- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Propagation and Purification of Chlamydia trachomatis Serovar L2 Transformants and Mutants
Published: Vol 9, Iss 24, Dec 20, 2019 DOI: 10.21769/BioProtoc.3459 Views: 4564
Reviewed by: Alka MehraLionel SchiavolinAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Mar 2019
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Chlamydia trachomatis (C.t.) is an obligate intracellular pathogen that cannot be cultured axenically and must be propagated within eukaryotic host cells. There are at least 15 distinct chlamydial serovariants that belong to 2 major biovars commonly referred to as trachoma and lymphogranuloma venereum (LGV). The invasive chlamydia LGV serovar L2 is the most widely used experimental model for studying C.t. biology and infection and is the only strain with reliable genetic tools available. New techniques to genetically manipulate C.t. L2 have provided opportunities to make mutants using TargeTron and allelic exchange as well as strains overexpressing epitope-tagged proteins, in turn necessitating the regular purification of transformant and mutant clones. Purification of C.t. is a labor-intensive exercise and one of the most common reagents classically used in the purification process, Renografin, is no longer commercially available. A similar formulation of diatrizoate meglumine called Gastrografin is readily available and we as well as others have had great success using this in place of Renografin for chlamydial purifications. Here, we provide a detailed general protocol for infection, propagation, purification, and titering of Chlamydia trachomatis serovar L2 with additional notes specifically pertaining to mutants or recombinant DNA carrying clones.
Keywords: ChlamydiaBackground
Chlamydia trachomatis (C.t.) is the etiological agent of blinding trachoma and the most prevalent sexually transmitted disease in the world. Chlamydiae are gram-negative obligate intracellular bacteria that have evolved to occupy a niche within the host cytoplasm derived from a modified endocytic vesical called an inclusion. Chlamydia initiates entry into the host cell by binding to the sialic acid moieties of proteins on the host cell surface. The precise mechanisms regulating entry of C.t. into the host cell are poorly understood but several studies have implicated clathrin mediated endocytosis (Hodinka et al., 1988; Wyrick et al., 1989; Majeed and Kihlstrom, 1991), while others have suggested caveola mediated entry is involved (Boleti et al., 1999). C.t. is taken up into the cell within an endocytic vesicle which it heavily modified to form the chlamydial inclusion. Fusion of this compartment with lysosomes is blocked and the bacterium secretes numerous effector proteins that subvert host resources and redirect them to the inclusion to promote chlamydial proliferation. The Chlamydial lifecycle is biphasic, consisting of different infectious and proliferative stages. The infectious but metabolically inactive form is referred to as an elementary body (EB) while the metabolically active but non-infectious form is called the reticulate body (RB) (Figure 1). Chlamydial EB’s bind to sialic acid residues on cell surface receptors and through an unknown mechanism induce their uptake by the cell into an endocytic vesicle. The inclusion avoids fusion with lysosomes and degradative compartments while trafficking along microtubules to the peri Golgi region. By about 4 h post-infection, most of the EBs have converted to RBs. Following chlamydial cell division, the RB’s undergo asynchronous conversion to EB’s. Depending on stress, cellular environment, and nutrient availability the chlamydial replicative cycle takes 48-72 h to complete and chlamydia exits via host cell lysis or through extrusion. Of note, the EB is the form usually purified (Caldwell et al., 1981) although techniques have been described to purify RBs (Skipp et al., 2016). For C.t. purifications, the EB is most desirable, since it is the stage associated with infectivity.
Figure 1. Transmission electron micrograph of wild-type C.t. L2 at 36h post-infection. Scale bar is 1 µm.
Although one study reported that a medium had been developed that could support the axenic metabolic and biosynthetic activity from both EBs and RBs, to the best of our knowledge no lab has had success in using this media to support propagation of chlamydia (Omsland et al., 2012). Thus, in the research laboratory, C.t. is typically cultivated in HeLa 229, Hep2, McCoy, or Vero cells although other cell-types can be used they are less common. Until recently, C.t. was largely intractable to genetic manipulation but advances in tools to generate site-specific mutations and to overexpress proteins during infection have revolutionized the field and labs are now making recombinant C.t. clones regularly. In the past, large preparations of C.t. EBs could carry the lab for quite some time. Now, with the generation of new strains occurring on a regular basis, C.t. preparations are a much more common occurrence in the chlamydia laboratory. This is certainly a good thing but cultivating, purifying, and titering new C.t. strains on a regular basis can be a costly and time-consuming endeavor. Not to mention that a key reagent that has been used for years, Renografin, has been made extremely difficult to obtain due to Mallinckrodt ceasing production. Fortunately, a similar product has been identified and new sources discovered (this information will be provided in the materials section).
