- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Analysis of EGF Receptor Endocytosis Using Fluorogen Activating Protein Tagged Receptor
Published: Vol 9, Iss 24, Dec 20, 2019 DOI: 10.21769/BioProtoc.3463 Views: 5298
Reviewed by: Samantha E. R. DundonLinlin SunAnonymous reviewer(s)
Original research article
The authors used this protocol in:

May 2019

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Functional activities of many transmembrane proteins are controlled by their endocytosis. One of the most studied experimental models is the epidermal growth factor (EGF) receptor (EGFR). However, endocytic trafficking of EGFR has been predominantly analyzed using labeled EGF, whereas quantitative analyses of the endocytosis of the receptor itself have been sparse. The fluorescence microscopy methods described here are designed to directly quantify EGFR internalization in living cells without labeled EGFR ligands or antibodies. These methods utilize an engineered EGFR chimera in which the fluorogen activating protein (FAP) is fused to the receptor extracellular domain (FAP-EGFR). Binding of malachite green (MG) based dyes to FAP results in a strong far-red fluorescence of MG, thus efficiently labeling FAP-EGFR. In particular, binding of the cell impermeant MG-Bis-SA dye to FAP produces the pH-sensitive dual-excitation fluorescence, which allows differentiation of the cell-surface and internalized pools of FAP-EGFR. Two modifications of the methodology are described: 1) single-cell three-dimensional confocal imaging; and 2) high-throughput assay in multi-well plates. These methodologies can be adopted to study endocytosis of any other transmembrane protein extracellularly tagged with FAP.
Keywords: EGF receptorBackground
Although EGFR internalization has been studied for decades and is often used as a prototypical experimental model to investigate the mechanisms of clathrin-dependent and -independent endocytosis, our knowledge about these mechanisms is still incomplete. EGFR internalization has been mostly studied by monitoring fluorescently or radiolabeled EGF (Sorkin and Duex, 2010; Pinilla-Macua and Sorkin, 2015). Limited quantitative information is available about EGFR internalization induced by other ligands (that are technically difficult to label) and other stimuli, as well as about constitutive internalization of the receptor. Conventional cell-surface biotinylation and antibody-based methods have been used to monitor EGFR internalization (Burke and Wiley, 1999; Burke et al., 2001). However, these methods either do not have sufficient sensitivity to detect endocytosis induced by physiological-relevant low ligand concentrations or have other caveats related to antibody bi-valency and selective recognition of different receptor conformations, or do not allow monitoring the time-course of EGFR endocytosis in living single cells.
Here we describe methods to visualize EGFR endocytosis and measure the rate of EGFR internalization. These methods are based on the expression of the EGFR chimeric protein in which the fluorogen activating protein (FAP) is genetically fused to the receptor amino-terminus (FAP-EGFR). Binding of cell impermeant malachite green fluorogen (MG-B-Tau) to FAP results in a strong far-red fluorescence (excitation at 640 nm; emission at 680-700 nm), which labels the cell-surface FAP-EGFR. Binding of cell impermeant MG-Bis-SA, a pH sensitive Cy3-MG tandem dye, to FAP-EGFR, results in a single far red emission by dual-excitation at 561 nm and 640 nm. In this system, MG fluorescence results from both the direct excitation (640 nm) and the fluorescence resonance energy transfer (FRET) from the pH-sensitive Cy3 (excitation at 561 nm), which is minimal at the neutral pH of the extracellular medium but increases several fold at the acidic pH environment of endosomes and lysosomes (pH 6.5-4.5). The ratio of MG fluorescence excited at 561 nm and 640 nm (the “561/640” ratio) is proportional to the ratio of the internalized and total MG-Bis-SA-bound FAP-EGFR and is, therefore, used to determine the extent of FAP-EGFR endocytosis. The principles of the ratiometric assay in cells expressing FAP-tagged receptors are described in detail in our previous studies (Perkins et al., 2018; Larsen et al., 2019).
