- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

cDNA Display Mediated Immuno-PCR (cD-IPCR): A Novel PCR-based Antigen Detection Method


Published: Vol 9, Iss 24, Dec 20, 2019 DOI: 10.21769/BioProtoc.3457 Views: 6526
Reviewed by: Alessandro DidonnaTanxi CaiRubul Mout
Original research article
The authors used this protocol in:

Aug 2019

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Immuno-PCR (IPCR) is a powerful method in antigen detection where a PCR-amplifiable DNA reporter is conjugated to a specific antibody or an aptamer for the target molecule. In the development and application of IPCR, successful conjugation of a protein (an antibody) with a reporter DNA becomes challenging. To address this issue, we recently demonstrated the feasibility of IPCR based on cDNA display, a 1:1 covalent complex of a polypeptide and its encoding cDNA at the single molecule level. The cDNA display molecule for IPCR is generated first by transcribing the DNA that encodes the detection antibody into an mRNA by in vitro transcription. A puromycin DNA linker is then ligated to the mRNA and then in vitro translation and reverse-transcription are performed to generate the cDNA display molecule. The molecule is then directly used in antigen detection and subsequent qPCR. This method can be applied to detect various antigens in biological samples, if sequences of their single-domain antibodies (VHHs) or peptide aptamers are known.
Keywords: Immuno-polymerase chain reactionBackground
Immuno-PCR, often abbreviated as IPCR, is an extremely powerful method in detection and quantification of low abundance biomarkers that exist in biological samples (e.g., serum and urine). It acts as a bridge between immuno-reaction and signal amplification. In most cases, IPCR relies on the use of a detection antibody which has been conjugated to a reporter oligonucleotide, followed by the quantification of the analytes using real-time PCR of the reporter (Sano et al., 1992). However, in the development and application of IPCR, the difficulty associated with appropriately conjugating antibodies to oligonucleotides has become the most challenging issue (van Buggenum et al., 2016). Strategies available to conjugate the antibody and the DNA include non-covalent conjugation such as coupling via biotin-streptavidin (Sano et al., 1992) and covalent conjugation using chemical crosslinkers (Hendrickson et al., 1995). However, the tetrameric nature of streptavidin leads to the formation of heterogeneous DNA-antibody conjugates, which may decrease the reproducibility of IPCR. At the same time, conventional crosslinking chemistry reacts with the cysteine/lysine in the antibody, and the modification may compromise binding affinity of the antibody.
To avoid these problems, Guo et al. developed a new immuno-assay protocol with the aid of phage display: phage display mediated immuno-PCR (PD-IPCR) (Guo et al., 2006). In PD-IPCR, an engineered recombinant phage particle which expresses a single-chain antibody for the analyte is used as a ready reagent for IPCR instead of a monoclonal antibody (mAb) and chemically bound DNA. However, in PD-IPCR, the specificity of the DNA-protein linkage is compromised due to the difficulty in controlling of the number of exposed engineered coat proteins on the phage surface. Further, since these bacteriophages are genetically engineered viruses, experiments must be performed under strict legal regulations, which may be a hurdle for real-world applications.
cDNA display is one of the simplest genotype-phenotype linking methods, where cDNA is covalently fused to its coding polypeptide (which can act as the affinity probe for the target) via a uniquely designed puromycin linker (Yamaguchi et al., 2009; Mochizuki et al., 2011). So far, we and others have successfully applied cDNA display for in vitro selection of high affinity peptides/proteins against various targets (Suzuki et al., 2018; Terai et al., 2019). Stimulated by PD-IPCR, we envisioned that cDNA display molecules can also be regarded as antibody-DNA conjugates that are used for IPCR. Thus, we recently demonstrated the feasibility of IPCR based on cDNA display (cD-IPCR, Anzai et al., 2019). The cD-IPCR, which takes advantage of the structural characteristics of cDNA display, proved to work in detection of target proteins both in direct and sandwich-type formats. The cD-IPCR method offers several merits over the conventional IPCR and PD-IPCR, which include 1) based on a covalent homogeneous DNA-antibody conjugation, 2) non-viral, and 3) almost a ready to use reagent, 4) easy to perform by biologists without expertise in organic chemistry. The overall procedures for cD-IPCR are shown in Figure 1. Refer to the original paper "Anzai et al. (2019)" for further clarifications about the newly developed method. Here in this article, we describe step by step preparation of cDNA display molecule and its application in detecting analytes in a sandwich-type manner, which should be most general. To demonstrate the above steps, sandwich-type detection of green fluorescent protein (GFP) using anti-GFP VHH (Single variable domain on a heavy chain antibody) (Fridy et al., 2014) and commercially available polyclonal anti-GFP antibody is explained here. Of course, the readers can detect other target proteins with this method if appropriate VHH or peptide aptamers as well as their immobilization antibodies are available. Current article takes advantage from our previously published "Anzai et al. (2019)" but different from original paper as this article includes all the experimental details that even novices could easily reproduce the experiment. We hope this article will help to ensure the high reproducibility of our newly developed protocol, and also lend a helping hand to researchers who are interested in the procedures and want to use themselves.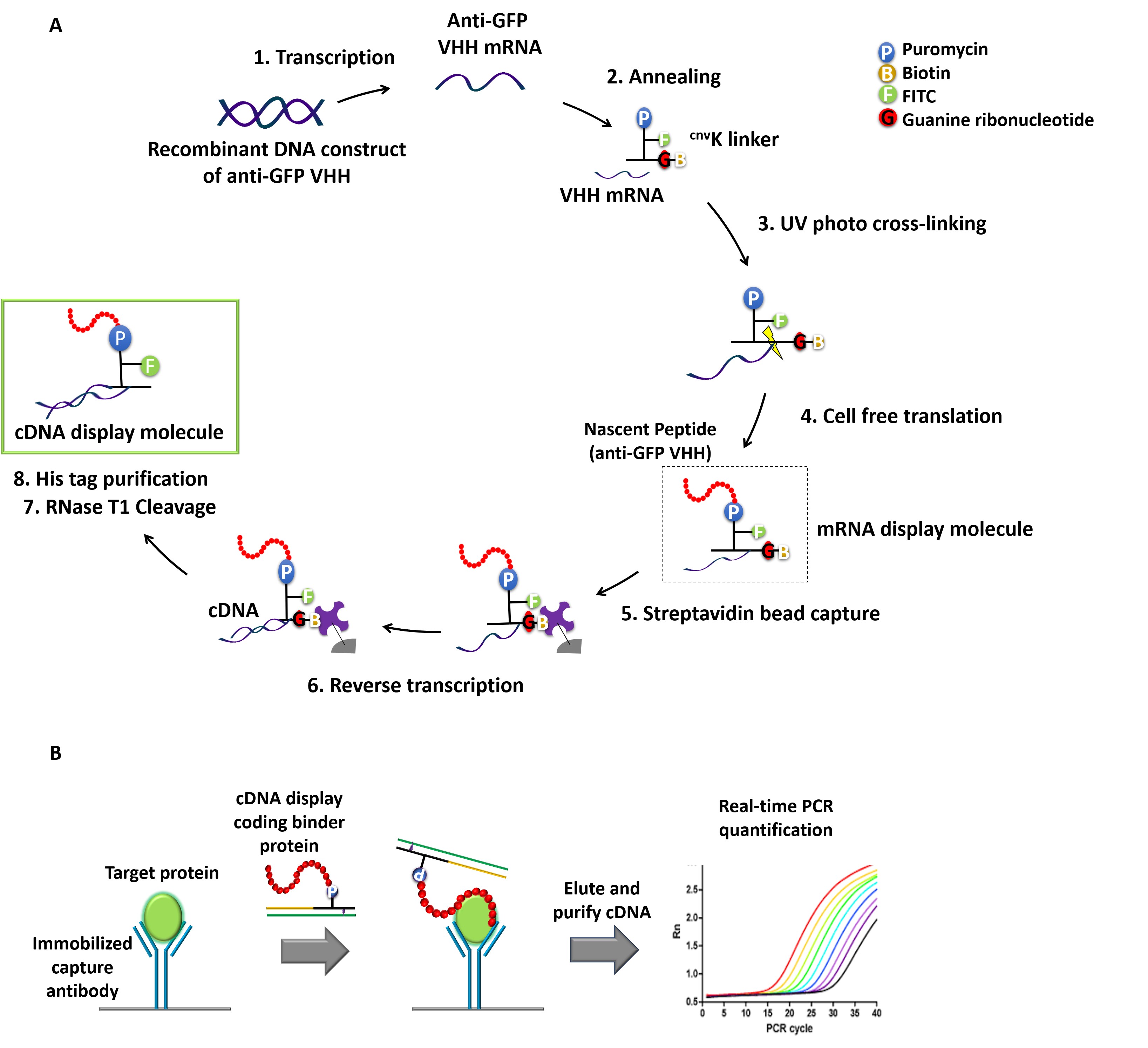
Figure 1. Schematic representation of cDNA displays mediated immuno-PCR (cD-IPCR). A. Preparation of cDNA display (schematic representation). B. Schematic diagram of cD-IPCR (sandwich-type detection). A target protein in a biological sample is captured on solid phase using a capture antibody. After washing, cDNA display of a polypeptide/VHH that has affinity to the target is added. Unreacted display molecules are washed away, and resulting cDNA is quantified by qPCR.