Materials and Reagents
- Consumables
- Tissue culture flask T175 (Fisher Scientific, catalog number: 07-000-384)
- Basix 5 ml serological pipette (Fisher Scientific, catalog number: 14-955-233)
- Basix 10 ml serological pipette (Fisher Scientific, catalog number: 14-955-234)
- Basix 25 ml serological pipette (Fisher Scientific, catalog number: 14-955-235)
- Basix 50 ml conical tube (Fisher Scientific, catalog number: 14-955-239)
- Beckman 250 ml centrifuge bottle (Fisher Scientific, catalog number: NC9304619)
- Oak Ridge tube (VWR, catalog number: 76324-812)
- Thickwall polycarbonate ultracentrifuge tube (Fisher Scientific, catalog number: NC9439275)
- 18 Gauge Canula (Thomas Scientific, catalog number: 8957G34)
- 10 ml syringe (Fisher Scientific, catalog number: 14-841-54)
- 1.5 ml conical screwcap tube (USA Scientific, catalog number: 1415-8700)
- 24-well plate (Fisher Scientific, catalog number: 07-000-030)
- N-95 mask
- Tissue culture flask T175 (Fisher Scientific, catalog number: 07-000-384)
- Experimental models: cell lines
- HeLa cells ATCC CCL-2
- HeLa cells ATCC CCL-2
- Bacterial strains
- Chlamydia trachomatis serovar L2 ATCC VR-902B
- Chlamydia trachomatis serovar L2 ATCC VR-902B
- Antibodies
- Chlamydia trachomatis LPS antibody, mouse monoclonal Novus NBP1-28820
- Rabbit anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 (Thermo Fisher, catalog number: A-11059)
- Chemicals, peptides, recombinant proteins, and miscellaneous reagents
- Gastrografin (Bracco, catalog number: 0270-0445-40)
- RPMI 1640 medium with L-glutamine (Thermo Fisher, catalog number: 11875-093)
- NaHCO3 (Sodium bicarbonate) (Thermo Fisher, catalog number: 25080094)
- Gentamicin (Thermo Fisher, catalog number: 15710072)
- Trypsin-EDTA 0.5% (Thermo Fisher, catalog number: 15400054)
- Heat-inactivated Fetal Bovine Serum (VWR, catalog number: 89510-186)
- 1x PBS without calcium chloride and magnesium (Thermo Fisher, catalog number: 10010-049)
- 1x HBSS without calcium chloride and magnesium (Thermo Fisher, catalog number: 14170112)
- Cycloheximide (MilliporeSigma, catalog number: C4859-1ML)
- KH2PO4 (Fisher Scientific, catalog number: P285-500)
- K2HPO4 (Fisher Scientific, catalog number: P288-500)
- KCl (Fisher Scientific, catalog number: P217-500)
- NaCl (Fisher Scientific, catalog number: S271-500)
- L-Glutamic Acid (Fisher Scientific, catalog number: A125-100)
- DEAE Dextran (MP Biomedicals, catalog number: ICN19513380)
- Methanol (Fisher Scientific, catalog number: A412-500)
- Sucrose
- Na2HPO4 (Sodium phosphate dibasic anhydrous)
- NaH2PO4·H2O (Sodium phosphate monobasic monohydrate)
- Blood agar plate
- Yeast peptone dextrose (YPD) agar plate
- 10x K36 (see Recipes)
- 1x K36 (see Recipes)
- Sucrose Phosphate Glutamate buffer (SPG) (see Recipes)
- 30% Gastrografin (see Recipes)
- 54% Gastrografin (see Recipes)
- 44% Gastrografin (see Recipes)
- 40% Gastrografin (see Recipes)
- 10x DEAE dextran (see Recipes)
- 1x DEAE dextran (see Recipes)
Equipment
- P200 Pipette
- Shaker
- Humidified CO2 cell culture incubator set to 37 °C
- Floor Centrifuge Avanti JXN-30 (Beckman, catalog number: B38624)
- Rotor: JLA-16.250 (Beckman, catalog number: 363934)
- Rotor: JLA-25.50 (Beckman, catalog number: 363058)
- Rotor: JS-24.15 (Beckman, catalog number: 362396)
- Table top Centrifuge Allegra X-14R (Beckman, catalog number: A99465)
- Microcentrifuge Microfuge20R (Beckman, catalog number: B31599)
- Sonicator Q-sonica Q500 (VWR, catalog number: 89207-036)
- Sonicator Probe CL-334 (VWR, catalog number: 89207-036)
- Fluorescent microscope
- Biosafety cabinet
- -80 °C freezer
- Large cell scraper (Fisher Scientific, catalog number: 08-100-241)
Procedure
Note: As Chlamydia trachomatis is a BSL2 pathogen, all steps must be carried out in a biosafety cabinet (BSC). Ensure all solutions are sterile prior to beginning the purification.
