size control 45 mer (100 nmol):
(5’-AUGUACACGGAGUCGACCCGCAACGCGAUGUACACGGAGUCGACC) - RT primer NI-NI-9 (250 nmol, DNA oligo, 96 bases, PAGE purified Ultramer)
(/5Phos/AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGTAGATCTCGGTGGTCGC/iSP18/CACTCA/iSp18/TTCAGACGTGTGCTCTTCCGATCTATTGATGGTGCCTACAG-3) - PCR primer Forward (100 nmol; not gel purified):
(5’-AATGATACGGCGACCACCGAGATCTACAC) - PCR primer Reverse (100 nmol; not gel purified):
(5’-CAAGCAGAAGACGGCATACGAGATNNNNNNGTGACTGGAGTTCAGACGTGTGCTCTTCCG)
- Isopropanol
- Ethanol
- Dry ice
- EGTA
- MilliQ H2O
- Sodium acetate
- PEG 8000
- Chloroform
- NaOH
- MgCl2
- NH4Cl
- CaCl2
- Triton X-100
- NP-40
- SDS
- EDTA
- DTT
- NaOAc
- MOPS EZ Rich Defined Medium (Teknova, catalog number: M2105)
- Phenol, Saturated pH 4.5 (VWR, catalog number: 97064-714)
- Oligo Clean and Concentration kit (Zymo, catalog number: D4061)
- Ribo-ZeroTM Magnetic Kit (Gram-Negative Bacteria) (Epicentre, catalog number: MRZGN126)
- Agilent High Sensitivity DNA Kit (Bio Analyzer) (Agilent Technologies, catalog number: 5067-4626)
- GlycoBlue (Invitrogen Life Technologies, catalog number: AM9516)
- SYBR® Gold Nucleic Acid Gel Stain (Invitrogen Life Technologies, catalog number: S-11494)
- 1 M Tris, pH 8 (Life Technologies, catalog number: AM9856)
- D-Sucrose (Fisher, catalog number: BP220-1)
- 15% TBE Urea 1.0 mm (12 + 2) 45 µl gel (Bio-Rad, catalog number: 345-0091) (2-3 per experiment)
- 10% TBE Urea 1.0 mm (12 + 2) 45 µl gel (Bio-Rad, catalog number: 345-0088) (2-3 per experiment)
- 10% TBE Urea 1.0 mm (18 + 2) 30 µl gel (Bio-Rad, catalog number: 345-0089) (1 per experiment)
- 10% TBE Native 1.0 mm (12 + 2) 45 µl gel (Bio-Rad, catalog number: 345-0051) (4-6 per experiment)
- 10x Lysis Buffer (50 ml) (see Recipes)
- Pelleting Buffer (see Recipes)
- Resuspension Buffer (see Recipes)
- 10% Sucrose Gradient Buffer (see Recipes)
- 50% Sucrose Gradient Buffer (see Recipes)
- RNA Elution Buffer (50 ml) (see Recipes)
- DNA Elution Buffer (50 ml) (see Recipes)
Equipment
- Kontes 90 mm filtration apparatus
- Spex Sampleprep 6870 Freezer Mill
- Beckman Coulter Optima L-80 XP Ultracentrifuge
- SW41 Ti rotor
- Type 70 Ti rotor
- Polyclear Centrifuge tubes (Seton, catalog number: 7030)
- Polycarbonate bottle with cap for Ti 70
- Biocomp Gradient Maker and Fractionator
- Thermocycler
- Eppendorf Thermomixer R
- GE Typhoon Imager
- Agilent 2100 BioAnalyzer
Software
- Analysis of sequenced ribosome footprints was done using a custom python pipeline found in the link below: https://github.com/greenlabjhmi/2018_Bacterial_Pipeline_riboseq
Procedure
This protocol details every step of ribosome profiling from culturing cells up to PCR amplification of ribosome footprints in preparation for next-generation sequencing (NGS). A general outline of the workflow is described below:
Day 1
•Growth of bacterial cultures
•Harvest by whole culture freeze
•Harvest by filtration (for total RNA extraction for RNA-seq)
•Cell lysis by freezer mill
Day 2
•Pellet ribosomes over sucrose cushion
•Nuclease digestion
•Sucrose gradient and monosome isolation
Day 3
•Phenol chloroform extraction
•Size selection gel
Day 4 (can split into 2 days)
•Gel extraction
•Dephosphorylation
•Linker ligation
•rRNA depletion
•Reverse transcription
•RT gel
Day 5
•RT gel extraction
•Circularization
•Pilot PCR
•Pilot PCR gel
•Preparative PCR
•Preparative PCR gel
Day 6
•Preparative PCR gel extraction
•Sequencing and data analyses
Day 1
- Growth of bacterial cultures
Bacterial cultures should be grown in the appropriate medium and growth conditions. When preparing cultures, consider the volume necessary to obtain around 1 mg of total RNA. The following conditions are used for E. coli during exponential growth:- Inoculate an overnight starter culture (25 ml of MOPS EZ Rich media) from a single colony.
- The next day, prepare 300 ml of MOPS EZ Rich media and warm at 37 °C for 1 h prior to inoculation.
- Inoculate 300 ml of MOPS media with 1 ml of overnight culture.
