- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Retina Injury and Retina Tissue Preparation to Study Regeneration in Zebrafish
Published: Vol 9, Iss 24, Dec 20, 2019 DOI: 10.21769/BioProtoc.3466 Views: 5344
Reviewed by: Subhra Prakash HuiNobuhiko TachibanaAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Feb 2019

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Unlike mammals, primitive vertebrates have immense capability to regenerate almost all of their organs including the central nervous system. Among primitive organisms, zebrafish have been extensively used as a model system for regeneration studies. The retina is a part of the central nervous system and mammals lack the potential to repair any damage caused to it. Zebrafish have been used for retina regeneration studies because of ease in handling and maintenance. In zebrafish, Muller glia cells respond to damage and enter the regenerative cascade to maintain the retinal homeostasis. Zebrafish retinal damage can be induced by light, chemical or mechanical methods. Here we are describing the mechanical method of retinal injury, which ensures uniform damage to all retinal layers. Alongside this, we have also described in vivo manipulation strategies for the regeneration associated genes and preparation of retinal tissue for immunohistochemical analysis.
Keywords: ZebrafishBackground
Retina is the sensory part of the eye and any physical or physiological damage leads to impairment of vision. In the course of evolution higher vertebrates have lost the regenerative potential while primitive vertebrates have enormous capability to regenerate their lost vision. Studying the regenerative events in primitive organisms such as zebrafish can be a hope for mammalian regeneration studies. Zebrafish have been used for retina regeneration studies with various injury paradigms being used. The photobleaching method damages the photoreceptor cells, while chemical methods damage the ganglion cell layer. Mechanical injury by the stab wound method maintains a uniform injury to all retinal layers. Various approaches including transgenic approaches have been used to find the relevance of genetic events during retina regeneration. Here we are describing the in vivo method for mRNA transfection which allows manipulation of gene expression levels above endogenous levels.
Materials and Reagents
- 30G needle (BD Microlance, 30G ½”, 0.3 x 13 mm, catalog number: 304000)
- Centrifuge tubes (Tarsons, catalog number: 500010x)
- Paper towels (Scott SCOTTFOLD Towels, 31.4 cm x 19.9 cm, catalog number: 01960)
- Hamilton syringe (Hamilton Company, 10 µl, catalog number: MICROLITERTM #701)
- Staining box (Custom made by fixing two plastic pipettes parallel to each other at the bottom of a rectangular plastic box)
- Glass slides (Superfrost Plus Microscope Slides) (Fisher Scientific, catalog number: 12-550-15)
- pCS2+ plasmid (David Turner, University of Michigan, Ann Arbor)
- Ethyl 3-aminobenzoate, methane sulfonic acid salt, Tricaine methanesulfonate (Acros, catalog number: 118000500)
- 2x HBSS (Diluted from 10x solution, see Recipes)
- mMESSAGE mMACHINE SP6 kit (Thermo Fisher, catalog number: AM1340)
- Lipofectamine messenger max reagent (Invitrogen, catalog number: LMRNA001)
- DABCO (1,4 diazabicyclo [2.2.2] octane, Sigma-Aldrich, catalog number: D27802)
- Na2HPO4 (Sodium phosphate dibasic, Sigma-Aldrich, catalog number: 255793)
- NaH2PO4 (Sodium phosphate monobasic, Sigma-Aldrich, catalog number: 33198)
- KCl (Potassium chloride, Himedia, catalog number: MB043)
- HEPES (Sigma-Aldrich, catalog number: H3375)
- Glucose (Sigma-Aldrich, catalog number: G8270)
- Tissue-Plus O.C.T. compound (Fisher Health care, catalog number: 4585)
- BSA (Bovine serum albumin fraction-V, Himedia, catalog number: GRM105)
- Triton X-100 (Sigma-Aldrich, catalog number: T8787)