- Seeding and preparing HeLa Cells
- Seed 8 T175 flasks with early passage HeLa cells (< P10) at a cell concentration sufficient to reach confluency in 1-2 days.
Note: We have propagated C.t. in immortalized primary epithelial cells of several lineages (vaginal, cervical, conjunctival, foreskin), but have not carried out large scale purifications from these cells lines, primarily due to the exorbitant cost of the reagents needed to propagate these cells. Theoretically, the purifications would proceed identical to what we describe in this protocol with the only changes being the media for each respective cell type. - Decant the media from 8 T175 flasks of confluent HeLa cells.
- Gently wash the cells using 5 ml of 1x Phosphate Buffer Saline (PBS) per flask. Ensure most of the wash is removed using a 5 ml serological pipette.
- Add 5 ml Trypsin to each flask, gently swirl to ensure it covers the entire bottom of the flask, and incubate cells at 37 °C for 5-10 min or until cells detach.
- Add 5 ml RPMI 1640 with 10% FBS to each flask. Combine cells from 8 flasks into 1 flask.
- Prepare 20 new T175 flasks by adding 30 ml RPMI 1640 with 10% FBS.
- Add 4 ml diluted cell mixture (Step A4) to each new flask. Seed enough cells to obtain confluency (1 x 107/flask).
- Incubate cells at 37 °C with 5 % CO2 for 24 h or until HeLa monolayers are confluent.
- Seed 8 T175 flasks with early passage HeLa cells (< P10) at a cell concentration sufficient to reach confluency in 1-2 days.
- Infection (20 Flask Batch)
- Remove aliquot of Chlamydia trachomatis L2 from -80 °C freezer and let thaw at room temperature. Prepare Chlamydia trachomatis inoculum in 5 ml RPMI 1640 with 10% FBS and 10 μg/ml cycloheximide using a Multiplicity of Infection (MOI) 2.5 assuming 2 x 107 cells/flask at confluency. Use 100 ml total for 20 flasks.
Note: For mutants with a lysis or toxic phenotype, reduce MOI to 0.5. Include any relevant antibiotic for mutant selection in the RPMI 1640. - Decant the media from each flask.
- Add 5 ml of inoculum from Step B1 to each flask and incubate at 37 °C with slow shaking or rocking for 30 min-1 h.
- Add 25 ml RPMI 1640 with 10% FBS and 10 μg/ml cycloheximide to each flask.
Note: Cycloheximide prevents host cell synthesis to facilitate bacterial replication - Incubate for 36-48 h at 37 °C with 5% CO2.
- Remove aliquot of Chlamydia trachomatis L2 from -80 °C freezer and let thaw at room temperature. Prepare Chlamydia trachomatis inoculum in 5 ml RPMI 1640 with 10% FBS and 10 μg/ml cycloheximide using a Multiplicity of Infection (MOI) 2.5 assuming 2 x 107 cells/flask at confluency. Use 100 ml total for 20 flasks.
- Purification
- Decant the media from each of the 20 flasks.
- Add 8 ml ice-cold 1x K36 (Recipe 2) to each of the 20 flasks.
- Scrape each flask using a large cell scraper. Rinse the back of each flask with the 1x K36-cell mix and transfer cell suspension from 10 flasks to a 250 ml centrifuge bottle on ice. Repeat for the next 10 flasks with another 250 ml centrifuge bottle.
Note: The cell suspension from 10 flasks should be added to each of two 250 ml centrifuge bottles. Splitting the 20 flasks of cell suspension into two 250 ml centrifuge bottles will provide adequate space for mixing and a balance for all centrifugation steps moving forward. A total of four 250 ml centrifuge bottles will be needed for the entire purification. Two 250 ml centrifuge bottles will be used through Step C6 and two will be used starting on Step C7. - Rinse the back of the T175 flask with 5 ml K36, transfer to another T175 flask. Continue to rinse and transfer for 5 flasks in total. After washing the 5th flask, add the cell suspension to the 250 ml centrifuge bottle on ice. Discard the first 5 T175 flasks. Repeat procedure with remaining flasks.