- Grow to an OD600 = 0.3-0.5 (~2 h).
- Harvest by whole culture freeze
This method of harvesting captures ribosomes without inducing artifacts from stress seen from harvesting by filtration or centrifugation. Under the growth conditions above, only 100 ml of the 300 ml will be flash frozen. The rest can be filter harvested to collect RNA for RNA-seq or frozen as backup.- While cells are growing, prepare a liquid nitrogen bath:
- Line an ice bucket with 2-3 layers of aluminum foil (see Figure 1A).
- Fill bucket halfway with liquid nitrogen.
- When cells are at the correct OD600, use a 50 ml serological pipet and spray 50 ml of the culture directly into the liquid nitrogen bath. Make sure not to spray in one area. Instead make circles in the N2 (see Figures 1B and 1C).

Figure 1. Liquid nitrogen bath used to freeze cultures. A. Line a standard lab ice bucket with 2-3 layers of aluminum foil. B. Add 2 inches of liquid nitrogen, then drip 100 ml of the bacterial culture into the liquid nitrogen. C. Drips should form ice pellets about 0.25 to 0.5 inches in diameter. - Repeat Step B2 to collect a total of 100 ml of culture.
- If collecting cells for RNA-seq, immediately run the rest of the culture (200 ml) through the filtration harvest protocol below to collect cell pellets.
- Lift the aluminum foil and pour off the liquid nitrogen from the sample. Scrape the frozen pellets into several 50 ml conical tubes on dry ice. Avoid thawing.
- While cells are growing, prepare a liquid nitrogen bath:
- Harvest by filtration (for total RNA extraction for RNA-seq)
Note: This step is needed to create a sample matched RNA-seq library and can be skipped if RNA-seq is not necessary.- Set up the Kontes 90 mm filtration apparatus with Whatman cellulose nitrate 0.45 µm filters during cell growth.
- Pour ~200 ml of culture into the Kontes 90 mm filtration apparatus and turn on the vacuum.
- While the media is filtering, gently but quickly scrape with a plastic spatula along the surface until you get a good clump of cells. Freeze in liquid nitrogen.
Note: You should be able to do this twice in about 60 s. The sample should never go dry–the dryness and cold that comes with losing all the media stresses the cells. - Place pellets in a 50 ml conical tube and store at -80 °C, do not allow any thawing.
- RNA can be extracted using RNA extraction kits or phenol extraction from the cell pellet.
- Extracted RNA can then be used to crate RNA-seq using commercially available kits.
- Cell lysis by freezer mill
Lysis is done through mechanical grinding of the frozen culture. The lysis buffer used contains 150 mM MgCl2 to inhibit translation in the lysate.- Pre-Chill Mixer Mill grinding cylinders and 3 x 50 ml conical tubes on dry ice.
- Measure 50 g of frozen culture, making sure to avoid thawing. Add 5.5 ml of 10x lysis buffer to large grinding cylinder and allow it to freeze.
- Transfer 50 g frozen culture into the grinding cylinder.
- Grind using the following cycles: 10 cycles, 10 Hz, 5 min precool, 1 min run, 1 min cool.
Note: The grinding protocol used here was optimized to maximize RNA yield while maintaining polysome integrity visualized by sucrose gradient sedimentation. We recommend revalidating grinding settings if using a different model Mixer Mill or a different organism. - Transfer the pulverized cells into the pre-chilled 50 ml conical tubes.
- Store at -80 °C for later use, or continue.
Day 2
- Pelleting ribosomes over sucrose cushion
- Thaw lysate on benchtop until thawed (1-2 h, mixing every 10 min). Place on ice immediately after thawed.
- Pre-clear lysate by spinning for 10 min at 9,000 x g at 4 °C to remove cellular debris.
- Prepare two sucrose cushions per sample:
- Transfer 3 ml of pre-chilled pelleting buffer into a Ti 70 polycarbonate tube x 2.
- Layer 22 ml lysate gently on top of cushion x 2.
- Spin at 370,000 x g (60,000 rpm) for 1.5 h at 4 °C in a Ti 70 rotor to pellet ribosomes.
- Remove supernatant from the ribosome pellet and rinse each pellet with 150 µl of pre-chilled Resuspension Buffer without disturbing pellet.
- To resuspended pellet:
- Add 100 µl of Resuspension Buffer to each pellet.
- Gently vortex for 5 min, making sure the ribosome pellet is fully dissolved.
- Combine the 2 tubes for a total of 200 µl resuspended pellet.
- Measure A260 of a 1:100 dilution using water.
- Calculate the amount needed for 12.5 AU (roughly 0.5 mg RNA), where AU = 1 ml solution with a given A260.
Note: If sample is less than 12.5 AU, lyse and pellet the other 50 ml of frozen culture. Resuspend with the first 200 µl collected. As little as 5 AU can be used for library preparation, but this is not recommended.- 12.5 AU/(A260 * 100/1,000) = µl needed (this takes into account the 100-fold dilution)
- Use the calculation above to calculate the volume needed for 12.5 AU, and add Resuspension Buffer to bring volume to 200 µl.
- Freeze at -80 °C to store or continue.
- Nuclease digestion
Use 12.5 AU of sample in 200 µl that was pelleted for nuclease digestion.