Materials and Reagents
Note: All reagents should be of molecular biology grade to avoid contamination of ribonuclease.
- Polystyrene microtiter plate (MICROLON, 96 Well Single-Break Strip Plate, PS) (Greiner Bio-One, catalog number: 705071)
- PCR tubes (End point PCR tubes; TreffLab Laboratory Consumables, catalog number: 96.09852.9.01, qPCR strip tubes; SSIBio, catalog number: 3248-00)
- cnvK-rG Puromycin-linker, store at -20 °C
Note: The components of cnvK-rG puromycin-linker can be obtained from custom DNA synthesis service. Please refer (Nemoto et al., 2018 or Terai et al., 2019) for the detailed protocol of formation. - DNA oligos can be obtained from custom DNA synthesis service. Eurofins Genomics (Ota-ku, Tokyo, Japan), Tsukuba Oligo Service (Tsuchiura, Ibaraki, Japan), and Hokkaido System Science (Sapporo, Hokkaido, Japan), store at -20 °C
- Nuclease-free ultra-pure distilled water (UPDW) (various suppliers)
- PrimeSTAR HS DNA polymerase (Takara, catalog number: R010A), store at -20 °C
- FavorPrep PCR Clean-Up Mini Kit (Favorgen), store at room temperature
- RiboMAXTM Large-Scale RNA Production System T7 (Promega, catalog number: P1300), store at -20 °C
- RNA Clean-Up Kit (Favorgen, catalog number: FAPCK001), store at 4 °C
- Nuclease treated Rabbit Reticulocyte Lysate System (Promega, catalog number: L4960). Amino acid mixtures for translation are included. Store at -80 °C
- RNasin® Ribonuclease Inhibitor (Promega; catalog number: N2111), store at -20 °C
- Dynabeads MyOne streptavidin C1 (VERITAS; catalog number: DB65002), store at 4 °C
- ReverTra Ace® (Toyobo, catalog number: TRT-101). 5x RT Buffer and 2.5 mM each dNTP mix are included. Store at -20 °C
- His Mag Sepharose Ni (GE Healthcare, catalog number: 2896390)
- RNase T1 (1,000 U/L) (Thermo Fisher Scientific, catalog number: EN0541)
- RNase H (Takara, catalog number: 2150A, 10 U) and 10x NE buffer 2 (NEB)
- DL-Dithiothreitol ≥ 99.0% (RT) (Sigma, catalog number: 4381)
- Quick-Load 100 bp DNA Ladder (Biolabs, catalog number: N0467), NEB
- SYBRTM Gold Nucleic Acid Gel Strain (Invitrogen, catalog number: S11494)
- THUNDERBIRD SYBR qPCR Mix (Toyobo, catalog number: QPS-201)
- 10x PBS (Phosphate-buffered saline-) (Wako, catalog number: 163-25265)
- Anti-GFP (Green Fluorescent Protein) pAb (MBL Life Science, catalog number: 598)
- Skim milk (Wako, catalog number: 190-12865)
- NaCl (Wako, catalog number: 191-01665)
- Tris-HCl (Wako, catalog number: 208-14691)
- KCl (Wako, catalog number: 163-03545)
- MgCl2 (Wako, catalog number: 136-03995)
- EDTA (Invitrogen, catalog number: 15575-038)
- Urea (Wako, catalog number: 217-01215)
- Bromophenol Blue (BPB) (Wako, catalog number: 021-02911)
- Xylene Cyanol (XC) (Wako, catalog number: 244-00461)
- Sucrose (Wako, catalog number: 196-00015)
- UltraPure DNase/RNase-Free Distilled Water (UPDW) (Invitrogen, catalog number: 10977-015)
- Ammonium persulfate (Wako, catalog number: 012-20503)
- Acrylamide (Nacalai Tesque, catalog number: 00807-05)
- Tetramethylethylenediamine (TEMED) (Wako, catalog number: 205-06313)
- Boric acid (Wako, catalog number: 021- 02195)
- Tween20 (Sigma-Aldrich, catalog number: P9416)
- Dibasic sodium phosphate (Wako, catalog number: 042-29445)
- Imidazole (Wako, catalog number: 095-05392)
- Glycine (Wako, catalog number: 077-00735)
- Hydrochloric acid (HCl) (Sigma-Aldrich, catalog number: 320331)
- Tris-borate-EDTA (TBE) Buffer (10x) (Invitrogen, catalog number: B52)
- Tris-EDTA (TE) buffer (Invitrogen, catalog number: AM9849)
- Tris base (Wako, catalog number: 011-20095)
- SDS (Wako, catalog number: 194-13985)
- APS (Wako, catalog number: 016-08021)
- BIS (N, N’-Methylene-bis (acrylamide) (Wako, catalog number: 130-06031)
- 1x SYBR Gold Nucleic Acid Gel Strain (stored at 4 °C, see Recipes)
- 2x loading dye (see Recipes)
- 4% Denaturing urea polyacrylamide gel electrophoresis (PAGE) 10 ml (see Recipes)
- 5x TBE buffer 1 L (see Recipes)
- 2x Binding buffer (see Recipes)
- Ni-NTA binding/wash buffer (see Recipes)
- Ni-NTA elution buffer (see Recipes)
- SDS-page gel (prepare and allow to set 2-3-hour prior to the experiment) (see Recipes)
10 ml of 6% separating gel and 5 ml of 4% stacking gel - 0.2 M Glycine-HCl buffer (see Recipes)
- 40% (w/v) Acrylamide (see Recipes)
Equipment
- Thermal cycler (Biometra, Model: TRIO48)
- Biomolecular imager (GE Healthcare, Model: Typhoon FLA9500 )
- NanoDrop Spectrophotometer (Thermo Fisher Scientific, Model: 1000 V3.3)
- NanoDrop Spectrophotometer (Thermo Fisher Scientific, Model: 3300 V2.7, FITC)
- Heat block (ANATECH, Model: cool stat 5200)