- Sonicate at 60-100 watts for 20 s, immediately return centrifuge bottle to ice. Repeat sonication step for two sonication cycles total per 250 ml centrifuge bottle
Note: An N-95 mask should be worn during the sonication and sonication should be conducted in the BSC. - Centrifuge at 500 x g for 10 min at 4 °C.
- Transfer each supernatant to a new pre-chilled 250 ml centrifuge bottle on ice.
- Resuspend each pellet in 100 ml ice-cold 1x K36.
- Sonicate at 60-100 watts for 20 s, immediately return centrifuge bottle to ice. Repeat sonication step for two cycles in total per 250 ml centrifuge bottle.
- Centrifuge at 500 x g 10 min at 4 °C.
- Add supernatant to the pre-chilled 250 ml centrifuge bottles from Step C7.
- Centrifuge at 30,000 x g for 20 min at 4 °C.
- Remove the supernatant from each 250 ml centrifuge bottle (there are 2) and resuspend each pellet with 30 ml 1x K36. Disperse using an 18 ga cannula on a 10 ml syringe. Solution should be slowly dispersed to prevent frothing and should be passed 3-5 times or until the solution is homogenous.
- Add 8 ml of 30% Gastrografin (Recipe 4) to a thick-walled polycarbonate ultracentrifuge tube.
Note: Diatrizoate meglumine and diatrizoate sodium solutions allow for optimal density gradient isolation of chlamydial elementary bodies with minimal host debris and negligible loss of bacteria or toxicity. They are normally used as contrasting agents in radiography due to the iodine content. - Gently layer the suspension from Step C13 over the Gastrografin. Top the tubes with 1x K36.
- Centrifuge at 40,000 x g for 30 min at 4 °C.
- Decant supernatant and resuspend pellet in 1x K36 using 1 ml per T175 flask (10 ml per pellet for a 20-flask prep).
Note: To prepare 30% seed stock instead of doing the full purification, resuspend pellet in SPG (Recipe 3). Transfer to an oak ridge tube, and centrifuge at 30,000 x g for 20 min at 4 °C. Resuspend pellet in 5 ml ice-cold SPG, aliquot and store at -80 °C. - Prepare Gastrografin density gradient by very slowly layering the following in order into a thick-walled polycarbonate ultracentrifuge tube: 8 ml 54%, 12 ml 44%, and 5 ml 40% Gastrografin.
- Gently add the suspension from Step C17 to the gradient and top with K36.
- Centrifuge at 40,000 x g for 60 min at 4 °C.
- Collect the white elementary body band between the two lower layers (54% and 44%) using a cannula and transfer it to a pre-chilled oak ridge tube on ice (Figure 2A).
- Add approximately 10 volumes of ice-cold SPG.
- Centrifuge at 30,000 x g for 20 min at 4 °C. Resuspend pellet in 1 ml/flask of SPG. Use a cannula to disperse if necessary. Aliquot (50-100 μl) into sterile O-ring tubes using a P200 pipette and store at -80 °C.
- Test sterility by inoculating a blood agar plate, yeast peptone dextrose (YPD) agar plate, and RPMI 1640 media with 10 μl of purified EBs.
- Titers
- Seeding HeLa Cells for titer plates
- Decant the media from T175 flask of confluent HeLa cells.
- Gently wash the cells using 5 ml of 1x PBS per flask. Ensure most of the wash is removed using a serological pipette.
- Add 5 ml Trypsin to each flask, gently swirl to distribute along the back of the flask, and incubate cells at 37 °C for 5-10 min or until cells detach.
- Add 5 ml RPMI 1640 with 10% FBS to flask.
- Add 1 ml of cells to 20 ml of RPMI 1640 with 10% FBS.
- Add 1 ml of diluted cell mixture to each well of a 24-well plate. Cells should be seeded to obtain 1 x 105 per well.
- Incubate the cells at 37 °C with 5% CO2 for 24 h. The cells should be confluent.
- Infect cells
- Thaw an aliquot of purified EBs.
- Add 30 μl EBs to 270 μl 1x Hanks buffered saline solution (HBSS). Vortex to mix.