Note: If sample is less than 12.5 AU, scale the MNase accordingly.- Add 6 µl of SUPERase-In.
- Add 2 µl of 375 U/µl MNase (total of 750 U).
- Incubate at 25 °C for 1 h shaking at 1,400 rpm (thermomixer).
- Add 2 µl of 0.5 M EGTA pH 8 to quench the reaction (EGTA chelates calcium to quench MNase, a calcium dependent enzyme).
- Return to ice until next step.
- Sucrose gradient and monosome isolation
- Fill SW41 Polyallomer tubes with 6 ml of 50% Sucrose Gradient Buffer and layer on top ~6 ml of 10% Sucrose Gradient Buffer (all the way to the top of the tube).
- Put on rubber caps carefully to allow any air bubbles to escape through the hole in the cap.
- Rotate the tubes on the Biocomp gradient maker using the following settings:
Time = 1:48 s
Angle = 81.5°
Speed = 17 rpm - Remove rubber caps and 150 µl off the top of the gradient to make room for the sample.
- Gently layer 200 µl of the sample on top of gradient and balance in rotor tubes (sample should be approximately 210 µl).
- Spin in ultracentrifuge at 201,000 x g (35,000 rpm) for 2.5 h at 4 °C using acceleration and deceleration setting of 7.
- Prepare sucrose gradient fraction collector following the manufacturer's instructions.
- Set fraction collection so that each fraction has approximately 350 µl.
- Fractionate the sucrose gradient. Keep ~3 monosome fractions (~1,050 µl) (Figure 2A).
- Freeze fractions in liquid nitrogen and store at -80 °C.
Day 3
- Phenol chloroform extraction
Note: Use screwcap tubes and pipette in the fume hood. Spin steps are done on a tabletop centrifuge at > 10,000 x g at room temperature (RT) unless otherwise specified.- Pre-warm 750 µl of phenol, saturated pH 4.5 at 65 °C in thermomixer.
- Add 57.1 µl of 20% SDS to 1 ml of sample.
- Add sample to heated phenol.
- Incubate and shake for 5 min at 65 °C at 1,400 rpm on the thermomixer. Occasionally pull the tubes during this period and vortex on a full speed bench vortexer.
- Chill samples on ice for 5 min.
- Spin for 2 min.
- Keep the bottom aqueous layer and add to a tube containing 700 µl of phenol, saturated pH 4.5 at room temperature.
Important: Due to the high concentration of sucrose, there is layer inversion and the aqueous layer is on the bottom. - Vortex for 5 min at room temperature.
- Spin for 2 min.
- Keep the bottom aqueous layer and add to 600 µl of chloroform.
- Vortex for 30 s.
Note: If the samples become cloudy, warm to ~30 °C. This is likely the SDS falling out of solution. - Spin for 2 min.
- Keep the top aqueous layer (~1 ml) and place in a 15 ml conical tube.
- Add 1 volume (1 ml) of MilliQ H2O to dilute the sucrose.
- Add 3 M of sodium acetate (pH 5.5) to a final concentration of 0.3 M (~200 µl).
- Add an equal volume of isopropanol and mix well.
- Precipitate the RNA (30 min on dry ice, or overnight -20 °C).
- Centrifuge in Eppendorf 5804R at 12,000 x g (~9,000 rpm) for 30 min. Set temperature to 4 °C.
- Remove as much liquid as possible without disturbing the pellets.
- Air dry samples in the hood.
- Resuspend in 40 µl of MilliQ H2O or 10 mM Tris pH 8 and transfer to a 1.7 ml tube.
- Measure A260.
- You will run approximately 20 µg on the size selection gel.
- Store samples at -80 °C.
- Size selection gel
- Prepare samples:
20 µg Sample RNA + 2x RNA Loading Dye (NEB)
Ladder (oligos: 15 nt and 45 nt) = Mix 3 µl (10 µM stock) of each marker size + 6 µl 2x RNA Loading Dye per lane. Load 6 µl in one lane (enough for 2 gels) - Heat samples and ladder at 80 °C for 2 min, then place on ice.
- Prepare 15% TBE Urea 1.0 mm (12 + 2) 45 µl gel.
- Load samples carefully with at least one empty lane between samples.
- Run gel(s) until bromophenol blue is ¾ down the gel.
If running at 15 W:
1 gel = 17 min
2 gels = 30 min - Remove gels from plates and place in SYBR-gold stain (100 ml of 1x TBE + 10 µl of SYBR-gold) for 5 min.
- Remove from stain and place on transparency film, then cover with saran wrap.
- Scan on the GE Typhoon Imager using the fluorescence setting for SYBR Gold. Use a 350-400 PMT.
- Print the gel as actual size and use printout as a cutting guide to cut sections out of the gel. Cut from the top of the upper marker (45 nt) to bottom of lower marker (15 nt) (Figure 2B).
- Cut out 15 nt and 45 nt markers and treat as a control sample. This can be used to assess proper ligation and reverse transcription later on.
- Rescan gel to confirm proper cutting.
- For elution, crush gel by doing the following:
- Place gel cutout into a 0.5 ml tube with holes poked through the bottom (18 G needle).
- Place the 0.5 ml tube in a 1.7 ml tube to collect the gel.