Here we describe two experimental protocols that take advantage of the MG: FAP-EGFR system. 1) A single-cell internalization assay using MG-B-Tau or MG-Bis-SA: FAP-EGFR endocytosis is monitored on a pixel-by-pixel basis in single living cells by labeling the surface-localized receptor with either MG-B-Tau or pH-sensitive MG-Bis-SA dyes followed by time-lapse three-dimensional (4-D) spinning disk confocal microscopy imaging. Imaging of MG-B-Tau and MG-Bis-SA labeled FAP-EGFR are used to obtain qualitative information about the extent and rate of FAP-EGFR endocytosis. Labeling with MG-B-Tau can be combined with the stimulation of cells with the labeled ligand (e.g., Rhodamine labeled EGF, EGF-Rh), so that subcellular localization of the ligand and the receptor can be simultaneously monitored. Labeling with MG-Bis-SA is performed to distinguish FAP-EGFR located in endosomes from the cell-surface localized FAP-EGFR. 2) High-throughput internalization assay in 96-well plates: FAP-EGFR is labeled with MG-Bis-SA, followed up by automated two-channel 2D imaging to obtain the mean values of the 561/640 ratio for individual wells of the 96-well plate, increases in which correspond to movement of the receptor into acidic endosomes. This method is designed to quantify FAP-EGFR endocytosis under multiple experimental conditions. In particular it is useful for dose-response assays as well as screening siRNA, small-compounds and other libraries.
These methods require cells expressing extracellularly FAP-tagged EGFR. Although we have developed these assays using HeLa cells, in which endogenous EGFR was fused to FAP using CRISPR/Cas9 gene editing, the same methods are applicable to cells transiently or stably expressing recombinant FAP-EGFR. However, we believe that the endogenous FAP-EGFR system offers the highest sensitivity and ensures the physiological relevance. Technical information provided in the parental paper (Larsen et al., 2019) should facilitate gene-editing of endogenous EGFR in other cell lines of interest. Furthermore, the same approach can be used to study endocytosis of a multitude of other transmembrane proteins.
Materials and Reagents
- 96-well polystyrene plates (Thermo Fisher, Fisher Scientific, catalog number: 12565501)
- 55 ml reagent reservoir basin, non-sterile, clear PVC (Green Bioresearch, catalog number: 250052)
- Micropipette tips (Thermo Fisher, Fisher Scientific, catalog number: 02-707-425)
- Microscope cover glass 18-CIR2, thickness #2 (0.17-0.25 mm) (Thermo Fisher, Fisher Scientific, catalog number: 12-546)
- 12-well cell culture plate (Corning, catalog number: 3513)
- 50 ml polypropylene conical tube (Falcon, catalog number: 352070)
- 50 ml PS reservoir basin, sterile, multi-pack (Life Science Products, catalog number: 730-006)
- Optical: μCLEAR, black sterile 96-well plate for cell culture (Greiner Bio-One, catalog number: 655090)
- FAP-EGFR expressing HeLa (Larsen et al., 2019) or other cells
- Human recombinant EGF (BD Bioscience, 354052, stock at 1 μg/μl in H2O, store at -20 °C, once thawed keep at 4 °C)
- Bovine serum albumin (BSA) (Sigma-Aldrich, catalog number: A2153, stock solution at 10% in H2O, keep both powder and stock solution at 4 °C)
- Fetal bovine serum (FBS) (Thermo Fisher, Gibco, catalog number: 26140079, keep at -20 °C)
- Water repellent Rain-X
- Phosphate buffer saline (PBS) (Thermo Fisher, Gibco, catalog number: 14190144, keep at 4 °C)
- DMEM (Thermo Fisher, Gibco, catalog number: 11965092, keep at 4 °C)
- Trypsin-EDTA (0.25%), phenol red (Thermo Fisher, Gibco, catalog number: 25200056, keep 4 °C)
- MG-Bis-SA (Bruchez laboratory, stock 1 mM in ethanol 1% acetic acid, working stock 300 μM in ethanol 1% acetic acid, store at 4 °C)
- MG-B-Tau (Bruchez laboratory, stock 1 mM in H2O 1% acetic acid, working stock 150 μM in H2O 1% acetic acid, store at 4 °C)
- NaCl
- KCl
- MgCl2·6H2O
- CaCl2·2H2O
- HEPES
- Buffer solutions with pH 4.8-8.1 (see Recipes)
- Buffer pH 8 (see Recipes)
Equipment
- 8 and 12 channels multichannel pipettes (Biohit)
- Stainless steel manifolds (with PTFE stoppers) (CPLabSafety, catalog number: WH-851381 or similar)
- Microscope Stage adaptor (ALA Scientific instruments, catalog number: MS-508D or similar)
- Confocal laser microscope with A1 resonant scanner and 40x 1.15NA ELWD water immersion objective (Nikon ECLIPSE Ti-E)
- Spinning disk confocal microscope system with 63x 1.4 NA oil immersion objective equipped with environmental chamber to ensure 37 °C, humidity and 5% CO2 during imaging (Intelligent-imaging Innovation, Inc. (3i), Denver, CO, USA)
Software
- NIS Elements (Nikon Instruments Inc., www.nikon.com)
- Slidebook 6 (3i, www.intelligent-imaging.com)
- Microsoft Office Excel (Microsoft, www.microsoft.com)
- Prism 8 (GraphPad Software, Inc., www.graphpad.com)
Procedure
- Single-cell internalization assay using MG-B-Tau
- Day 1: Prepare cells
Plate FAP-EGFR expressing cells at ~20% confluence in a 12-well plate with 18 mm diameter glass coverslips in 1 ml DMEM containing 10% FBS per well. - Day 2: Serum-starve cells overnight to minimize the FAP-EGFR activity
- Aspirate the media and wash each well with 0.5 ml of 37 °C prewarmed PBS.