- Serially dilute 1:10 in 1x HBSS.
- Remove media from the 24-well plate using a serological pipette.
- Add 200 μl of each dilution in triplicate to 3 wells of the 24-well plate.
- Add 1 ml RPMI 1640 with 10% FBS and 10 μg/ml cycloheximide. Incubate at 37 °C with 5% CO2 for ~24 h.
- Determine titer
- Remove media from the 24-well plate using a serological pipette.
- Wash each well with 0.5 ml 1x PBS twice.
- Add 0.5 ml methanol to each well and incubate at room temperature for 20 min.
- Remove methanol and discard in methanol waste container.
- Wash each well with 0.5 ml 1x PBS twice.
- Add 0.25 ml of anti-LPS diluted 1:1,000 in 1x PBS. Incubate on a shaker at 70 rpm for ~1 h at room temperature.
- Wash each well with 0.5 ml 1x PBS three times.
- Add 0.25 ml of anti-mouse 488 diluted 1:1,000 in 1x PBS. Incubate on a shaker at 70 rpm for ~1 h at room temperature.
- Wash each well with 0.5 ml 1x PBS three times.
- Leave 0.5 ml 1x PBS in each well after the final wash.
Note: Plate can be stored at 4 °C till counts are complete. - Count 10 fields of view per well using 20x object on fluorescent microscope (Figure 2B).
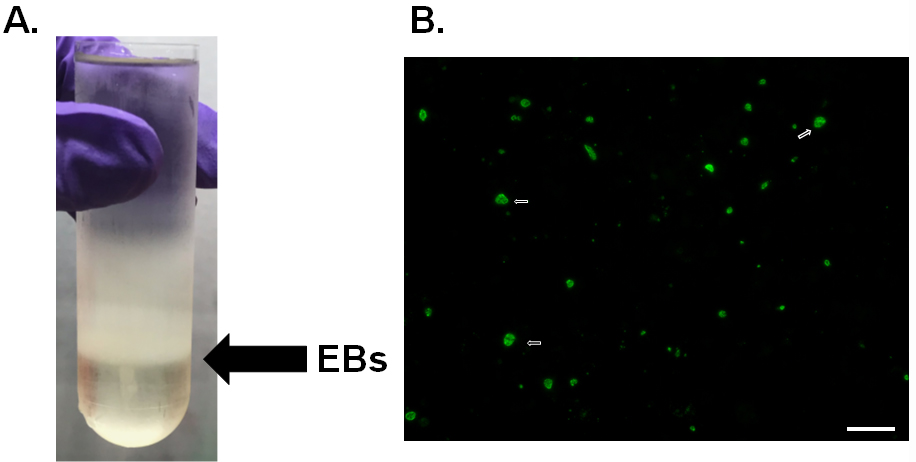
Figure 2. Density gradient purification of EBs. A. Gradient purification of C.t. L2. Black arrow denotes the location of EBs at the 54% and 44% interface. B. Immunofluorescence of chlamydia titer plates. C.t. was stained using anti-LPS (green) and the white arrow denotes examples of the chlamydia inclusions. Scale bar is 50 µm.
- Seeding HeLa Cells for titer plates
Data analysis
Determine the titer using the following equation:
Titer = (X/10) x 196 x 5 x D
where,
X is the total number of inclusions from the 10 fields counted;
196 is the number of fields of view on a 24-well plate using a 20x objective;
5 is the fraction of a ml plated;
D is the dilution factor counted;
Example: If 232 inclusions were observed in 10 fields of view using a sample diluted 104. The titer is (232/10) x 196 x 5 x 10,000 = 2.27 x 108.
Notes
- This protocol involves purification of L2 EBs, however EBs from other C. trachomatis serovars can be accomplished using this protocol with a few minor modifications (Table 1) regarding the length of time from infection to harvest and pretreatment with DEAE-dextran (Sabet et al., 1984). Cells are pretreated prior to infection by adding 5 ml 1x DEAE-dextran to each flask and are incubated at 37 °C for 30 min. The dextran is removed and cells are ready to be infected.
- A common question for this procedure is how EBs are separated from RBs during the purification. Sonication is sufficient to lyse RBs along with the host cell while leaving EBs intact. All debris is then separated from the relatively dense EBs via gradient centrifugation.
- Purification of chlamydia must be carried out in a Biosafety Level 2 laboratory.