- Spin for 5 min, gel should extrude from the 0.5 ml tube into the bottom 1.7 ml tube.
- Make sure to collect any remaining gel from the 0.5 ml tube.
- Prepare samples:
- Gel extraction
- Add 500 µl of RNA Elution Buffer to each sample.
- Add 2.5 µl of SUPERase-Inhibitor to each sample.
- Shake overnight in a thermomixer, 1,400 rpm at 4 °C.
- The next day, briefly spin samples.
- Transfer gel slurry into Spin-X columns (using P1000 tips with the ends cut off).
- Spin for 3 min at 20,000 x g at room temperature.
- Put eluate into a new 1.7 ml tube.
- Add 2 µl of GlycolBlue to each sample.
- Add 650 µl of isopropanol.
- Precipitate (40 min–dry ice), spin for 30 min at > 10,000 x g, 4 °C.
- Remove supernatant, and gently wash with ~100 µl of 80% cold ethanol.
- Dry pellets by speed vac (10 min).
- Resuspend pellet in 5 µl of MilliQ H2O.
- Dephosphorylation
Reaction Mix (per sample)
1 µl of 10x PNK buffer
1 µl of SUPERase-In
2 µl of MilliQ H2O- Heat samples collected from J13 at 80 °C for 2 min and then place on ice.
- Add 4 µl of Reaction Mix to each sample.
- Add 1 µl of PNK to each sample (10K U/µl).
- Incubate at 37 °C for 1 h.
- Linker ligation
Reaction Mix (per sample):
1 µl of 10x Ligation Buffer
0.4 µl of 50 µM Universal miRNA cloning linker
7.6 µl of 50% PEG 8000
Note: Ligation buffer and PNK buffer are nearly identical, so half of the required ligation buffer is used.- Add 9 µl of Reaction Mix to each sample. Any white precipitate is assumed to be glycogen coming out as a result of the PEG.
- Add 1 µl of T4 RNA ligase 2 truncated to each sample.
- Incubate at 37 °C for 3 h.
- Clean up with oligo and concentration kit and elute in 15 µl of MilliQ H2O.
- rRNA depletion
Deplete rRNA for all samples except the 15 nt and 45 nt controls.- Follow Ribo-Zero kit instructions for rRNA depletion.
- Clean up with oligo and concentration kit and elute in 15 µl of MilliQ H2O.
- Reverse transcription (RT)
Reaction Mix (per sample):
4 µl of 5x RT buffer
1 µl of 10 mM dNTPs
1 µl of 0.1 M DTT
1 µl of SUPERase-In- Use 10.75 µl of sample for RT, save the other 4.25 µl.
- Add 1.25 µl of RT primer NI-NI-9 (10 µM stock) and heat sample to 80 °C for 2 min, then chill on ice.
- Add 7 µl of Reaction mix to each sample.
- Add 1 µl of Superscript III RT to each sample (Invitrogen 56575).
- Incubate at 48 °C for 30 min.
- Add 2.2 µl of 1 M NaOH to each sample.
- Heat to 98 °C for 20 min.
- Add 28 µl of MilliQ H2O.
- Clean up with oligo and concentration kit (Zymo) and elute in 10 µl of MilliQ H2O.
- RT Gel
- Prepare Samples:
10 µl of sample cDNA + 10 µl of 2x RNA Loading Dye
5 µl of 10 bp O'RangeRuler ladder
Note: The sample containing the 15 and 45 nt markers will be used to indicate proper ligation and reverse transcription, and act as size markers for gel excision. - Heat to 80 °C for 2 min and then put on ice.
- Prepare 10% TBE Urea 1.0 mm (18 + 2) 30 µl gel.
- Load samples carefully with at least one empty lane between samples.
- Run gel(s) until xylene cyanol is run off the gel.
If running at 15 W:
1 gel = 40 min
2 gels = 60 min - Remove gels from plates and place in SYBR-gold stain (100 ml 1x TBE + 10 µl SYBR-gold) for 5 min.
- Remove from stain and place on transparency film, then cover with saran wrap.
- Scan on the GE Typhoon Imager using the fluorescence setting for SYBR Gold. Use a 350-400 PMT.
- Print the gel as actual size and use printout as a cutting guide to cut sections out of the gel. Cut from the top of the upper marker (45 nt) to bottom of lower marker (15 nt) (Figure 2C).
Important: Avoid excess RT primer band (band below 15 nt marker, 110 nt in size determined by the O'RangeRuler ladder). - Rescan to confirm proper cutting.
- For elution, crush gel by doing the following:
- Place gel cutout into a 0.5 ml tube with holes poked through the bottom (18 G needle).
- Place the 0.5 ml tube in a 1.7 ml tube to collect the gel.
- Spin for 4 min, gel should extrude from the 0.5 ml tube into the bottom 1.7 ml tube.
- Prepare Samples:
- RT Gel Extraction
- Add 500 µl DNA Elution Buffer to each sample.
- Shake overnight in thermomixer 1,050 rpm at 25 °C (room temperature).
- The next day, briefly spin samples.
- Transfer gel slurry into Spin-X columns (using P1000 tips with the ends cut off).
- Spin for 3 min.
- Put eluate in new 1.7 ml tube.
- Add 2 µl of GlycoBlue to each sample.