- Add 1 ml of 37 °C prewarmed starvation media (DMEM containing 0.1% BSA) to each well.
- Day 3: Treat and perform fluorescence microscopy imaging
If inhibitors are to be tested, pretreat as required and then proceed as below:- Prepare an aliquot of intermediate MG-B-Tau solution (1:3 dilution of 150 μM working stock in H2O) and add 1 μl to each well to a final MG-B-Tau concentration of 50 nM.
- Incubate at room temperature for 30-60 s protected from light.
- Treatment:
For time course experiments, mount the coverslip into a 37 °C prewarmed microscope stage adaptor. Fill it with 0.9 ml of the same media. Place the adaptor on the microscope stage. Perform time-lapse imaging using spinning disk confocal system with 63x N.A. 1.4 oil immersion objective through the 640 nm channel (excitation 640 nm, emission 680 nm) at 37 °C in 5% CO2 atmosphere. Acquire a Z-stack of two-dimensional (512 x 512 pixels) confocal images at 300 nm intervals for each time point. Acquire at least one Z-stack before adding 100 μl of the treatment to be tested prepared in starvation media (final volume is 1 ml). Acquire all images using the same parameters.
Notes:- If labeled ligand is used as treatment, imaging through an additional filter channel is performed. Different fluorescently labeled EGF conjugates are commercially available. We recommend EGF-rhodamine (EGF-Rh) as rhodamine is a bright and photostable fluorophore directly conjugated to EGF (Pinilla-Macua and Sorkin, 2015). EGF-Rh is detected through the 561 nm channel (excitation at 561 nm; emission at 580 nm).
- Because x-axis resolution in our system is ~600-700 nm, z-stack of images were acquired at 300-nm steps, which approximately corresponds to Nyquist sampling. The speed of image acquisition is limited by the minimal exposure time (typically hundreds of milliseconds) necessary to obtain an image with the high signal-to-noise ratio.
- The image acquisition parameters (laser power, exposure times, imaging times, etc.) are established using the experimental conditions of maximum FAP-EGFR endocytosis, e.g., maximal accumulation of FAP-EGFR in vesicular structures and minimal FAP-EGFR fluorescence at cell edges (plasma membrane). In our experiments, the EMCCD Evolve camera with the linear detection range is used. Images are acquired with the maximum fluorescence intensity of less than ~90% of the detection saturation. Typically, specific fluorescence intensity is more than 5-fold higher than the background at the plasma membrane and more than 30-fold in endosomal compartments. Certainly, other camera- or photomultiplier-based systems can be utilized in this protocol.
- Day 1: Prepare cells
- Single-cell internalization assay using pH-sensitive MG-Bis-SA
- Day 1: Prepare cells
Plate FAP-EGFR expressing cells at ~20% confluence in a 12-well plate with 18 mm diameter glass coverslips in 1 ml DMEM containing 10% FBS per well. - Day 2: Serum-starve cells overnight to minimize the FAP-EGFR activity
- Aspirate the media and wash each well with 0.5 ml of 37 °C prewarmed PBS.
- Add 1 ml of 37 °C prewarmed starvation media (DMEM containing 0.1% BSA) to each well.
- Day 3: Treat and perform fluorescence microscopy imaging
If inhibitors are to be used, pretreat as required and then proceed as below:- Prepare an intermediate solution of MG-Bis-SA (1:3 dilution of the 300 μM working stock in H2O). Add 1 μl to each well to achieve the final concentration of 100 nM.
- Incubate at room temperature for 30-60 s protected from light.