- UV Crosslinker (UVP, Model: CL-1000)
- Vortex mixer (IWAKI, Model: TM 2000)
- Magnetic separator (Invitrogen, Model: 12320D)
- Thermo block rotator (NISSIN, Model: SNP 24B)
- StepOne Real-Time PCR System (Thermo Fisher Scientific, Model: 4376600)
- High speed refrigerated microcentrifuge (Tomy Tech, Model: KITMAN MX-301)
- Gel electrophoresis apparatus (PAGE/SDS-PAGE) (ATTO, Model: AE 6510)
- Pipettes (Gilson Pipetman, Model P2- P1000)
Software
- Quantity One 1-D Analysis Software (Bio-Rad, Version 4.6.6)
- Primer Express (Thermo Fisher Scientific, Version 3.01)
Procedure
- Synthesis of cnvK-rG puromycin-linker (Figure 2)
Please refer (Nemoto et al., 2018 or Terai et al., 2019) for the detailed protocol of formation of the cnvK-rG puromycin-linker.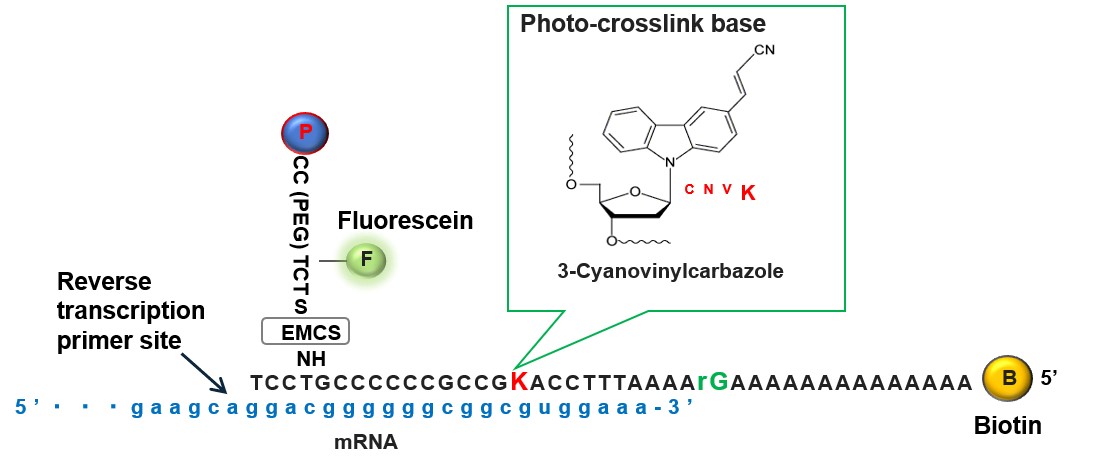
Figure 2. Schematic diagram of puromycin-linker DNA. The puromycin-linker DNA comprises four parts: a photo cross-linking site for mRNA (cnvK), a primer region for reverse transcription, a biotin moiety for immobilization onto SA beads for purification, and a restriction site for release from SA beads. In addition, the linker has puromycin moiety and FITC moiety for detection. - Construction anti-GFP VHH DNA templates and their PCR amplification
- Prepare the Anti-GFP VHH coding DNA construct as follows. If you want to use other VHHs, please change “Anti-GFP VHH” part. The DNA construct can be chemically synthesized as an artificial gene on a plasmid or overlap extension PCR from several fragments. Please ensure that there is no stop codon before cnvK NewYtag region.
GATCCCGCGAAATTAATACGACTCACTATAGGGGAAGTATTTTTACAACAATTACCAACAACAACAACAAACAACAACAACATTACATTTTACATTCTACA14-33 T7 34-36 5′ cap 37-107 Ω 110-114 kozak 118-498 Anti-GFP VHH 499-522 GGGSGGGS 523-540 His Tag 541-549 GGS 550-571 cnvK NewYtag
ACTACAAGCCACCATGGCCCAGGTGCAGCTGGTTGAAAGCGGTGGCCGTCTGGTGCAGGCGGGTGATAGCCTGCGTCTGAGCTGTGCCGCAAGCG
GTCGCACCTTTAGCACCAGCGCCATGGCATGGTTTCGTCAGGCCCCGGGCCGTGAACGCGAATTTGTGGCGGCCATTACCTGGACCGTTGGTAACAC
CATCCTGGGCGATAGCGTGAAAGGTCGTTTTACCATTAGCCGTGATCGCGCCAAAAACACCGTGGATCTGCAGATGGATAATCTGGAACCGGAAGATA
CCGCGGTTTATTATTGTAGCGCCCGTAGCCGCGGTTATGTGCTGAGCGTTCTGCGCAGCGTTGATAGCTATGATTATTGGGGTCAGGGCACCCAGGTT
ACGGTCAGCGGCGGCGGCTCGGGCGGCGGTTCCCATCATCATCATCATCATGGCGGAAGCAGGACGGGGGGCGGCGTGGAAA - Perform PCR amplification to generate appropriate DNA concentrations as follows: Mix 2 µl of DNA sample (~50 ng), 1 µl of 10 µM Forward primer, 1 µl of 10 µM Reverse primer, 4 µl of 2.5 mM dNTP Mix, 10 µl of 5x PrimeSTAR buffer (Mg2+), 0.5 µl of PrimeSTAR HS DNA polymerase (2.5 U/µl), and UPDW up to 50 µl in PCR tube.
- The PCR program is as follows: 98 °C for 1 min, 98 °C for 10 s, 64 °C for 5 s, 72 °C for 37 s (extension time is dependent on the DNA length), repeat 25 cycles of these three steps, followed by 72 °C for 2 min. Forward primer: 5′-GATCCCGCGAAATTAATACGACTCACTATAGGG-3’ and Reverse primer: 5′-TTTCCACGCCGCCCCCCGTCCT-3′.
- Analyze the amplification of DNA on 4% denaturing urea polyacrylamide gel electrophoresis (PAGE) using a gel imager as follows.
Note: Ensure there are no sub bands during amplification and if so, adjust the PCR cycles or cut the correct gel band and purify the DNA. - Mix 0.5 µl of DNA sample, 1.5 µl of UPDW and 2 µl of 2x loading buffer. Use 100 bp DNA Ladder as the marker (Recipe 1).
- Denature the DNA by heating at 85 °C for 5 min before loading.
- Perform PAGE analyses for 20 min at 60 °C, 200 V using gels (1 mm thickness) containing 8 M urea (Recipe 2), with 0.5x TBE as a running buffer (Recipe 3).
- Purify the DNA using FavorPrep DNA purification kit. Use UPDW as eluent.
- Measure the DNA concentration using NanoDrop.
- Prepare the Anti-GFP VHH coding DNA construct as follows. If you want to use other VHHs, please change “Anti-GFP VHH” part. The DNA construct can be chemically synthesized as an artificial gene on a plasmid or overlap extension PCR from several fragments. Please ensure that there is no stop codon before cnvK NewYtag region.
- Synthesis of cDNA display molecules
- In vitro transcription
- Prepare the transcription reaction solution as follows: mix 4 µl of 5x T7 transcription buffer, 6 µl of 25 mM each rNTP mix [mix equal volumes of four individual 100 mM rNTPs (rATP, rCTP, rGTP, and rUTP)], 200 ng of the template DNA, 2 µl of T7 Enzyme Mix, and add nuclease-free water up to 20 µl.
- ncubate at 37 °C for 2-4 h on a heat block.
Note: The amount of mRNA will depend on the incubation time. - Add 2 µl of RQ-1 RNase-free DNase and incubate at 37 °C for 15 min on a heat block.