- Add 650 µl of isopropanol.
- Precipitate (40 min–dry ice), spin for 30 min at > 10,000 x g, 4 °C.
- Remove supernatant, and gently wash with ~100 µl 80% cold ethanol.
- Dry pellets by speed vac (10 min).
- Resuspend pellets in 15 µl of 10 mM Tris pH 8.
- Circularization
Reaction mix (per sample):
2 µl of CircLigaseTM buffer
1 µl of 1 mM ATP (from CircLigaseTM Kit)
1 µl of 50 mM MnCl2 (from CircLigaseTM Kit)- Add 4 µl of Reaction Mix to each sample
- Add 1 µl of Circ ligase to each sample.
- Incubate at 60 °C for 60 min.
- Incubate at 80 °C for 10 min, then put on ice.
- Pilot PCR
The goal of the PCR step is to amplify the sample while minimizing PCR amplification bias and over-amplification. To estimate the cycles of PCR needed for proper amplification, several pilot cycles are run to visualize the point at which over-amplification occurs.
Reaction Mix (6 samples)
40 µl of HF Buffer (F-518)
4 µl of 10 mM dNTPs
1 µl of 100 µM PCR primer Forward
2 µl of Phusion polymerase (F-530)
133 µl of MilliQ H2O- Prepare Reaction mix.
- Combine in a 1.7 ml tube:
27 µl Reaction mix
1.5 µl barcoded PCR primer Reverse (10 µM)
1.5 µl Sample - Divide this into 3 x 9 µl aliquots into 0.5 ml tubes.
- Run on PCR protocol:
Step 1: 98 °C–30 s
Step 2: 98 °C–10 s
Step 3: 65 °C–10 s
Step 4: 72 °C–5 s
Step 5: go to step 2–8 times
Step 6: 4 °C–forever
End
Each sample will have 3 aliquots in the PCR machine. Take one aliquot from each sample out of the PCR machine at the end of 5, 7, and 9 PCR amplification cycles.
Notes:- Pause the machine at 72 °C, wait for 5 s.
- Quickly remove tubes and place directly on ice.
- Quickly close the lid and continue the program.
- Each sample will now have 3 PCR products made from 3 different PCR cycle numbers.
- Proceed to Pilot PCR gel.
- Pilot PCR gel
- Prepare Samples:
9 µl of PCR sample + 2 µl of 6x DNA Loading Dye
5 µl of 10 bp O'RangeRuler ladder - Prepare 10% TBE Native 1.0 mm (12 + 2) 45 µl gel.
- Load samples carefully, grouping each sample together with the cycle numbers in ascending order.
- Run gel(s) until xylene cyanol is run off the gel.
If running at 15 W:
1 gel = 40 min
2 gels = 60 min - Remove gels from plates and place in SYBR-gold stain (100 ml of 1x TBE + 10 µl of SYBR-gold) for 5 min.
- Remove from stain and place on transparency film, then cover with saran wrap.
- Scan on the GE typhoon Imager using the fluorescence setting for SYBR Gold. Use a 350-400 PMT (Figure 2D).
- Determine how many cycles are needed to obtain a good band around 150-190 bp without any larger molecular weight products.
Note: The ideal number of cycles gives you a nice product band but no trace of higher molecular weight products. Be very careful to avoid this because when you see higher molecular weight products, bias is likely being introduced. After determining the appropriate number of cycles from the Pilot PCR, run the Preparative PCR at 1 fewer cycles to ensure that no high weight bands (usually a smear) emerge. - Proceed to Preparative PCR.
- Prepare Samples:
- Preparative PCR
Determine the proper cycle count for each sample. Follow the same steps as the pilot PCR, but do not divide the reaction.- Combine in 1.7 ml tube:
27 µl of Reaction mix
1.5 µl of barcoded PCR primer (10 µM) (can use either Ni or IDX primers)
1.5 µl of Sample - Run PCR using the cycle count determined from the pilot PCR.
- Combine in 1.7 ml tube:
- Preparative PCR gel
- Prepare Samples:
30 µl of PCR sample + 6 µl of 6x DNA Loading Dye
5 µl of 10 bp O'RangeRuler ladder - Prepare 10% TBE Native 1.0 mm (12 + 2) 45 µl gel.
- Load samples carefully, leaving one lane empty between samples.
- Run gel(s) until xylene cyanol is run off the gel.
If running at 15 W:
1 gel = 40 min
2 gels = 60 min - Remove gels from plates and place in SYBR-gold stain (100 ml of 1x TBE + 10 µl of SYBR-gold) for 5 min.
- Remove from stain and place on transparency film, then cover with saran wrap.
- Scan on the GE Typhoon Imager using the fluorescence setting for SYBR Gold. Use a 350-400 PMT.
- Print the gel as actual size and use printout as a cutting guide to cut sections out of the gel. Cut from the 150 bp marker to the 200 bp marker (Figure 2E).
Important: Avoid the excess RT primer band (runs at 140 bp mark). - Rescan to confirm proper cutting.
- For elution, crush gel by doing the following:
- Place gel cutout into a 0.5 ml tube with holes poked through the bottom (18 G needle).
- Place the 0.5 ml tube in a 1.7 ml tube to collect the gel.