- Treatment:
- For time course experiments, mount the coverslip into a prewarmed microscope stage adaptor. Fill it with 0.9 ml of the same media. Place the adaptor on the microscope stage. Perform time-lapse imaging using spinning disk confocal system with the 63x 1.4 N.A. oil immersion objective through the FRET channel (excitation at 561 nm; emission at 680 nm) and 640 nm channel (excitation 640 nm, emission 680 nm) at 37 °C in 5% CO2 atmosphere. Acquire z-stack of two-dimensional confocal images (512 x 512) at 300 nm intervals at each time point. Acquire at least one z-stack before adding 100 μl of treatment prepared in starvation media (final volume is 1 ml). Acquire all images with the same parameters.
- For single-time point imaging add 100 μl of treatment prepared in starvation media for a desired time (final volume 1.0 ml). Stop EGFR trafficking by placing the 12-well plate on ice. Wash cells with 0.5 ml of cold PBS per well and add 1 ml of cold Buffer pH 8; place the cells on ice (cells cannot be fixed otherwise pH difference between compartments disappears). Image immediately as described above for time course imaging.
Note: The image acquisition parameters (laser power, exposure times, imaging times, etc.) are established using the experimental conditions of maximum FAP-EGFR endocytosis, e.g., maximal accumulation of FAP-EGFR in vesicular structures and minimal FAP-EGFR fluorescence at cell edges (plasma membrane). In our experiments, the EMCCD Evolve camera with the linear detection range is used. Images are acquired with the maximum fluorescence intensity of less than ~90% of the detection saturation. Typically, specific fluorescence intensity for both 640 nm and 561 nm channels can be more than 30-fold higher than background in endosomal structures. The specific fluorescence signal of cell-surface FAP-EGFR for the 640 nm channel is at least 5-fold higher than the background whereas the signal through the 561 nm channel typically does not exceed 2-fold over the background signal. Certainly, other camera- or photomultiplier-based systems can be used in this protocol.
- Day 1: Prepare cells
- Internalization assay in 96-well plates
- Day 1: Prepare cells
Plate FAP-EGFR expressing cells in an optical 96-well plate at 30% confluency (~9,000 cells per well). Because the duplication time of HeLa cells is ~24 h, they typically reach 100% confluency (~30,000 cell/well–required for consistent results) after 2 days of growth including serum starvation overnight. Other cell lines with different growth rates can be plated at different initial confluency.
Note: For siRNA studies instead of seeding cells on Day 1, perform reverse transfection of cells on Day 0 following manufacturer’s instructions. - Day 2: Serum-starve cells overnight
- Aspirate the media and wash with 100 μl of 37 °C prewarmed PBS.
- Replenish with 100 μl of 37 °C prewarmed starvation media (DMEM containing 0.1% BSA) to minimize basal FAP-EGFR activity.
- Day 3: EGFR internalization assay
If inhibitors are to be tested, pretreat as required and then proceed as below:- Aspirate media with stainless steel manifold.
- For each 96-well plate, prepare 6 ml DMEM containing 2 μl MG-Bis-SA of 300 μM working stock solution (final concentration 100 nM).
- Add 50 μl per well of the 100 nM MG-Bis-SA solution using an eight-channel pipette in the dispense mode. Keep at least 2 wells without MG-Bis-SA for background measurements. Those wells are to be incubated with 50 μl DMEM alone.
- Incubate at room temperature for 30-60 s protected from light. Aspirate the medium with the stainless-steel manifold and wash each well once with 100 μl of 37 °C prewarmed PBS.
Note: For pH calibration curve: Apply series of buffer solutions (100 μl per well) with pH ranging from 4.8 to 8.1 at room temperature (see Recipes below). Use at least 2 wells for each pH. Image immediately. See an example of the calibration curve in our recent paper (Larsen et al., 2019) and Figure 2. - Apply 100 μl per well of starvation media containing EGF and/or other compounds using the 12-channel pipette.
Note: If inhibitors are used, add them again in the “treatment” medium. - Incubate at 37 °C for a desired time. A maximum accumulation of EGFR in endosomes is typically after 15-30 min of continuous internalization in EGF-stimulated cells, and thus such incubation times are most optimal for the measurements of internalization rates. At later times, contribution of the degradation and recycling of EGF:EGFR complexes into the relative amount of internalized EGFR is significant.
Notes:- For time-course assays, start incubations by adding EGF and/or other compounds to wells that are to be incubated for the longest times and finish with adding treatment media to the wells that are to be incubated for the shortest time. In this way, all incubations are finished at the same time, which allows simultaneous termination of endocytosis and imaging of all wells (see below).