- Purify the transcript (mRNA) with RNA Clean-Up kit.
- Determine its concentration by absorbance at 260 nm using NanoDrop.
Note: The concentration of mRNA should be over 2 pmol/µl and the ratio of 260/280 nm should be over 1.8. - Store the mRNA at -80 °C until use.
- Photo-crosslinking between mRNA and cnvK-rG puromycin-linker
- Prepare the photo-crosslinking reaction solution as follows: mix 4 µl of 1 M NaCl, 4 µl of 0.25 M Tris-HCl (pH 7.5), 20 pmol of cnvK-rG puromycin-linker, 20 pmol of mRNA, and nuclease-free water up to 20 µl.
Note: It is convenient to prepare working solution of the ligation product (mRNA-puromycin-linker) as 1 pmol/µl. - Incubate at 90 °C for 1 min followed by lowering the temperature to 70 °C at a rate of 0.4 °C/s and incubate for 1 min and then cool down to 25 °C at a rate of 0.08 °C/s using a thermocycler.
- Irradiate (in the same tube) with UV light at 365 nm for about 30 s using a UV cross linker.
- Prepare the samples for gel electrophoresis the same as described above in Procedure B.
- Analyze the photo-crosslinked product on a 4% denaturing PAGE containing 8 M urea and visualize the FITC motif in Puromycin-linker using a fluorescence gel imager (Data analysis A).
Note: Binding efficiency can be estimated by analyzing the FITC band intensity ratios of bound and unbound puromycin-linker.
After that, stain the same gel with SYBR Gold (Recipe 9) for ~1 min, wash with water, and take an image. - Store the ligated sample at -80 °C until use. Keep 0.5 µl of the sample separately for subsequent SDS-PAGE analysis (Procedure D).
- Prepare the photo-crosslinking reaction solution as follows: mix 4 µl of 1 M NaCl, 4 µl of 0.25 M Tris-HCl (pH 7.5), 20 pmol of cnvK-rG puromycin-linker, 20 pmol of mRNA, and nuclease-free water up to 20 µl.
- In vitro translation (synthesis of mRNA-VHH fusion molecule)
- Prepare the translation mixture as follows: mix 6 pmol/6 µl of photo-crosslinked product, 35 µl of Rabbit Reticulocyte Lysate, 1 µl of Amino Acid Translation Mixture, 1 µl of RNasin® Ribonuclease Inhibitor, and nuclease-free water up to 50 µl.
Note: As the working solution of the mRNA-Puromycin-linker is 1 pmol/µl, 6 µl equals to 6 pmol. The reaction volume should be less than 50 µl, because of higher translation efficiency. If the reaction needs to be scaled up, prepare 50 µl of the reaction solution in several test tubes. - Incubate the mixture at 30 °C for 20 min on a heat block.
- Add 24 µl of 3 M KCl and 6 µl of 1 M MgCl2 to each tube. Incubate at 37 °C for 60 min.
- Add 18 µl of 0.5 M EDTA and 98 µl (same volume to the mixture) of 2x binding buffer (Recipe 4). Incubate at 37 °C for 5 min.
- Collect 16.3 µl of translation reaction sample into a separate tube for subsequent SDS-PAGE analysis (Procedure D).
- Prepare the translation mixture as follows: mix 6 pmol/6 µl of photo-crosslinked product, 35 µl of Rabbit Reticulocyte Lysate, 1 µl of Amino Acid Translation Mixture, 1 µl of RNasin® Ribonuclease Inhibitor, and nuclease-free water up to 50 µl.
- Immobilization of mRNA-VHH fusion molecule on streptavidin magnetic beads
- Take 10 µl suspension of streptavidin (SA) magnetic beads for 1 pmol of the photo-crosslinked product (i.e., 60 µl for 6 pmol) and wash the beads twice with 200 µl of 1x binding buffer.
Note: All washing steps should be performed at room temperature. - Resuspend the beads with rest of the translation reaction sample.
Note: If the reaction volume is too large for one tube, use several tubes with beads. It is not necessary to cut tips during pipetting beads. - Incubate the mixture in a thermo block rotator at 25 °C for 30 min.
Note: Keep the beads in suspension during the incubation to avoid the aggregation of beads. - Place the tube on a magnetic separator for 1 min and collect 16.3 µl of supernatant into a separate tube for subsequent SDS-PAGE analysis (Procedure D).
- Discard the rest of the supernatant and wash the beads with 200 µl of 1x binding buffer for three times.
- Take 10 µl suspension of streptavidin (SA) magnetic beads for 1 pmol of the photo-crosslinked product (i.e., 60 µl for 6 pmol) and wash the beads twice with 200 µl of 1x binding buffer.
- Reverse transcription to form cDNA display molecules.
- Wash the mRNA-VHH fusion molecules immobilized on SA beads (from above procedures) with 100 µl of 1x RT Buffer.
- Prepare the reverse transcription (RT) reaction solution for 60 µl of SA beads as follows: mix 10 µl of 5x RT Buffer, 25 µl of 2 mM dNTP mix, 14 µl of nuclease-free water, and 1 µl of ReverTra Ace® (100 U/µl).
- Resuspend the SA beads in RT reaction solution and incubate at 42 °C for 30 min using a thermo block rotator.
Note: Keep the beads in suspension during the incubation to avoid the aggregation of beads. - Place the tube on a magnetic separator for 1 min and discard the supernatant, wash the beads twice with 200 µl of 1x binding buffer and once with 100 µl of 1x Ni-NTA binding/wash buffer (Recipe 5).
- Resuspend the above beads with 29.8 µl of 1x Ni-NTA binding/wash buffer and add 0.2 µl of RNase T1 (1,000 U/µl). Incubate using the rotator at 37 °C for 15 min.
Note: Keep the beads in suspension during the incubation to avoid the aggregation of beads. - Place the tube on a magnetic separator for 1 min and collect the supernatant containing cDNA display molecules.
- Save 2.7 µl of the aliquots for SDS-PAGE analysis (Procedure D).
- His-tag affinity purification of cDNA display
- Take 20 µl of His Mag Sepharose Ni beads into a tube and wash twice with 100 µl of the 1× Ni-NTA binding/wash buffer.
- Resuspend the beads in mRNA/cDNA-VHH fusion molecule solution (supernatant of RNase T1 cleavage) and incubate at 25 °C for 120 min using the rotator.
Note: Adjust the incubation temperature accordingly with incubation time to prevent DNA degradation. - Place the tube on a magnetic separator for 1 min and collect 2.7 µl of supernatant into a separate tube for subsequent SDS-PAGE analysis (Procedure D).
- Discard the rest of the supernatant and wash the beads twice with 100 µl of 1x Ni-NTA binding/wash buffer.
- Resuspend the beads in 25 µl Ni-NTA elution buffer (Recipe 6) and incubate using the rotator at 25 °C for 20 min.
Note: Elution volume can be adjusted as needed and can be used for immunoassay after appropriate dilutions. - Place the tube on a magnetic separator for 1 min and collect the supernatant containing purified cDNA display molecules.
- Save 2.5 µl of the aliquots for SDS-PAGE analysis (Procedure D).
- In vitro transcription
- Confirmation of cDNA display formation by SDS-PAGE
- Aliquots collected during the above process (Procedure C) correspond to 0.5 pmol of molecules each.