- BrdU (5-Bromo-2’-deoxyuridine, Sigma-Aldrich, catalog number: B5002)
- PFA (Paraformaldehyde, Sigma-Aldrich, catalog number: P6148)
- PVA (Polyvinyl alcohol, Sigma-Aldrich, catalog number: P8136)
- Glycerol (Sigma-Aldrich, catalog number: G7757)
- Tris Base (Trizma base, Sigma-Aldrich, catalog number: T1503)
- HCl (Hydrochloric acid, Sigma-Aldrich, catalog number: 320331)
- Sucrose (Sigma-Aldrich, catalog number: S0389)
- Tris-HCl (1 M, pH 7.5, 100 ml) (see Recipes)
- Tricaine methanesulfonate solution (100 ml) (see Recipes)
- Phosphate buffer (PB, 1 M, pH 7.4, 100 ml) (see Recipes)
- Phosphate buffered saline (PBS, 10x, pH 7.4, 1 L) (see Recipes)
- HBSS (Hanks balanced salt solution, 10x, pH 7.14, 100 ml) (see Recipes)
- 4% PFA (50 ml) (see Recipes)
- DABCO (2.5%, 50 ml) (see Recipes)
- Sucrose solutions (50 ml) (see Recipes)
Equipment
- Forceps (McPherson Suture Tying Forceps Straight-With Tying Platform 10cm (4”) 5 mm size, jaw length, Surtex Instruments, catalog number: FR-780-10)
- Stereomicroscope (Carl ZeissTMStemiTM DV4 Series Stereomicroscopes with LED Illumination)
- Electroporator (Electro Square Porator, BTX Harvard Apparatus, model: ECM 830)
- Electrodes (Platinum Tweezertrode, 5 mm Diameter with 45-0204 cables, BTX, catalog number: 45-0489)
- Cryostat (Leica, model: CM3050 S)
- Rotospin Rotary Mixer (Tarsons)
- Shaking water bath (Stuart, model: SBS40)
- Centrifuge (Labocene, catalog number: ScanSpeed 1736R)
- Nikon Ni-E fluorescence microscope and Nikon A1 confocal imaging system (Nikon A1-SHS, catalog number: 10225)
Procedure
- Retina Injury (Kaur et al., 2018; Mitra et al., 2018; Mitra et al., 2019; Sharma et al., 2019)
- Prepare 50 ml of 1x tricaine methane sulfonate solution in the fish water.
- Anesthetize the fish by putting in tricaine solution till the gill’s movement slows down.
- Keep the anesthetized fish on a wet paper towel bedding with the right side facing upward.
- Focus the zebrafish eye under a stereomicroscope and tilt the dorsal side of eyeball gently with forceps.
- Poke one edge of the ventral side of the eye with 30 G needle by stab wound injury and then injure the other edge of the eye.
- Repeat the injuries on the dorsal side of the eye by tilting it gently from the ventral side so that a total of four injuries are made.
- Let the fish revive in the system water (see Video 1 for details).
 Video 1. Retinal injury in zebrafish
Video 1. Retinal injury in zebrafish
- In vivo mRNA transfection (Mitra et al., 2018; Sharma et al., 2019)
In vivo overexpression of genes is achieved by mRNA transfection in the retina. It involves the following steps (see Video 2 for details): Video 2. Intravitreal injection and morpholino electroporation- Preparation of transfection reagent
- Clone GFP or gene of interest into pCS2+ plasmid. Linearize the vector containing insert from the 3’ end. Prepare mRNA with a sp6 message machine in vitro transcription kit. Precipitate mRNA and dissolve in nuclease-free water to make a stock of 2,000 ng/µl.
- Prepare transfection mixture (8 µl) with lipofectamine 2,000 transfection reagent as follows:
- Mix 2 µl each of mRNA (2,000 ng/µl) and 2x HBSS solution at room temperature.
- Mix 2 µl of lipofectamine with an equal volume of 2x HBSS solution at a ratio of 1:1 and keep the solution at room temperature.
- Let both the solutions stand at room temperature for five minutes.
- Mix the solutions (i) and (ii), dropwise at a ratio of 1:1 and keep the mixture the room temperature for 30 min. Use the final transfection mixture containing 500 ng/µl of mRNA for intravitreal injection.
- In vivo overexpression by intravitreal injection
- Anesthetize the fish and injure the right eye by making four stab wounds with a 30 G needle.
- From the fourth poke inject 1 µl of transfection mixture using Hamilton syringe.