- For dose-response assays, keep at least 2 wells (labeled with MG-Bis-SA) untreated by adding 100 μl of the starvation media without stimuli or inhibitors. These wells are used to measure basal internalization rates.
- Place the cells on ice to stop endocytosis.
- Aspirate the treatment media with the stainless-steel manifold and add 200 μl of cold Buffer pH 8 to each well (mild extracellular alkalization maximizes the pH difference between the plasma membrane and endosomal FAP-EGFR). Keep cells on ice until imaging.
- Image immediately by automated confocal imaging using 40x 1.15 NA water immersion objective. Capture and stich nine (3 x 3) image fields per well using both FRET channel (excitation 561 nm; emission 700/75 nm) and 640 nm channel (excitation 640 nm, emission 700/75 nm), and pinhole of 5.
Notes:- This assay is based on a single-plane confocal image (2-D) of the large area of cell monolayer. A large pinhole (5) ensures a high signal-to-noise ratio (SNR) while reduced z-axis resolution, thus preventing small changes in the focal plane that might occur during automated multi-well imaging. Reduced x-y-axis resolution when using pinhole of 5 is not detrimental to these measurements.
- The image acquisition parameters (laser power, exposure times, etc.) are set using the experimental conditions of maximum FAP-EGFR endocytosis, e.g., maximum high 561/640 ratio (see data analysis below). The signal counts should be in the order of thousands.
- For each 96-well plate prepare 6 ml DMEM containing 6 μl MG-Bis-SA of 300 μM working stock solution.
- Add 50 μl per well of the MG-Bis-SA solution using an eight-channel pipette in the dispense mode (final volume 150 μl, final concentration 100 nM). Keep at least 2 wells without MG-Bis-SA for background measurements. Add 50 μl DMEM alone to those wells.
- Incubate at room temperature for 30-60 s protected from light.
- Aspirate the medium with the stainless-steel manifold.
- Proceed as indicated above in Step C3e.
- Day 1: Prepare cells
Data analysis
- Single-cell internalization assay using MG-Bis-SA:
This assay yields images that allow visually distinguish plasma membrane (relatively low 561/640 ratio) from endosomal FAP-EGFR (relatively high 561/640 ratio). Therefore, this assay is considered qualitative rather than quantitative.
The data can be presented as a ratiometric image of 561/640 in pseudocolor:- Select an area of the image where there are no cells. Draw an ROI (R of interest). Use it to subtract background.
- Adjust the Lookup table (Lut) of both 561 nm and 640 nm channels of all images to be the same.
- Divide the 561 nm (FRET channel) image (numerator) by the 640 nm image (denominator) using SlideBook6 software ratio module. Adjust the scale from 0 to 1.
- Show the ratio image in pseudocolor so the value of the ratio is displayed stretched according to a temperature-based lookup table with blue (cold) indicating low values and red (hot) indicating high values.
- Modulate the ratio image obtained in (d) by the 640 nm image to eliminate distracting data from regions outside the cells (Figure 1).
Note: The 640 nm channel is used as a saturation channel, so it gates the pseudocolor ratio image based on intensity. Pixels with high ratio values but low 640 nm intensity are shown in dim red. Pixels with low ratio values but high 640 nm intensity will appear as bright blue whereas pixels with 640 nm values below the low threshold are displayed in black. - See an example image in Figure 1 and in our parental publication (Larsen et al., 2019).
- Internalization assay in 96-well plates: quantification of internalized/total EGFR.
- Create a mask for the entire image, stitched from 9 image fields, by drawing an ROI that contains the entire montage and obtain the average intensity values for each of the imaging channels (excitation at 561 nm or 640 nm; emission at 700 nm) in each well. The Nikon NIS Elements HCA JOBS module automatically calculates these values.
Note: For the 640 nm channel fluorescence, the mean intensity is typically ~4-fold higher than in the background designated wells. For the 561 nm channel, the mean intensity can be 1-4-fold higher than in the background wells depending on the extent and rate of FAP-EGFR internalization. - Average the mean intensity values from background wells (no MG-Bis-SA added) in both channels.
- Subtract the background values obtained in step 2b from raw mean intensity values calculated in step 2a to obtain specific fluorescence intensity of MG-Bis-SA in two channels.
- Divide background-subtracted (specific) intensity values of the 561 nm channel by the background-subtracted values (specific) of the 640 nm channel to obtain the value of the 561/640 ratio for individual wells.