(1) mRNA-puromycin linker 0.5 µl (2) Input (mRNA-VHH fusion molecules after translation) 16.3 µl (3) Supernatant of SA-beads immobilization 16.3 µl (4) Elution of RNase T1 treatment (crude cDNA display) 2.7 µl (5) Supernatant of Ni beads immobilization 2.7 µl (6) Ni-NTA elution (purified cDNA display) 2.5 µl - Incubate sample (4), (5) and (6) with 0.2 µl RNase H, 1 µl 10x NE buffer 2 and nuclease-free water (adjust the final volume up to 10 µl) at 37 °C for 30 min to digest the RNA/cDNA duplex.
- For sample (1), add 1.5 µl of UPDW and 2 µl of 2x SDS sample buffer
- For samples (4), (5) and (6) after RNase H incubation, add 10 µl of 2x SDS sample buffer.
- For samples (2) and (3), add 16.3 µl of 2x SDS sample buffer.
- Incubate all samples for 5 min at 95 °C in a thermoblock before loading on a gel.
- Analyze the samples in SDS-PAGE containing 8 M urea (4% stacking-6% separating gel, 0.02 A, 2 h) and visualize the FITC motif in puromycin-linker using a fluorescence scanner (Recipe 7).
- Calculate the band intensity ratio of (1) and (6) to estimate the cDNA display formation efficiency (Data analysis B).
- Aliquots collected during the above process (Procedure C) correspond to 0.5 pmol of molecules each.
- Sandwich cD-IPCR for target protein (sfGFP) detection
- Coat a polystyrene microtiter plate with 100 µl of anti-GFP pAb [anti-GFP pAb in PBS (dilution = 1:450)] and keep at 4 °C overnight.
- Wash the plates 3 times with 200 µl of PBS each.
- Add 200 µl of 1% skim milk in PBS (w/v) and incubate for 2 h at 25 °C to block the wells.
- Washed out the plates with 200 µl of PBS.
- Prepare 10-fold serial dilutions of GFP protein in PBS from 100 µg/ml to 1 ng/ml as well as the negative control (no GFP). It is recommended that all the samples should be prepared in triplicate or more.
Note: In this study, we used superfolder GFP (sfGFP) as the target protein. The amino acid sequence and the methods of expression and purification of the protein is described in the original paper (Pédelacq et al., 2006). - Add 100 µl of each dilution of GFP protein to the wells and incubate at 25 °C for 2 h.
- Wash with 200 µl of PBS, 8 times.
- Dilute the cDNA display coding anti-GFP VHH (prepared as described in Procedure C) up to 65-fold with PBS-T buffer.
Note: The dilution value should be changed and decided empirically depending on the target. - Add 100 µl of cDNA display (corresponded to ~1.3 fmol/sample) to wells and incubate at 25 °C for 2 h.
Note: It is not necessary to shake the plates during incubation. - Wash with 200 µl of PBS-T, 8 times. (0.05% Tween 20 in PBS)
- Elute the bound cDNA display with 100 µl of 0.2 M glycine/HCl buffer (pH 2.2) for 15 min at 25 °C (Recipe 8).
- Neutralize the elute DNA with 20 µl of 1 M Tris-base solution and purify the cDNA display using a Favorprep kit.
- Use 2-8 µl of the purified cDNA display as template for the following qPCR.
- Perform the qPCR. The step program for PCR was as follows: 95 °C for 1 min, followed by 40 cycles of 95 °C for 15 s and 66 °C for 30 s. Forward primer: AACACCATCCTGGGCGATAG, Reverse primer: GTGTTTTTGGCGCGATCAC. The PCR mixture was prepared as follows: 10 µl of 2x reaction buffer (including enzyme and dNTP), 0.6 µl of each primer (10 µM), 2 µl of template, 0.4 µl of reference dye (ROX) and 6.4 µl of water.
Note: These primers are specific to anti-GFP VHH. So, if another VHH or peptide aptamer is used, design an appropriate primer set using software such as Primer Express 3.01. - When performing qPCR, use anti-GFP VHH DNA to prepare the standard curve (100 pM, 1 pM, 10 fM,100 aM), and include a primer only control to verify the quality of amplification.
- Calculate the difference of Ct values between the samples and the negative control containing no GFP (Data analysis C).
Data analysis
- Photo-crosslinking of mRNA and cnvK-rG puromycin-linker
- Save the image data and open the file with Quantity One 1-D Analysis Software. Representative images of a PAGE gel of photo-ligation samples are shown in Figure 3.
Note: The digital images should not be over-exposed for optimal quantification.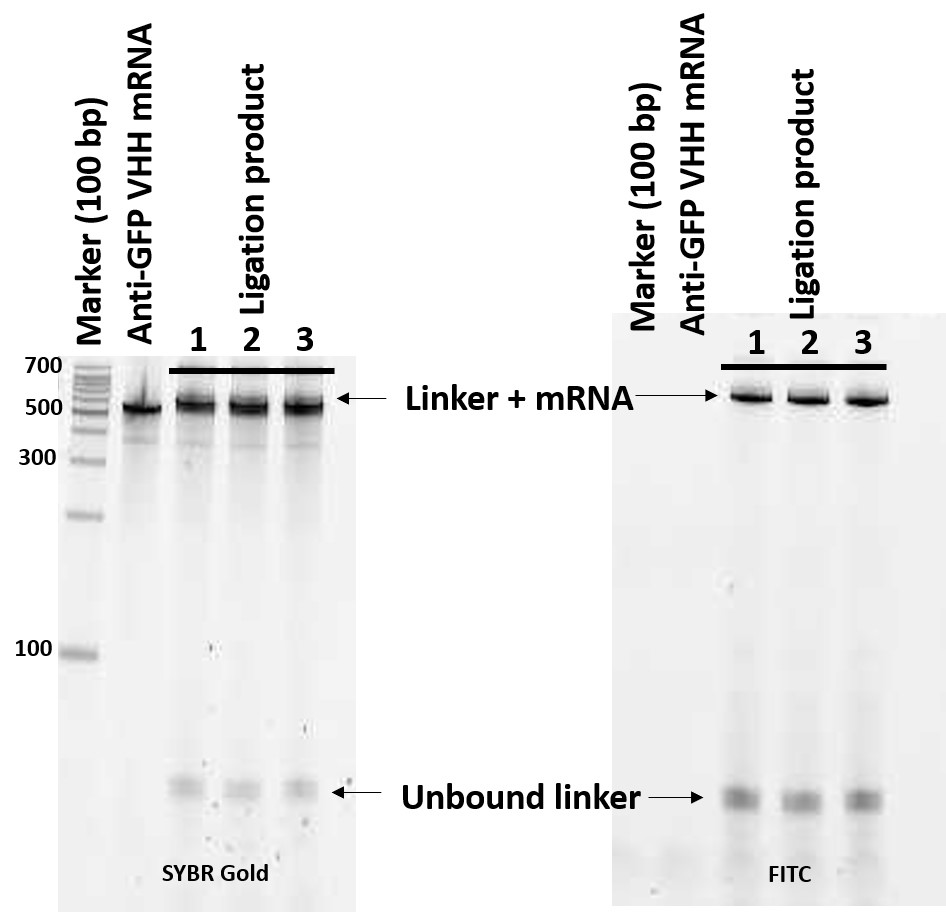
Figure 3. Visualization of photo-crosslinked product on denaturing PAGE using the FITC motif in puromycin-linker (right) and after SYBR Gold staining (left) - Confirm the position of ligated mRNA (linker + mRNA, it should appear at slightly higher position than intact mRNA) and that no sub band in SYBR Gold-stained gel.
- Quantify the band intensity of ligated mRNA and unbound linker in FITC-imaged gel. If more than about 80% of the linker is ligated, proceed to the next step (in vitro translation). If not, change the stoichiometry of mRNA and linker (in the range between 1:2 and 2:1) and perform photo-ligation again.