- Electroporate the fish by placing a negative electrode on the right eye and positive electrode on the other side.
- Electroporate with five pulses at 70 V of 50 milliseconds duration with a gap period of 950 milliseconds between the pulses.
- Harvest the eyes on 4 days post-injury as described in sections E and F.
- In gene overexpression retina check the proliferation in terms of BrdU positive cells in comparison to GFP transfected retinae (see Procedures E, F and G).
- Check the transfection by immunostaining for GFP and subsequent imaging which shows GFP in the whole retina. Two representative images to show GFP expression at the injury site and away from the injury site are appended here for reference (Figure 1).
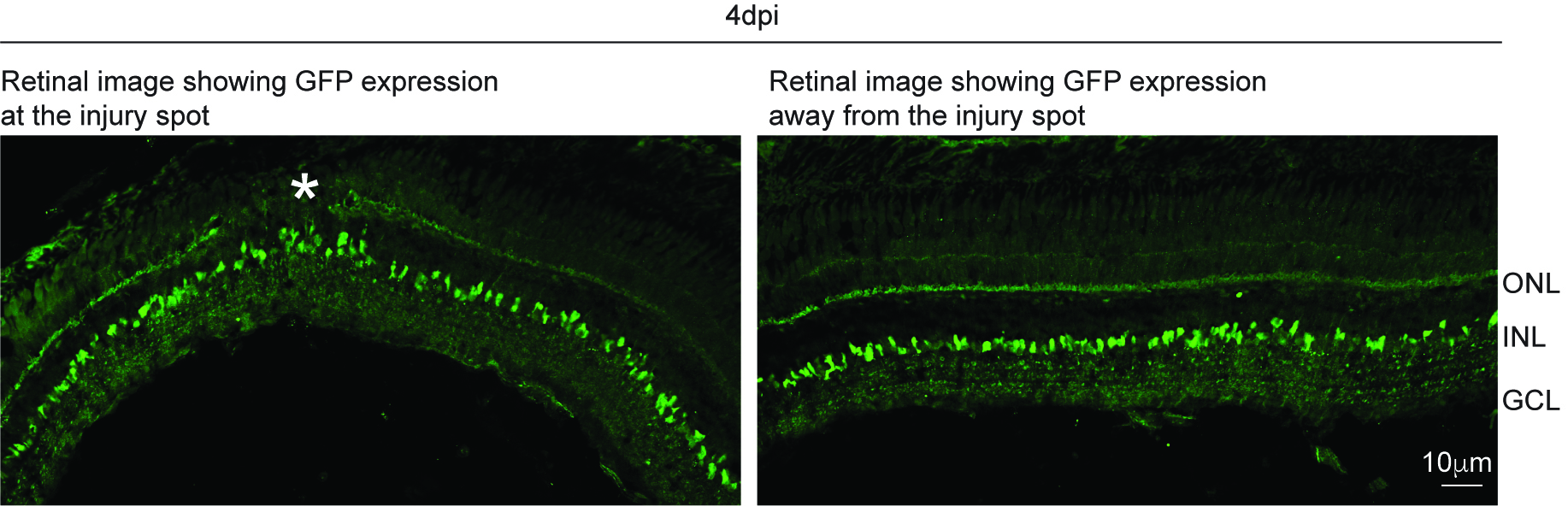
Figure 1. Showing GFP expression in zebrafish retina after GFP mRNA transfection. Asterisk marks the injury spot. ONL−outer nuclear layer; OPL−outer plexiform layer; INL−inner nuclear layer; IPL−inner plexiform layer; GCL−ganglion cell layer.
- Preparation of transfection reagent
- Drug delivery and Morpholino electroporation (Kaur et al., 2018; Mitra et al., 2018; Mitra et al., 2019; Sharma et al., 2019)
To study the localized effect of drugs, proteins, and gene knockdown approaches, intravitreal injections are made.- Preparation of solution for injection:
- Dissolve the lyophilized morpholino (300 ng) in 300 µl of autoclaved Milli-Q water to make a stock of 1 mM. Use neat for 1 mM or dilute with Milli-Q water to make stocks of 0.5 or 0.25 mM for injection.