- Spin for 5 min, gel should extrude from the 0.5 ml tube into the bottom 1.7 ml tube.
- Prepare Samples:
Day 6
- Preparative PCR gel extraction
- Add 500 µl of DNA Elution Buffer to each sample.
- Shake overnight in thermomixer–1,050 rpm at 25 °C (room temperature).
- The next day, briefly spin samples.
- Transfer gel slurry into Spin-X columns (using P1000 tips with the ends cut off).
- Spin for 3 min at 20,000 x g at RT.
- Put eluate in new 1.7 ml tube.
- Add 2 µl of GlycoBlue to each sample.
- Add 650 µl of isopropanol.
- Precipitate (40 min–dry ice), spin for 30 min at > 10,000 x g, 4 °C.
- Remove supernatant, and gently wash with ~100 µl of 80% cold ethanol.
- Dry pellets by speed vac (10 min).
- Resuspend pellets in 10 µl of 10 mM Tris pH 8.
- Check size and concentration using a BioAnalyzer.
- Sequencing and data analyses
- Dilute an aliquot of each library to 2 nM and combine together to multiplex.
- Submit 10 µl of 2 nM multiplexed sample for 50 cycles, single-end sequencing on the Illumina HiSeq 2500.
- Process the sequencing data with custom python 2.7 scripts available here:
https://github.com/greenlabjhmi/2018_Bacterial_Pipeline_riboseq. - In broad strokes, the Jupyter notebook Github-Ribo_Density.ipynb.
- Remove the linker CTGTAGGCACCATCAATAGATCGGAAGAGCACACGTCTGAACTCCA GTCA from the 3’-end of reads using Skewer version 0.2.2.
- Use Bowtie version 0.12.7 to align reads to indices including ladder oligos, tRNA, and rRNA; reads that do not map to these indices are then mapped to the E. coli genome (NC_000913.2) allowing two mismatches and requiring unique mapping to a single site.
- Write out ribosome density as a WIG file, as a binary file, and a size-separated dictionary using the 3’-end of reads to assign ribosome position.
- The Jupyter notebook Github-Ribo-Analysis.ipynb provides further analyses including plots of average ribosome density at start or stop codons, the calculation of pause scores at specific codons, reading frame, and the degree of drop-off across genes (their asymmetry scores).
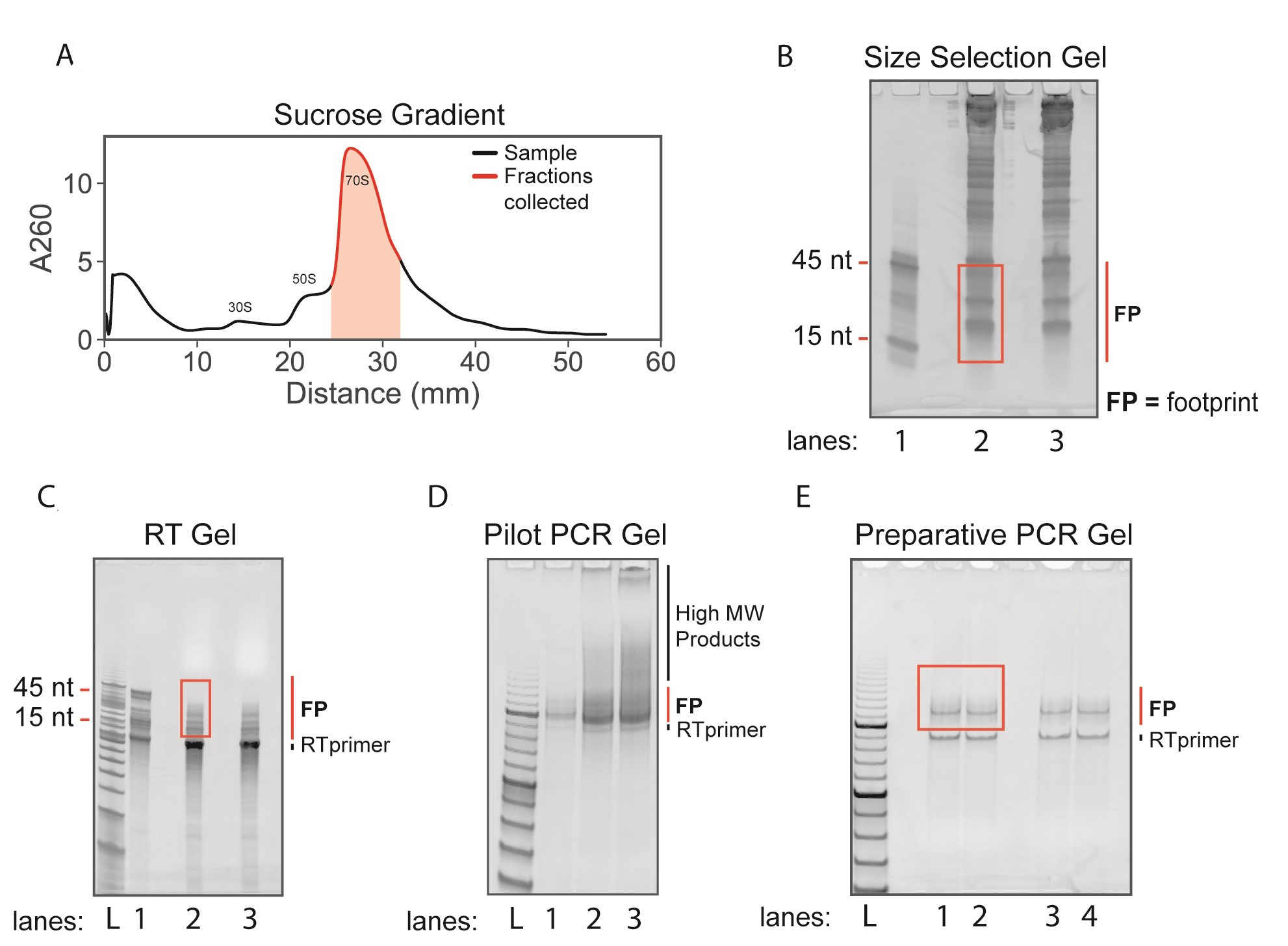
Figure 2. Ribosome profiling sucrose gradients and gels. A. Sucrose gradient trace from Step G9. A volume of ~1,050 µl was collected (highlighted in red) containing 70S ribosomes. B. Size selection gel from Step I9. Lane 1: 15 nt and 45 nt size markers. Lanes 2 and 3: RNA from fractionated monosomes. The red box indicates the excised region containing ribosome footprints (FP). C. Reverse transcription gel from Step O9. Lane 1: ligated and reverse transcribed 15 nt and 45 nt size markers. Lanes 2 and 3: ligated and reverse transcribed ribosome footprints. The red box indicates excised region which avoids the excess RT primer band. D. Pilot PCR gel from Step S7. Lanes 1-3: PCR amplified products after 5, 7, and 9 PCR cycles. Note the appearance of unwanted high molecular weight products after 7 and 9 PCR cycles. E. Preparative PCR gel from Step U8. The red box indicates excised region which avoids the RT primer band. FP = ribosome footprints. L = O’RangeRuler 10 bp ladder.