Note: It is recommended to have more than 80% of the linker to be ligated to mRNA because remaining free linkers in the protein translation solution may end up inefficient cDNA display formation.
- Save the image data and open the file with Quantity One 1-D Analysis Software. Representative images of a PAGE gel of photo-ligation samples are shown in Figure 3.
- Formation of cDNA display
- Visualize the gel of SDS-PAGE with FITC filter set. Save the data and open the file with Quantity One 1-D Analysis Software. A representative image is shown in Figure 4A. Please note that it corresponds to the gel of a different model protein, because formation efficiency of anti-GFP VHH is low and hard to analyze.
Note: If the readers want to check the data of anti-GFP VHH, please refer to Anzai et al., 2019. - Calculate the band intensity percentage of each band (with appropriate background subtraction) in the gel using the software. Use “volume rectangle” function to indicate regions of interest and “volume analysis report” function to show the results. Since all samples are corresponding to the same amount of moles, you can directly compare the band intensities with each other. Band intensities of a model protein is given in Figure 4B for better understanding.
Note: “Background” is the free gel space without any band. - The formation efficiency of cDNA display (i.e., yield of cDNA display from mRNA-linker) is the percentage value of band intensity ratio between sample (1) and (6). For example, when we compare the (1) mRNA-puromycin linker (whose intensity occupies 65.55% of all bands) and (6) Ni-NTA elution (purified cDNA display, 5.15%) intensities, the cDNA display formation efficiency is calculated to be 7.85%. Generally, the formation efficiencies of cDNA display for small polypeptides are approximately 5-25%.
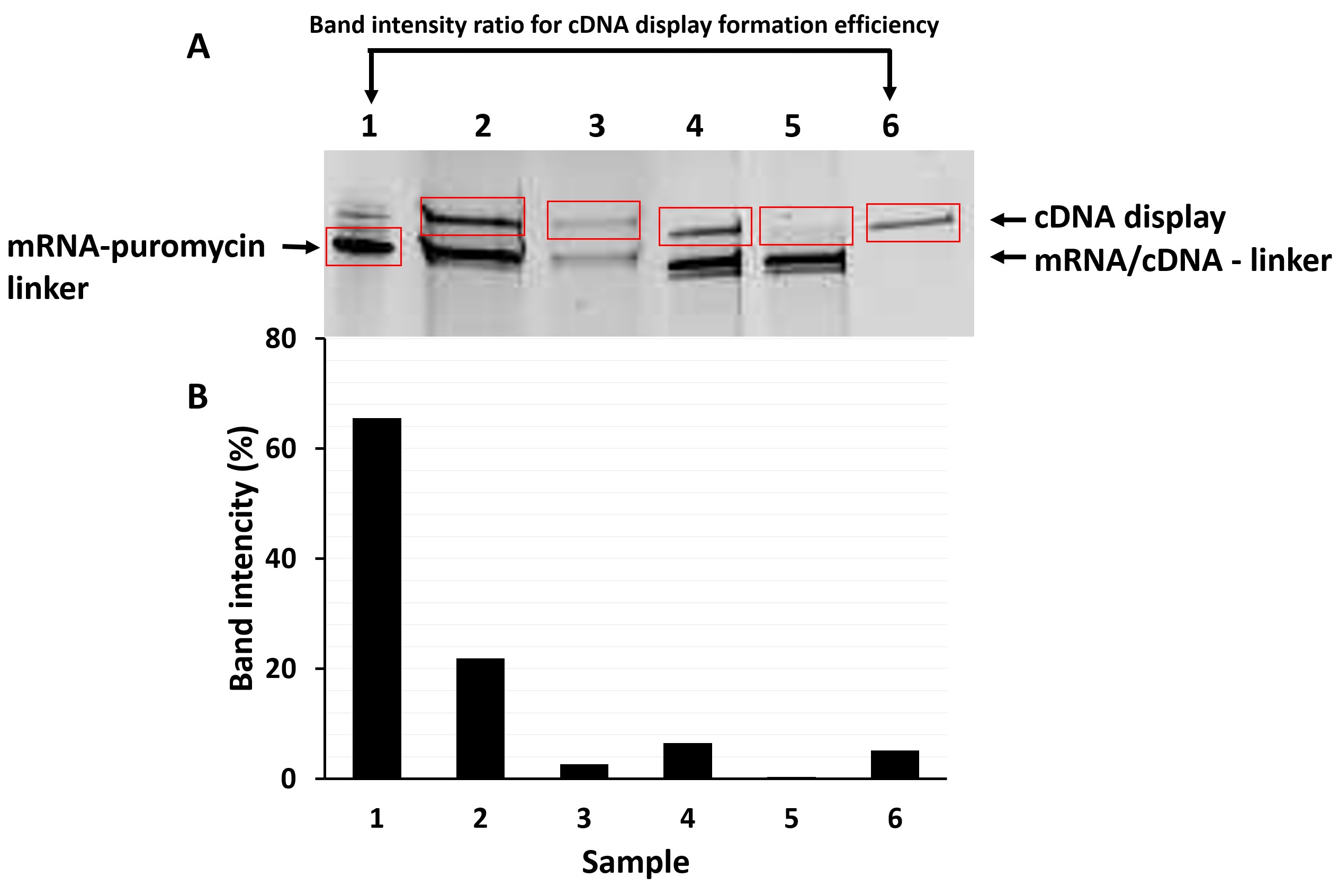
Figure 4. Confirmation of cDNA display formation of VHH. A. Example of an image of SDS-PAGE gel using FITC filter set. Upper bands are responsible for the correctly formed cDNA display. Note that the lower band corresponds to the mRNA-linker that was not conjugated to a peptide. Red rectangles indicate the ROIs used for intensity calculation. B. Corresponding band intensities calculated from data of Figure 4A. (1) mRNA-puromycin linker (65.55%), (2) Input (mRNA-VHH fusion molecules after translation) (21.88%), (3) Supernatant of SA-beads immobilization (2.59%), (4) Elution of RNase T1 treatment (crude cDNA display) (6.49%), (5) Supernatant of Ni beads immobilization (0.32%), (6) Ni-NTA elution (purified cDNA display) (5.15%).
- Visualize the gel of SDS-PAGE with FITC filter set. Save the data and open the file with Quantity One 1-D Analysis Software. A representative image is shown in Figure 4A. Please note that it corresponds to the gel of a different model protein, because formation efficiency of anti-GFP VHH is low and hard to analyze.
- Sandwich CD-IPCR detection of target protein GFP
- After performing a qPCR experiment, the software attached to the instrument automatically prepare amplification plot and calculate the Ct (threshold cycle) value of each sample (Figure 5). Representative data of Ct values are shown in Table 1.