- Dissolve the protein in the recommended solvent to make a stock solution and dilute further in 1x PBS or recommended diluent.
- Dissolve the drugs in 1 ml DMSO (Dimethyl sulfoxide) or recommended solvent to make a stock solution of final concentration depending upon the molecular mass and weight of the pharmacological inhibitor. Make the required working concentrations from the main stock by diluting with Milli-Q water.
- Injection of the solution:
- Injure one retina by stab wound injury as described above.
- From the fourth poke inject around 1 µl of the reagent with Hamilton syringe.
- In the case of morpholino delivery, electroporate the morpholino to make their entry in the retina.
- Place the positive electrode on the eye injected with morpholino and the negative electrode on the other eye.
- Electroporate at the conditions given above.
- Harvest the eyes on 4 days post retinal injury (dpi) as described below.
- Preparation of solution for injection:
- BrdU labeling
BrdU pulse labeling is done for 3 h before harvesting the eye on 4 dpi. It labels actively proliferating cells by incorporating thymidine analog (BrdU) in the replicating DNA.- Make 5 mM solution of BrdU by dissolving 15.35 mg of BrdU powder in 10ml of autoclaved Milli-Q water. Make 1 ml aliquots and store at -20 °C for later use.
- Bring the BrdU solution at room temperature before use and fill in the insulin syringe.
- Anesthetize the fish and hold on a wet paper towel under the microscope.
- Insert the insulin needle gently in the midway between pelvic fins and making needle parallel to the fish body taking care not to damage internal organs.
- Inject around one unit (15-20 µl) of BrdU solution and leave the fish for 3 h in the system water.
- Harvesting and tissue preparation
- Harvesting of eye (see Video 3 for details)
- Euthanize the fish by tricaine overdosing (prolonged immersion in 1x tricaine solution).
- Gently pull out the eye out of the socket with forceps.
- Keep the eye in chilled fixative in the Petri plate and focus under a stereomicroscope.
- Hold the eye with forceps and pierce the cornea with a needle. With the help of forceps tear the cornea and gently push the eye allowing the lens come out.
Video 3. Zebrafish euthanasia and harvesting of the eye - Tissue fixation
- Fix the eye overnight at 4 °C in 4% PFA with continuous rotation at 12 rpm on rotor spin.
- Remove the PFA and dehydrate the tissue in the sucrose gradient for 45 min each at room temperature with continuous rotation. Add the sucrose solutions as follows:
5% sucrose, 1 volume
5%:20% sucrose, 2:1 volume
5%:20% sucrose, 1:1 volume
5%:20% sucrose, 1:2 volume
20% sucrose, 1 volume - Finally, replace 20% sucrose with 500 µl of fresh sucrose solution and add an equal volume of OCT into it. Mix by rotation at room temperature for 30 min.
- Make cubic molds of aluminum foil by wrapping on cubits.
- Fill the mold till half with OCT, put a labeled flag at the top and place the eye dorsoventrally parallel to it.
- Freeze the positioned eye immediately by keeping at -80 °C.
- Harvesting of eye (see Video 3 for details)
- Cryo-sectioning and Immunostaining
- Cryo-sectioning
- Turn on the cryostat and keep eye blocks inside the chamber for 10 min.
- Fix the tissue on specimen disc with the help of OCT.
- Fix the specimen disc on the specimen head and label the glass slides.
- Remove the excess of OCT till retinal tissue starts appearing.
- Take 8-10 µm thin serial sections on glass slides.
- Allow the slides dry overnight at room temperature.
- Immunostaining
- Take one set of slides and lay them horizontally on the immunostaining rack.
- Give three washes with 1x PBS for 10 min each to remove OCT.
- Block the tissues with 5% BSA in PBST (1x PBS with 0.01% Triton X-100) for 2-3 h at room temperature.
- Remove the blocking solution and overlay the slides with the primary antibody at 4 °C overnight.
- Next day collect the primary antibody and wash the slides three times with PBST at room temperature for 10 min each.
- Overlay the slides with fluorescently labeled secondary antibody for 2-3 h at room temperature.