Recipes
Note: All concentrations listed are final concentrations.
- 10x Lysis Buffer (50 ml)
200 mM Tris pH 8
1.5 M MgCl2
1 M NH4Cl
50 mM CaCl2
4% Triton X-100
1% NP-40
MilliQ H2O - Pelleting Buffer
1.1 M Sucrose
20 mM Tris pH 8
500 mM NH4Cl
10 mM MgCl2
0.5 mM EDTA pH 8
MilliQ H2O - Resuspension Buffer
20 mM Tris pH 8
15 mM MgCl2
100 mM NH4Cl
5 mM CaCl2
MilliQ H2O - 10% Sucrose Gradient Buffer
Each sample uses 6 ml
20 mM Tris pH 8
15 mM MgCl2
100 mM NH4Cl
2 mM DTT
10% sucrose
MilliQ H2O - 50% Sucrose Gradient Buffer
Each sample uses 6 ml
20 mM Tris pH 8
15 mM MgCl2
100 mM NH4Cl
2 mM DTT
50% sucrose
MilliQ H2O - RNA Elution Buffer (50 ml)
300 mM NaOAc pH 5.5
1 mM EDTA pH 8
MilliQ H2O - DNA Elution Buffer (50 ml)
300 mM NaCl
1 mM EDTA pH 8
10 mM Tris pH 8
MilliQ H2O
Acknowledgments
Our work on ribosome profiling was supported by NIH grant GM110113. This protocol was adapted from the early bacterial profiling studies by Bukau and Weissman (Oh et al., 2011; Li et al., 2012; Becker et al., 2013) and incorporates recent improvements described by Ingolia for use in yeast and mammalian cells (McGlincy and Ingolia, 2017).
Competing interests
There are no conflicts of interest or competing interest.
References
- Baggett, N. E., Zhang, Y. and Gross, C. A. (2017). Global analysis of translation termination in E. coli. PLoS Genet 13(3): e1006676.
- Balakrishnan, R., Oman, K., Shoji, S., Bundschuh, R. and Fredrick, K. (2014). The conserved GTPase LepA contributes mainly to translation initiation in Escherichia coli. Nucleic Acids Res 42(21): 13370-13383.
- Becker, A. H., Oh, E., Weissman, J. S., Kramer, G. and Bukau, B. (2013). Selective ribosome profiling as a tool for studying the interaction of chaperones and targeting factors with nascent polypeptide chains and ribosomes. Nat Protoc 8(11): 2212-2239.
- Burkhardt, D. H., Rouskin, S., Zhang, Y., Li, G. W., Weissman, J. S. and Gross, C. A. (2017). Operon mRNAs are organized into ORF-centric structures that predict translation efficiency. Elife 6: e22037.
- Elgamal, S., Katz, A., Hersch, S. J., Newsom, D., White, P., Navarre, W. W. and Ibba, M. (2014). EF-P dependent pauses integrate proximal and distal signals during translation. PLoS Genet 10(8): e1004553.
- Guo, M. S., Updegrove, T. B., Gogol, E. B., Shabalina, S. A., Gross, C. A. and Storz, G. (2014). MicL, a new sigmaE-dependent sRNA, combats envelope stress by repressing synthesis of Lpp, the major outer membrane lipoprotein. Genes Dev 28(14): 1620-1634.