Table 1. Purification guidelines for Chlamydia trachomatis serovars
Recipes
- 10x K36
- Dissolve 68 g Potassium phosphate monobasic (500 mM KH2PO4), 87.1 g potassium phosphate dibasic (500 mM K2HPO4), 74.5 g potassium chloride (1 M KCl), 8.7 g sodium chloride (150 mM NaCl) in 900 ml distilled water. Adjust to pH 7.0 with 1 N KOH
- Adjust volume to 1 L using a graduated cylinder
- Aliquot in 100 ml sterile bottles
- Autoclave and store at 4 °C
- 1x K36
Add 100 ml 10x K36 to 900 ml sterile distilled water, store at 4 °C - Sucrose Phosphate Glutamate buffer (SPG)
- Dissolve 3.75 g sucrose (10 mM), 2.47 g sodium phosphate dibasic anhydrous (17.4 mM Na2HPO4), 0.36 g sodium phosphate monobasic monohydrate (2.5 mM NaH2PO4·H2O) and 0.72 g L-Glutamic Acid in 900 ml distilled water. Adjust to pH 7.4 with either 1 N HCl or 1 N KOH as needed. The pH should be very close to 7.4 already
- Using a graduated cylinder, adjust the volume to 1 L
- Filter sterilize and store at 4 °C
- 30% Gastrografin
30 ml Gastrografin
70 ml 1x K36 - 54% Gastrografin
54 ml Gastrografin
46 ml 1x K36 - 44% Gastrografin
44 ml Gastrografin
66 ml 1x K36 - 40% Gastrografin
40 ml Gastrografin
60 ml 1x K36 - 10x DEAE dextran
Dissolve 450 μg/ml of DEAE-dextran in100 ml distilled water to prepare 10x stock - 1x DEAE dextran
- Add 10 ml 10x Hanks Buffered Saline Solution (HBSS) and 10 ml 10x DEAE-dextran to 80 ml distilled water
- Adjust to pH 7.4 using 7.5% NaHCO3
- Filter sterilize solution and store at room temperature
Acknowledgments
This research was supported by startup funds from the University of Iowa Department of Microbiology and Immunology to M.M.W. This protocol was adapted from Caldwell et al., 1981.
Competing interests
The authors have no conflict of interest to declare.
References
- Boleti, H., Benmerah, A., Ojcius, D. M., Cerf-Bensussan, N. and Dautry-Varsat, A. (1999). Chlamydia infection of epithelial cells expressing dynamin and Eps15 mutants: clathrin-independent entry into cells and dynamin-dependent productive growth. J Cell Sci 112 (Pt 10): 1487-1496.
- Caldwell, H. D., Kromhout, J. and Schachter, J. (1981). Purification and partial characterization of the major outer membrane protein of Chlamydia trachomatis. Infect Immun 31(3): 1161-1176.
- Hodinka, R. L., Davis, C. H., Choong, J. and Wyrick, P. B. (1988). Ultrastructural study of endocytosis of Chlamydia trachomatis by McCoy cells. Infect Immun 56(6): 1456-1463.
- Majeed, M. and Kihlström, E. (1991). Mobilization of F-actin and clathrin during redistribution of Chlamydia trachomatis to an intracellular site in eucaryotic cells. Infect Immun 59(12): 4465-4472.
- Omsland, A., Sager, J., Nair, V., Sturdevant, D. E. and Hackstadt, T. (2012). Developmental stage-specific metabolic and transcriptional activity of Chlamydia trachomatis in an axenic medium. Proc Natl Acad Sci U S A 109(48): 19781-19785.
- Skipp, P. J., Hughes, C., McKenna, T., Edwards, R., Langridge, J., Thomson, N. R. and Clarke, I. N. (2016). Quantitative proteomics of the infectious and replicative forms of Chlamydia trachomatis. PLoS One 11(2): e0149011.
- Wyrick, P. B., Choong, J., Davis, C. H., Knight, S. T., Royal, M. O., Maslow, A. S. and Bagnell, C. R. (1989). Entry of genital Chlamydia trachomatis into polarized human epithelial cells. Infect Immun 57(8): 2378-2389.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Faris, R. and Weber, M. M. (2019). Propagation and Purification of Chlamydia trachomatis Serovar L2 Transformants and Mutants. Bio-protocol 9(24): e3459. DOI: 10.21769/BioProtoc.3459.
Category
Immunology > Immune cell function > Macrophage
Microbiology > Microbe-host interactions > Bacterium
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.