- Collect the secondary antibody and wash the slides thrice with PBST.
- Stop the reaction by washing with autoclaved water.
- Let the slides dry vertically in the dark for 10-15 min.
- Mount the slides with 60 µl DABCO and coverslip them.
- Let the slides dry overnight at room temperature in a dark chamber.
- Cryo-sectioning
- Image acquisition
Examine the slides under a fluorescence microscope and take images with a confocal imaging system with a Nikon Ni-E fluorescence microscope equipped with fluorescence optics and Nikon A1 confocal imaging system.
Data analysis
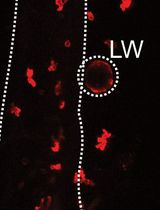
Count the BrdU labeled cells under a fluorescence microscope by observation of their fluorescence in retinal sections. In both experimental and treatment sets count the cells at the main injury spot and avoid the spots away from the main injury. The injury spot can be demarcated by discontinuous outer nuclear layer as shown below (Figure 2). Take a minimum of three eyes in each experimental setup. Count the labeled cells in all the main injury spots present in the three retinal sections. Exclude the outliers (extremely low and extremely high values) and calculate the average and standard deviation by analyzing the data in an Excel sheet.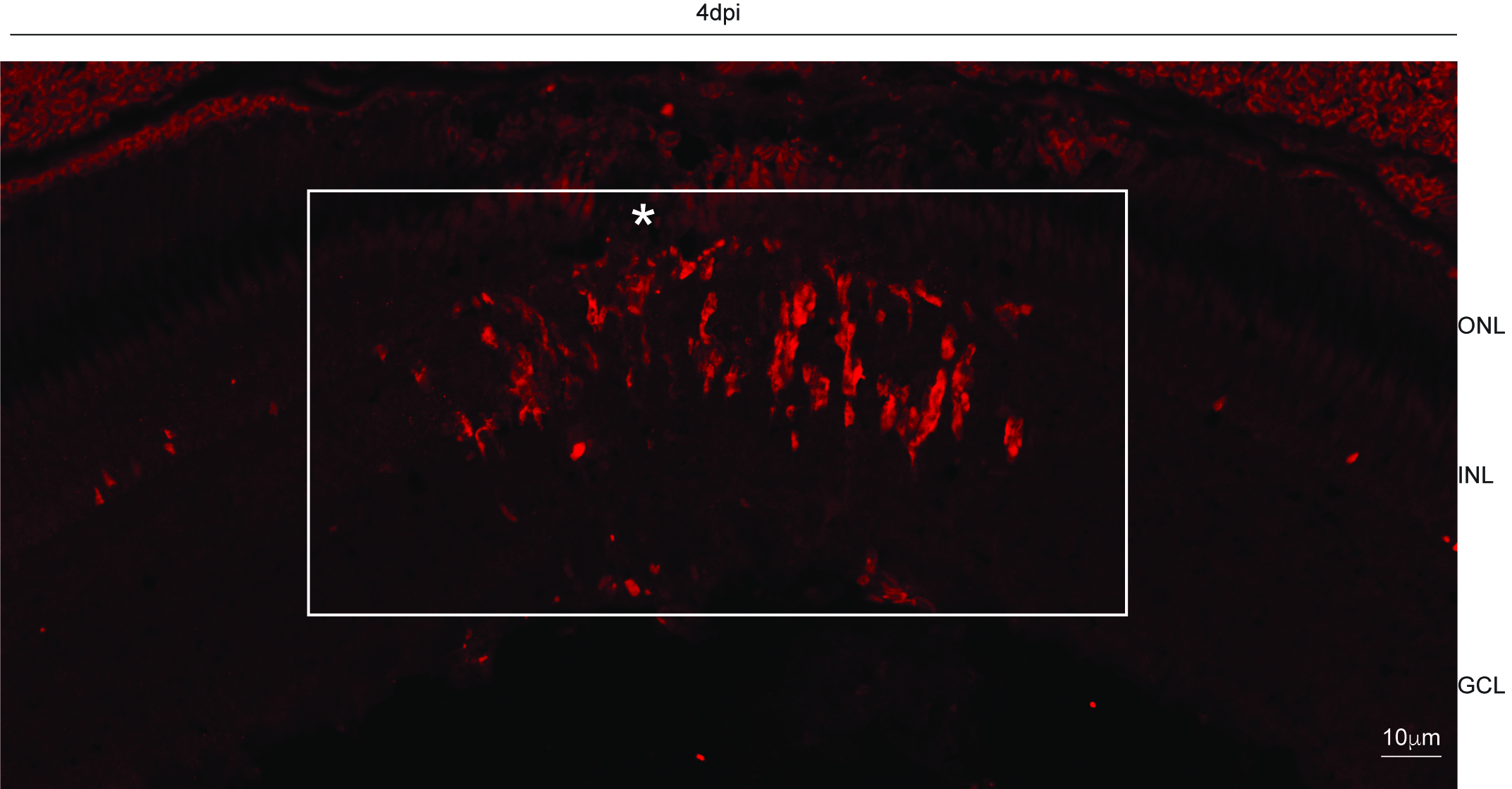
Figure 2. Showing injury spot (demarcated by an asterisk) with BrdU positive cells labeled in red. Square around the injury spot marking the area for cell count. Scale bar−10 µm; ONL−outer nuclear layer; INL−inner nuclear layer; GCL−ganglion cell layer.
Recipes
- Tris-HCl (1 M, pH 7.5, 100 ml)
- Make 1 M stock of Tris-HCl by dissolving 12.1 g of Tris base in 80 ml of Milli-Q water
- Adjust the pH to 7.5 with concentrated HCl solution and make the final volume to 100 ml with Milli-Q water
- Tricaine methane sulfonate solution (100 ml)
- Measure 2 g of Ethyl 3-aminobenzoate, methane sulfonic acid salt and dissolve it in 100 ml of 0.1 M Tris-HCl (pH 7.5) solution (Dilute the stock solution to 0.1 M by taking 10 ml of 1 M solution in 90 ml of Milli-Q water)
- Keep the solution at 4 °C and dilute it (1:100) in fish system water before use
- Phosphate buffer (PB, 1 M, pH 7.4, 100 ml)
- Weigh the following components:
Na2HPO4 10.9877 g NaH2PO4 2.711548 g - First, dissolve Na2HPO4 in 80 ml of Milli-Q H2O, then add NaH2PO4
- Adjust the pH to 7.4 and make up the volume to 100 ml with Milli-Q H2O
- Autoclave the solution and dilute to a working stock of 0.1 M with autoclaved Milli-Q water