- For dose-response assays, subtract the values obtained in control (ligand-untreated cells) from all treated cells and plot the resulting 561/640 ratio against log (10) of ligand concentration.
- For pH calibration curve, plot 561/640 ratio obtained against pH.
- See an example in Figure 2 and our previous paper (Larsen et al., 2019).
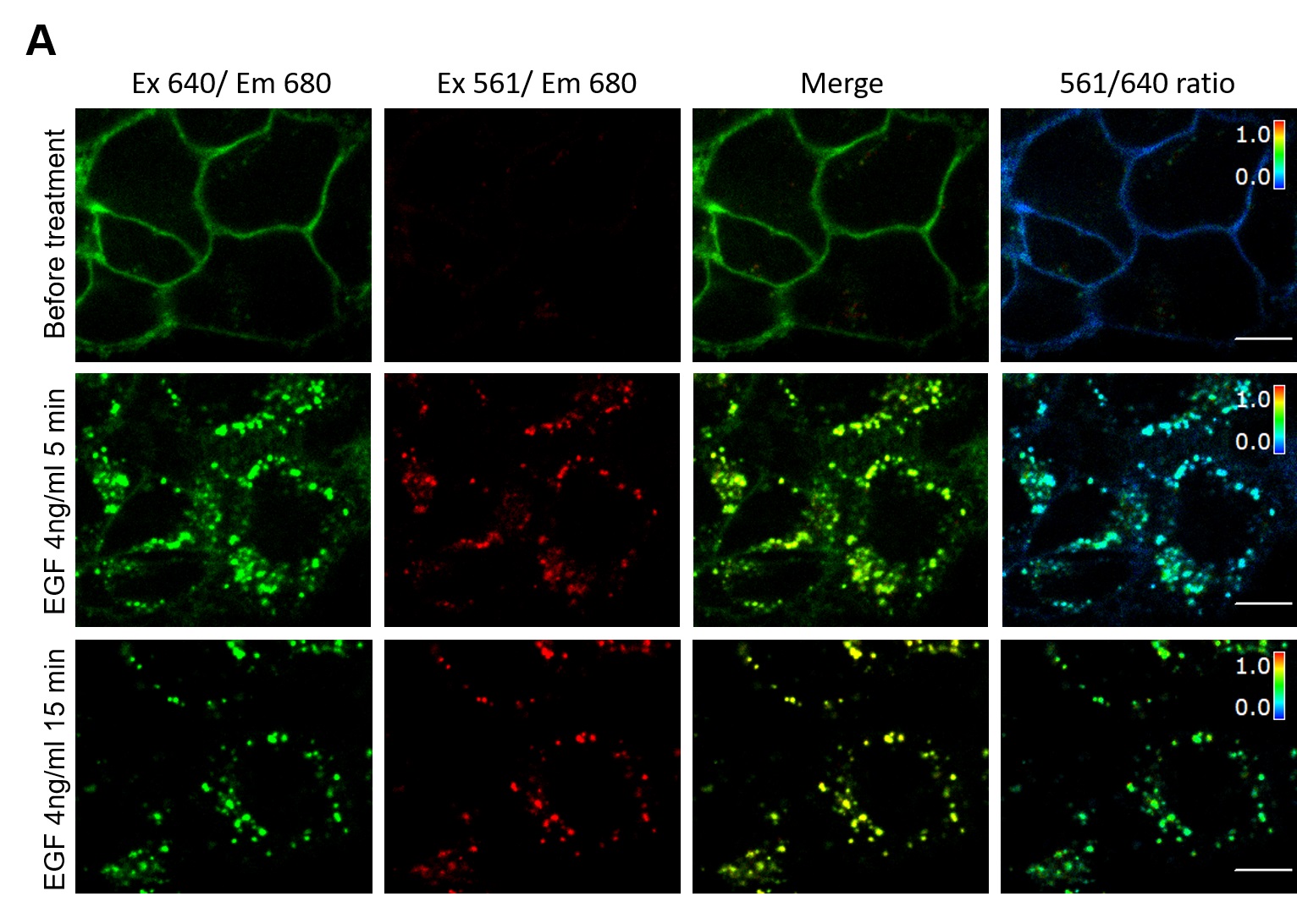
Figure 1. An example image of MG-Bis-SA labeled FAP-EGFR expressing cells (single-cell internalization assay). A. Cells were labeled with MG-Bis-SA and then treated with 4 ng/ml EGF. Images were acquired before treatment and after 5 and 15 min of EGF stimulation at 37 °C through the 640 nm channel (green; 640 nm excitation; 680 nm emission) and FRET channel (red; 561 nm excitation; 680 nm emission). On the right, the 561/640 ratio image is shown as the pseudocolor image modulated to the intensity of the 640 nm channel. All fluorescence intensities scales are identical between untreated and EGF-treated cells. Scale bars: 10 μm.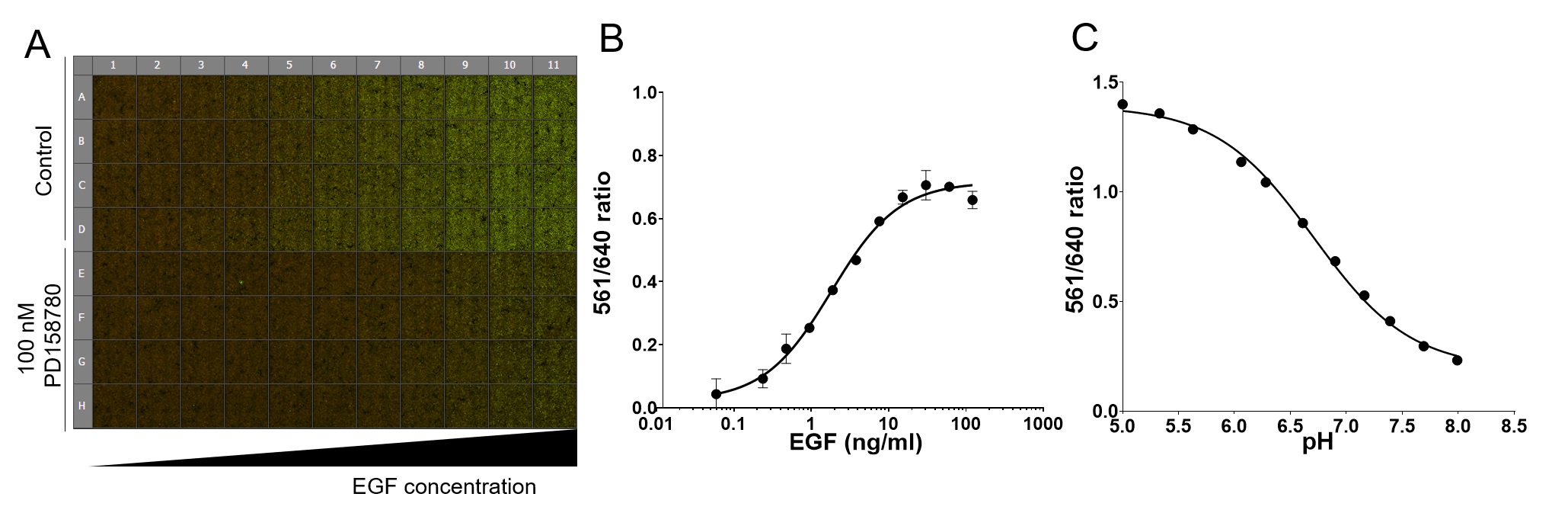
Figure 2. Example of the data obtained using the 96-well internalization assay. A. pH calibration curve raw data is shown. Cells were labeled with MG-Bis-SA and exposed to a series of buffer solutions with different pH (from 4.8-8.1). Last row (Bg) was not labeled with MG-Bis-SA and is used to calculate background. Each well of 96-well plate shown is a montage of 9 merged images of 561 nm excitation (FRET) channel (green) and 640 nm channel (red) acquired in each well. B. Quantification of A. C. Example of the quantification of the EGF dose-response. Cells were labeled first with MG-Bis-SA and then exposed for 15 min at 37 °C to a range of EGF doses (0.49-100 ng/ml). Mean values (± SEM) of duplicates are plotted against log (10) of EGF concentration.
- Create a mask for the entire image, stitched from 9 image fields, by drawing an ROI that contains the entire montage and obtain the average intensity values for each of the imaging channels (excitation at 561 nm or 640 nm; emission at 700 nm) in each well. The Nikon NIS Elements HCA JOBS module automatically calculates these values.
Notes
- Medium change by itself may induce EGFR internalization in some cells, e.g., HeLa. Therefore, in such cases, it is important to avoid or minimize the temperature and medium changes during initial steps in all protocols involving addition of inhibitors, dyes or other treatments. Other cell lines should be first tested to confirm that the medium change does not induce FAP-EGFR internalization. To this end, perform imaging for 15 min at 37 °C without any treatment. Adapt the protocol (minimizing medium changes) until basal internalization is negligible or minimal.