Table 1. Ct values from qPCR reaction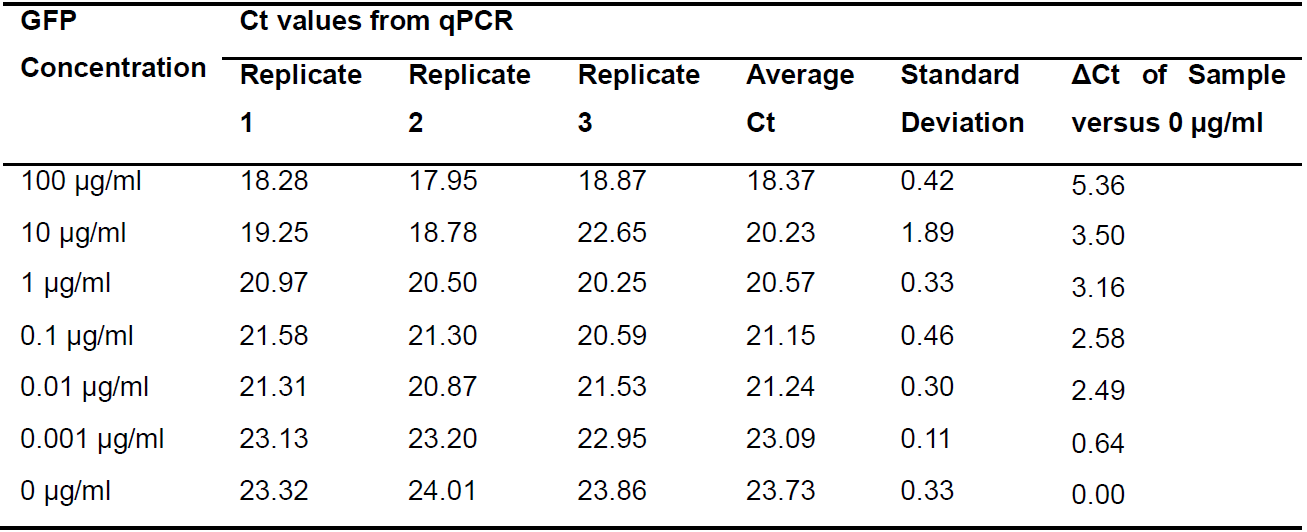
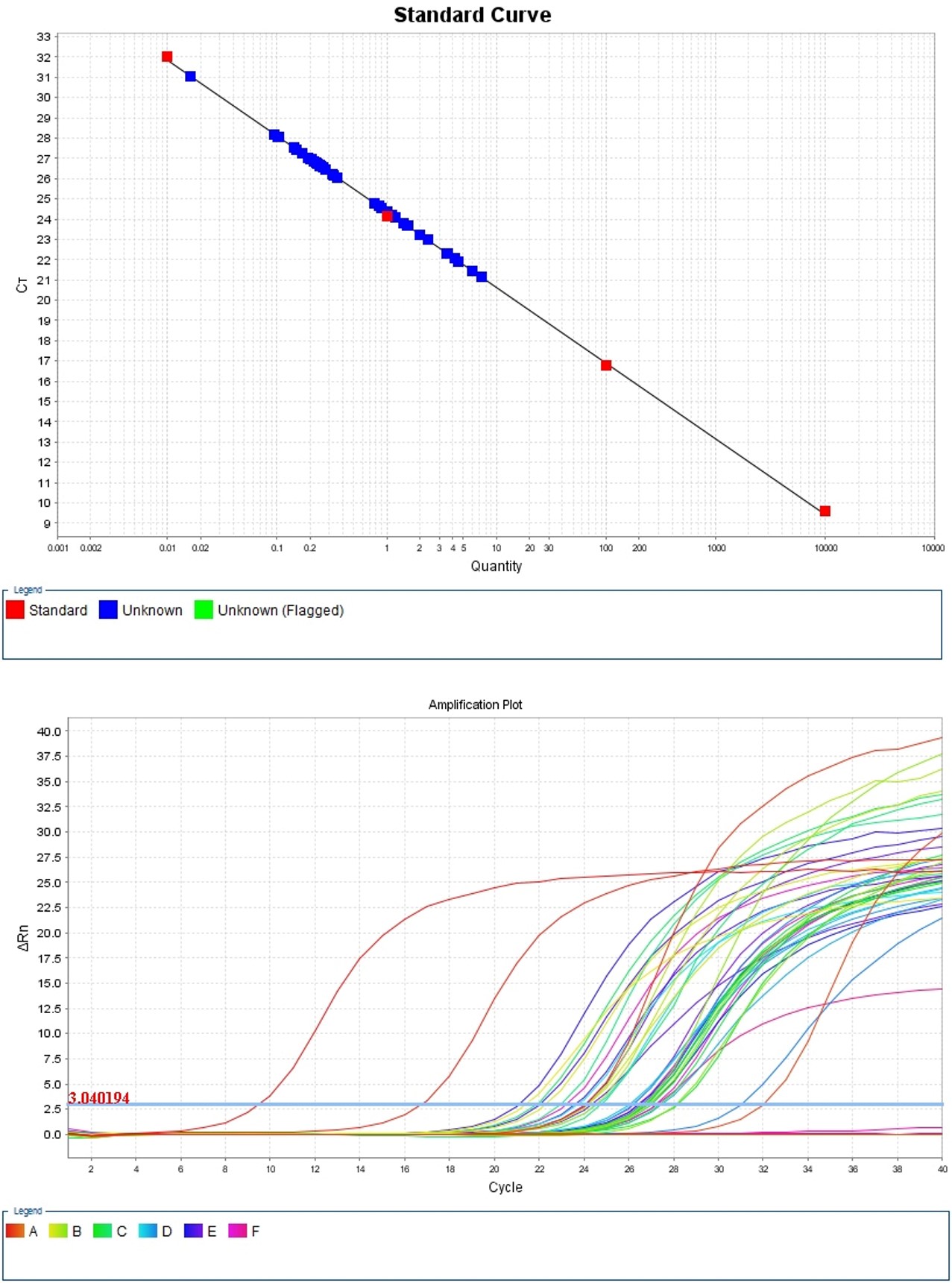
Figure 5. Standard curve and amplification plot for the qPCR reaction - Calculate the ΔCt values by substracting the Ct value of 0 µg/ml (no analyte) from sample Ct value for each GFP concentration (see Table 1).
- Compare the ΔCt values statistically using t-test. The ΔCt value is gradually increased with the GFP concentration (from 0.01 µg/ml to 100 µg/ml), and 0.01 µg/ml is appeared as the lowest detected concentration according to the statistical analysis (Figure 6).
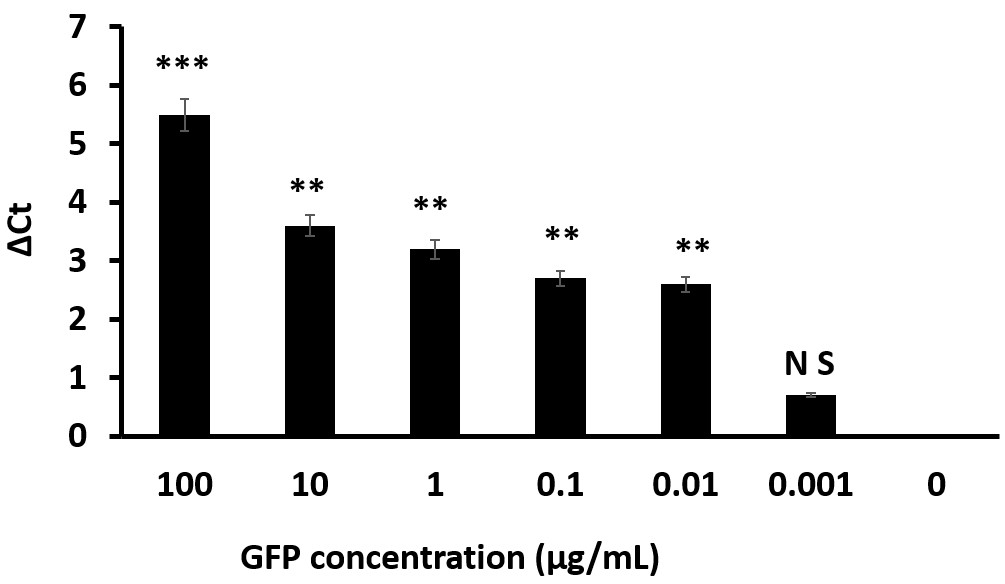
Figure 6. ΔCt values for sandwich CD-IPCR detection of target protein GFP. *** indicates P < 0.05, ** indicates P < 0.001. n = 3 (error bars indicate S.D.).