- Weigh the following components:
- Phosphate buffered saline (PBS, 10x, pH 7.4, 1 L)
- Measure the following components:
NaCl 75.97 g Na2HPO4 9.937 g NaH2PO4 3.59 g - Dissolve all the components in 800 ml of Milli-Q water by vigorous shaking or with magnetic beads
- Once all the components have dissolved adjust the pH to 7.4 and make up the final volume to 1,000 ml
- Autoclave the solution and use it as a 1x solution diluted with Milli-Q water
- Measure the following components:
- HBSS (Hanks balanced salt solution, 10x, pH 7.14, 100 ml)
NaCl 8 g KCl 0.354 g Glucose 1.351 g HEPES 5.466 g Na2HPO4 99.35 mg - Measure these components and dissolve in 80 ml of Milli-Q water
- Adjust the pH to 7.14 and make up the final volume to 100 ml
- Prepare 2x solution from 10x stock and store in 1 ml aliquots at -20 °C
- 4% PFA (50 ml)
- Measure 2 g of PFA in a 50 ml dry centrifuge tube and add 5 ml of 1 M PB into it
- Make the volume to 45 ml with autoclaved Milli-Q water and dissolve PFA by heating at 65 °C in a shaking water bath
- Take out the centrifuge tube in-between to mix settled powder by inverting vigorously. It takes around 15-30 min to dissolve completely
- After dissolving properly, adjust the volume to 50 ml with autoclaved Milli-Q water and keep immediately on ice. For long term storage, make small aliquots and store at -20 °C
- DABCO (2.5%, 50 ml)
- Measure 12 g of glycerol in a 50 ml centrifuge tube
- Measure 4.8 g of PVA and add it to glycerol. Mix well by gently inverting the tube till homogenous solution
- Add 12 ml of autoclaved Milli-Q water and mix overnight on rotor. Then spin at room temperature
- Add 24 ml of 0.2 M Tris-HCl, pH 8-8.5
- Make 100 ml of 1 M Tris-HCl solution by dissolving 12.1 g of Tris base in 80 ml of Milli-Q water
- Adjust the pH to 8.2 with a concentrated HCl solution and make up the final volume to 100 ml
- To make 0.2 M Tris-HCl take 6 ml of 1 M stock in 24 ml of Milli-Q water
- Heat the solution to 50 °C in a shaker water bath for 15-30 min and centrifuge at 5,000 x g for 15 min
- Carefully remove the supernatant and add 1.25 g of DABCO into the supernatant
- Centrifuge at 5,000 x g for 15 min
- Aliquot the supernatant in 1 ml tubes and store the at -20 °C for later use
- Sucrose solutions (50 ml)
- 5% sucrose: Measure 2.5 g of sucrose and dissolve it in 40 ml of 0.1 M PB solution. Adjust the volume to 50 ml with 0.1 M PB