- In experiments requiring several-hour-long incubations, considerable amounts of newly synthetized EGFR might be delivered to the cell surface during the incubation. Therefore, unbound MG dye present in the media can label these receptors. To avoid labeling of this EGFR pool by MG dyes during time-course experiments, replace MG dye containing medium with the fresh 37 °C prewarmed starvation media prior to cell stimulation or other treatments. To confirm that medium change does not induce FAP-EGFR internalization, perform imaging for 15 min at 37 °C in untreated cells.
Recipes
- Buffer solutions with pH 4.8-8.1
- Prepare one common solution (for 1 L):
Note: To cover the whole pH range, use two different buffers (MES for acidic and HEPES for alkaline part of the range) while Keeping the ion composition.
140 mM NaCl (8.18 g)
5 mM KCl (0.37 g)
1 mM MgCl2 (0.20 g of MgCl2·6H2O)
1 mM CaCl2 (0.15 g of CaCl2·2H2O) - Split into two (for 0.5 L)
- For pH < 6.8: 50 mM MES (4.88 g)
Take 7 aliquots of 50 ml. Adjust the pH of the aliquots at: 4.8, 5.1, 5.4, 5.7, 6.0, 6.3, 6.6 - For pH > 6.8: 50 mM HEPES (5.96 g)
Take 5 aliquots of 50 ml. Adjust the pH of the aliquots at: 6.9, 7.2, 7.5, 7.8, 8.1
- Prepare one common solution (for 1 L):
- Buffer pH 8
140 mM NaCl
5 mM KCl
1 mM MgCl2
0.1 mM CaCl2
25 mM HEPES, pH 8.0
Keep at 4 °C
Acknowledgments
A Sorkin and SC Watkins were supported by the NIH grants GM124186 and CA089151. Marcel P Bruchez was supported by NIH grants R01EB017268 and R01GM114075. This protocol was adapted from Larsen et al., 2019.
Competing interests
Dr. Bruchez is the Founder of Sharp Edge Laboratories, a company that licensed the FAP technology from CMU. The author has no other competing interests to declare.
References
- Burke, P., Schooler, K. and Wiley, H. S. (2001). Regulation of epidermal growth factor receptor signaling by endocytosis and intracellular trafficking. Mol Biol Cell 12(6): 1897-1910.
- Burke, P. M. and Wiley, H. S. (1999). Human mammary epithelial cells rapidly exchange empty EGFR between surface and intracellular pools. J Cell Physiol 180(3): 448-460.
- Larsen, M. B., Perez Verdaguer, M., Schmidt, B. F., Bruchez, M. P., Watkins, S. C. and Sorkin, A. (2019). Generation of endogenous pH-sensitive EGF receptor and its application in high-throughput screening for proteins involved in clathrin-mediated endocytosis. Elife 8: e46135.
- Perkins, L. A., Yan, Q., Schmidt, B. F., Kolodieznyi, D., Saurabh, S., Larsen, M. B., Watkins, S. C., Kremer, L. and Bruchez, M. P. (2018). Genetically targeted ratiometric and activated pH indicator complexes (TRApHIC) for receptor trafficking. Biochemistry 57(5): 861-871.
- Pinilla-Macua, I. and Sorkin, A. (2015). Methods to study endocytic trafficking of the EGF receptor. Methods Cell Biol 130: 347-367.
- Sorkin, A. and Duex, J. E. (2010). Quantitative analysis of endocytosis and turnover of epidermal growth factor (EGF) and EGF receptor. Curr Protoc Cell Biol Chapter 15: Unit 15.14.
Article Information
Copyright
![]() Perez Verdaguer et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Perez Verdaguer et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Perez Verdaguer, M., Larsen, M. B., Bruchez, M. P., Watkins, S. C. and Surve, S. (2019). Analysis of EGF Receptor Endocytosis Using Fluorogen Activating Protein Tagged Receptor. Bio-protocol 9(24): e3463. DOI: 10.21769/BioProtoc.3463.
- Larsen, M. B., Perez Verdaguer, M., Schmidt, B. F., Bruchez, M. P., Watkins, S. C. and Sorkin, A. (2019). Generation of endogenous pH-sensitive EGF receptor and its application in high-throughput screening for proteins involved in clathrin-mediated endocytosis. Elife 8: e46135.
Category
Cell Biology > Cell-based analysis > Endocytosis
Cell Biology > Cell imaging > Confocal microscopy
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.