- After performing a qPCR experiment, the software attached to the instrument automatically prepare amplification plot and calculate the Ct (threshold cycle) value of each sample (Figure 5). Representative data of Ct values are shown in Table 1.
Recipes
- 2x Loading dye (store at room temperature)
Urea 24 g
1% BPB 2.5 ml
1% XC 2.5 ml
0.5 M EDTA 0.1 ml
10x TBE buffer 1 ml
Sucrose 4 g
Up to 50 ml by UPDW - 4% Denaturing urea polyacrylamide gel electrophoresis (PAGE) 10 ml (prepare and allow to set 2-3-h prior to the experiment)
4.8 g 8 M Urea
1 ml 5x TBE buffer
1 ml 40% Polyacrylamide and UPDW up to 10 ml
Microwave the content for proper dissolving and let cool
Add 25 μl of 20% Ammonium Persulfate (APS) and 10 μl of Tetramethylethylenediamine (TEMED) immediately before pouring into gel mold - 5x TBE buffer 1 L (store at room temperature)
54 g of Tris base
27.5 g of Boric acid
20 ml of 0.5 M EDTA (pH 8.0)
The 0.5x working solution is 45 mM Tris-borate/1 mM EDTA - 2x Binding buffer (store at 4 °C)
20 mM Tris-HCl, pH 8.0
2 mM EDTA
2 M NaCl
0.2% Tween20
Prepare with nuclease-free water - Ni-NTA binding/wash buffer (store at 4 °C)
20 mM Sodium phosphate dibasic, pH 7.4, 50 mM Tris-Cl (pH 8.0)
0.5 M NaCl
5 mM Imidazole
0.05% Tween20 - Ni-NTA elution buffer (store at 4 °C)
20 mM Sodium phosphate dibasic, pH 7.4, 50 mM Tris-Cl (pH 8.0)
500 mM NaCl
250 mM Imidazole
0.05% Tween20 - SDS-page gel (prepare and allow to set 2-3-hour prior to the experiment)
10 ml of 6% separating gel
4.8 g of urea (final 8 M)
1.5 ml of acrylamide (40 %)
2.5 ml of 1.5 M Tris (pH 8.8)
100 µl of SDS (10%)
25 µl of APS (20%)
5 µl of TEMED
Up to 10 ml with UPDW
5 ml of 4% stacking gel
0.5 ml of acrylamide (40 %)
0.5 ml of 1 M Tris (pH 6.8)
50 µl of SDS (10%)
15 µl of APS (10%)
5 µl of TEMED
Up to 5 ml with UPDW - 0.2 M Glycine-HCl buffer (pH 2.2) (store at room temperature)
25 ml of 0.2 M Glycine (15.01 g in 1 L distilled water)
22 ml of 0.2 M HCl
Up to total of 100 ml distilled water - 1x SYBR Gold Nucleic Acid Gel Strain (store at 4 °C)
5 µl of 10,000x SYBRTM Gold Nucleic Acid Gel Stain
50 ml of 1x Tris-EDTA (TE) buffer - 40% acrylamide
Compound for PAGE for SDS-PAGE Acrylamide 190 g 194.8 g Bis-acrylamide 10 g 5.2 g UPDW 500 ml 500 ml
Acknowledgments
This work was in part supported by KAKENHI (15H05723 and 17K19471 to N.N., 16J01710, 18H04535, and 18H02103 to T.T.). T.T. was also supported by a JSPS fellowship. This protocol was adapted from our recent article (Anzai et al., 2019). We especially thank Mr. Hiroki Anzai and Mr. Takeru Suzuki for conducting the parts of the original work and also other members in Nemoto laboratory, Saitama University for valuable advice and support on this work.
Competing interests
The authors have filed a patent for cD-IPCR.
References
- Anzai, H., Terai, T., Jayathilake, C., Suzuki, T. and Nemoto, N. (2019). A novel immuno-PCR method using cDNA display. Anal Biochem 578: 1-6.
- Fridy, P. C., Li, Y., Keegan, S., Thompson, M. K., Nudelman, I., Scheid, J. F., Oeffinger, M., Nussenzweig, M. C., Fenyö, D., Chait, B. T. and Rout, M. P. (2014). A robust pipeline for rapid production of versatile nanobody repertoires. Nat Methods 11(12): 1253-1260.
- Guo, Y. C., Zhou, Y. F., Zhang, X. E., Zhang, Z. P., Qiao, Y. M., Bi, L. J., Wen, J. K., Liang, M. F. and Zhang, J. B. (2006). Phage display mediated immuno-PCR. Nucleic Acids Res 34(8): e62.
- Hendrickson, E. R., Truby, T. M., Joerger, R. D., Majarian, W. R. and Ebersole, R. C. (1995). High sensitivity multianalyte immunoassay using covalent DNA-labeled antibodies and polymerase chain reaction. Nucleic Acids Res 23(3): 522-529.
- Mochizuki, Y., Biyani, M., Tsuji-Ueno, S., Suzuki, M., Nishigaki, K., Husimi, Y. and Nemoto, N. (2011). One-pot preparation of mRNA/cDNA display by a novel and versatile puromycin-linker DNA. ACS Comb Sci 13(5): 478-485.
- Nemoto, N., Kumachi, S. and Arai, H. (2018). In vitro selection of single-domain antibody (VHH) using cDNA display. Methods Mol Biol 1827: 269-285.
- Pédelacq, J. D., Cabantous, S., Tran, T., Terwilliger, T. C. and Waldo, G. S. (2006). Engineering and characterization of a superfolder green fluorescent protein. Nat Biotechnol 24(1): 79-88.
- Sano, T., Smith, C. L. and Cantor, C. R. (1992). Immuno-PCR: very sensitive antigen detection by means of specific antibody-DNA conjugates. Science 258(5079): 120-122.
- Suzuki, T., Mochizuki, Y., Kimura, S., Akazawa-Ogawa, Y., Hagihara, Y. and Nemoto, N. (2018). Anti-survivin single-domain antibodies derived from an artificial library including three synthetic random regions by in vitro selection using cDNA display. Biochem Biophys Res Commun 503(3): 2054-2060.
- Terai, T., Anzai, H. and Nemoto, N. (2019). Selection of peptides that associate with Dye-conjugated solid surfaces in a pH-dependent manner using cDNA display. ACS Omega 4(4): 7378-7384
- van Buggenum, J. A. G. L., Gerlach, J. P., Eising, S., Schoonen, L., van Eijl, R. A. P. M., Tanis, S. E. J., Hogeweg, M., Hubner, N. C., van Hest, J. C., Bonger, K. M. and Mulder, K. W. (2016). A covalent and cleavable antibody-DNA conjugation strategy for sensitive protein detection via immuno-PCR. Sci Rep 6: 22675.
- Yamaguchi, J., Naimuddin, M., Biyani, M., Sasaki, T., Machida, M., Kubo, T., Funatsu, T., Husimi, Y. and Nemoto N. (2009). cDNA display: a novel screening method for functional disulfide-rich peptides by solid-phase synthesis and stabilization of mRNA-protein fusions. Nucleic Acids Res 37(16):e108.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Jayathilake, C., Terai, T. and Nemoto, N. (2019). cDNA Display Mediated Immuno-PCR (cD-IPCR): A Novel PCR-based Antigen Detection Method . Bio-protocol 9(24): e3457. DOI: 10.21769/BioProtoc.3457.
Category
Immunology > Antibody analysis > Antibody detection
Cancer Biology > Cancer biochemistry > Cancer metabolism
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.