- 20% sucrose: Measure 10 g of sucrose and dissolve it in 40 ml of 0.1 M PB solution. After dissolving it by rotation, adjust the volume to 50 ml with 0.1 M PB
- Store the solutions at 4 °C for long term use
Acknowledgments
P.S acknowledges postdoctoral fellowship support from Wellcome Trust/DBT India Alliance and IISER Mohali. P.S thanks Ajay Kumar, Undergraduate student, for video capturing and editing. This work was supported by the Wellcome Trust/DBT India Alliance Intermediate Fellowship (IA/I/12/2/500630) awarded to Rajesh Ramachandran. R.R also acknowledges research funding from Science Education and Research Board (SERB), DST, India (EMR/2017/001816), DBT India (BT/PR9407/BRB/10/12612013), (BT/PR17912/ MED/31/336/2016) and support from IISER Mohali.
Competing interests
The authors declare that they have no conflict of interest.
References
- Kaur, S., Gupta, S., Chaudhary, M., Khursheed, M. A., Mitra, S., Kurup, A. J. and Ramachandran, R. (2018). let-7 microRNA-mediated regulation of shh signaling and the gene regulatory network is essential for retina regeneration. Cell Rep 23(5): 1409-1423.
- Mitra, S., Sharma, P., Kaur, S., Khursheed, M.A., Gupta, S., Ahuja, R., Kurup, A.J., Chaudhary, M., and Ramachandran, R. (2018). Histone deacetylase-mediated Muller glia reprogramming through her4.1-lin28a axis is essential for retina regeneration in zebrafish. iScience 7: 68-84.
- Mitra, S., Sharma, P., Kaur, S., Khursheed, M.A., Gupta, S., Chaudhary, M., Kurup, A.J., and Ramachandran, R. (2019). Dual regulation of lin28a by Myc is necessary during zebrafish retina regeneration. J Cell Biol 218: 489-507.
- Sharma, P., Gupta, S., Chaudhary, M., Mitra, S., Chawla, B., Khursheed, M. A. and Ramachandran, R. (2019). Oct4 mediates Muller glia reprogramming and cell cycle exit during retina regeneration in zebrafish. Life Sci Alliance 2(5): pii: e201900548.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Sharma, P. and Ramachandran, R. (2019). Retina Injury and Retina Tissue Preparation to Study Regeneration in Zebrafish. Bio-protocol 9(24): e3466. DOI: 10.21769/BioProtoc.3466.
- Mitra, S., Sharma, P., Kaur, S., Khursheed, M.A., Gupta, S., Chaudhary, M., Kurup, A.J., and Ramachandran, R. (2019). Dual regulation of lin28a by Myc is necessary during zebrafish retina regeneration. J Cell Biol 218: 489-507.
Category
Stem Cell > Pluripotent stem cell > Regenerative medicine
Cell Biology > Cell imaging > Confocal microscopy
Neuroscience > Sensory and motor systems > Retina
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.