- Haft, R. J., Keating, D. H., Schwaegler, T., Schwalbach, M. S., Vinokur, J., Tremaine, M., Peters, J. M., Kotlajich, M. V., Pohlmann, E. L., Ong, I. M., Grass, J. A., Kiley, P. J. and Landick, R. (2014). Correcting direct effects of ethanol on translation and transcription machinery confers ethanol tolerance in bacteria. Proc Natl Acad Sci U S A 111(25): E2576-E2585.
- Ingolia, N. T., Ghaemmaghami, S., Newman, J. R. and Weissman, J. S. (2009). Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324(5924): 218-223.
- Kannan, K., Kanabar, P., Schryer, D., Florin, T., Oh, E., Bahroos, N., Tenson, T., Weissman, J. S. and Mankin, A. S. (2014). The general mode of translation inhibition by macrolide antibiotics. Proc Natl Acad Sci U S A 111(45): 15958-15963.
- Lalanne, J. B., Taggart, J. C., Guo, M. S., Herzel, L., Schieler, A. and Li, G. W. (2018). Evolutionary convergence of pathway-specific enzyme expression stoichiometry. Cell 173(3): 749-761 e38.
- Li, G. W., Burkhardt, D., Gross, C. and Weissman, J. S. (2014). Quantifying absolute protein synthesis rates reveals principles underlying allocation of cellular resources. Cell 157(3): 624-635.
- Li, G. W., Oh, E. and Weissman, J. S. (2012). The anti-Shine-Dalgarno sequence drives translational pausing and codon choice in bacteria. Nature 484(7395): 538-541.
- Liu, X., Jiang, H., Gu, Z. and Roberts, J. W. (2013). High-resolution view of bacteriophage lambda gene expression by ribosome profiling. Proc Natl Acad Sci U S A 110(29): 11928-11933.
- Marks, J., Kannan, K., Roncase, E. J., Klepacki, D., Kefi, A., Orelle, C., Vazquez-Laslop, N. and Mankin, A. S. (2016). Context-specific inhibition of translation by ribosomal antibiotics targeting the peptidyl transferase center. Proc Natl Acad Sci U S A 113(43): 12150-12155.
- McGlincy, N. J. and Ingolia, N. T. (2017). Transcriptome-wide measurement of translation by ribosome profiling. Methods 126: 112-129.
- Meydan, S., Marks, J., Klepacki, D., Sharma, V., Baranov, P. V., Firth, A. E., Margus, T., Kefi, A., Vazquez-Laslop, N. and Mankin, A. S. (2019). Retapamulin-Assisted Ribosome Profiling Reveals the Alternative Bacterial Proteome. Mol Cell 74(3):481-493.
- Mohammad, F., Green, R. and Buskirk, A. R. (2019). A systematically-revised ribosome profiling method for bacteria reveals pauses at single-codon resolution. Elife 8: 42591.
- Oh, E., Becker, A. H., Sandikci, A., Huber, D., Chaba, R., Gloge, F., Nichols, R. J., Typas, A., Gross, C. A., Kramer, G., Weissman, J. S. and Bukau, B. (2011). Selective ribosome profiling reveals the cotranslational chaperone action of trigger factor in vivo. Cell 147(6): 1295-1308.
- Shell, S. S., Wang, J., Lapierre, P., Mir, M., Chase, M. R., Pyle, M. M., Gawande, R., Ahmad, R., Sarracino, D. A., Ioerger, T. R., Fortune, S. M., Derbyshire, K. M., Wade, J. T. and Gray, T. A. (2015). Leaderless Transcripts and Small Proteins Are Common Features of the Mycobacterial Translational Landscape. PLoS Genet 11(11): e1005641.
- Subramaniam, A. R., Deloughery, A., Bradshaw, N., Chen, Y., O'Shea, E., Losick, R. and Chai, Y. (2013). A serine sensor for multicellularity in a bacterium. Elife 2: e01501.
- Weaver, J., Mohammad, F., Buskirk, A. R. and Storz, G. (2019). Identifying small proteins by ribosome profiling with stalled initiation complexes. MBio 10(2).
- Woolstenhulme, C. J., Guydosh, N. R., Green, R. and Buskirk, A. R. (2015). High-precision analysis of translational pausing by ribosome profiling in bacteria lacking EFP. Cell Rep 11(1): 13-21.
- Zhang, Y., Burkhardt, D. H., Rouskin, S., Li, G. W., Weissman, J. S. and Gross, C. A. (2018). A stress response that monitors and regulates mRNA structure is central to cold shock adaptation. Mol Cell 70(2): 274-286 e7.
Article Information
Copyright
![]() Mohammad and Buskirk. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Mohammad and Buskirk. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Mohammad, F. and Buskirk, A. R. (2019). Protocol for Ribosome Profiling in Bacteria. Bio-protocol 9(24): e3468. DOI: 10.21769/BioProtoc.3468.
- Mohammad, F., Green, R. and Buskirk, A. R. (2019). A systematically-revised ribosome profiling method for bacteria reveals pauses at single-codon resolution. Elife 8: 42591.
Category
Microbiology > Microbial genetics > Gene expression
Molecular Biology > RNA > mRNA translation
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.