- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Enhanced-ice-COLD-PCR for the Sensitive Detection of Rare DNA Methylation Patterns in Liquid Biopsies

Published: Vol 9, Iss 23, Dec 5, 2019 DOI: 10.21769/BioProtoc.3452 Views: 6429
Reviewed by: Nicoletta CordaniMarija MojicAnonymous reviewer(s)
Original research article
The authors used this protocol in:

May 2018
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics








Abstract
In the context of precision medicine, the identification of novel biomarkers for the diagnosis of disease, prognosis, predicting treatment outcome and monitoring of treatment success is of great importance. The analysis of methylated circulating-cell free DNA provides great promise to complement or replace genetic markers for these applications, but is associated with substantial challenges. This is particularly true for the detection of rare methylated DNA molecules in a limited amount of sample such as tumor released hypermethylated molecules in the background of DNA fragments from normal cells, especially lymphocytes.
Technologies for the sensitive detection of DNA methylation have been developed to enrich specifically methylated DNA or unmethylated DNA using among other methods: enzymatic digestion, methylation-specific PCR (often combined with TaqMan like oligonucleotide probes (MethyLight)) and co-amplification at lower denaturation temperature PCR (COLD-PCR).
E-ice-COLD-PCR (Enhanced-improved and complete enrichment-COLD-PCR) is a sensitive method that takes advantage of a Locked Nucleic Acid (LNA)-containing oligonucleotide probe to block specifically unmethylated CpG sites allowing the strong enrichment of low-abundant methylated CpG sites from a limited quantity of input. E-ice-COLD-PCRs are performed on bisulfite-converted DNA followed by Pyrosequencing analysis. The quantification of the initially present DNA methylation level is obtained using calibration curves of methylated and unmethylated DNA. The E-ice-COLD-PCR reactions can be multiplexed, allowing the analysis and quantification of the DNA methylation level of several target genes. In contrast to the above-mentioned assays, E-ice-COLD-PCR will also perform in the presence of frequently occurring heterogeneous DNA methylation patterns at the target sites. The presented protocol describes the development of an E-ice-COLD-PCR assay including assay design, optimization of E-ice-COLD-PCR conditions including annealing temperature, critical temperature and concentration of LNA blocker probe followed by Pyrosequencing analysis.
Background
In the context of precision medicine, the discovery of novel non-invasive biomarkers in clinical samples such as circulating cell-free DNA (ccfDNA) is crucial for improving the diagnosis, prognosis, predicting and monitoring the response to a patient-tailored treatment regimen against cancer and other diseases (Siravegna et al., 2017, Wan et al., 2017; Cabel et al., 2018; Barlebo Ahlborn and Østrup, 2019). The analysis of ccfDNA is challenging because the outcome depends on multiple pre-analytical steps and requires the development of methods for the detection of genetic and epigenetic variations that are present in limited quantity in highly fragmented samples (Lewis et al., 2015; Kumar et al., 2018).
Especially, methylated ccfDNA could be a promising non-invasive biomarker for cancer and potentially other diseases (Lehmann-Werman et al., 2016; Warton et al., 2016). In comparison to mutations and other genetic changes, DNA methylation changes occur early during carcinogenesis and are also present in a number of other complex non-neoplastic human diseases. Furthermore, DNA methylation changes are often restricted to a genomic region of limited size, for example the promoter associated CpG island or the transcription start site in contrast to genetic mutations that might be present all along the gene. The most commonly used methods for the analysis of DNA methylation patterns are based on a bisulfite reaction that converts cytosine bases into uracil bases while 5-methylcytosine bases are not converted (Frommer et al., 1992). Bisulfite conversion thus translates a DNA methylation difference into a sequence change. DNA methylation patterns are then read-out using next generation sequencing or microarrays for genome-wide analysis or Pyrosequencing, methylation-sensitive or methylation-specific PCR (MSP) methods for locus-specific analysis (Tost, 2016).
Different DNA methylation enrichment approaches for the analysis of low DNA methylation levels have been developed based on digestion or oligonucleotide probes (Liu et al., 2017; Campan et al., 2018; Distler, 2019). For example, the HeavyMethyl method which is based on enrichment of methylated DNA by blocker probes competing with the amplification primers on the target site, is used for a commercially available non-invasive blood based test (Molnár et al., 2015; Jung et al., 2018).
While MSP and MethyLight will specifically amplify methylated molecules in the presence of a 1,000 to 10,000 fold excess of unmethylated molecules, the efficient amplification relies on a pre-defined consistent methylation profile underlying the bindings sites for the amplification primers and the hydrolysis probe (for MethyLight), usually completely methylated molecules (Candiloro et al., 2011). However, in the presence of heterogeneous DNA methylation patterns, the assays fail to amplify partially methylated molecules and significantly underestimate the proportion of methylated samples (Alnaes et al., 2015).
Co-amplification at lower denaturation temperature PCR (COLD-PCR) approaches have been developed to enrich and analyze rare mutations including full, fast, ice (improved and complete enrichment), E-ice (Enhanced-ice) and temperature tolerant COLD-PCR and have been combined with several read-out technologies (Mauger et al., 2017). These protocols use a critical temperature (Tc) to selectively denature wild-type mutant heteroduplexes to allow the enrichment of rare mutations. More recently, fast-COLD-MS-PCR and E-ice-COLD-PCR, have been shown to allow the enrichment of unmethylated DNA and methylated DNA, respectively, from bisulfite-converted DNA (Castellanos-Rizaldos et al., 2014; Mauger et al., 2018).
E-ice-COLD-PCR uses a blocker probe that contains several LNA bases which selectively hybridizes to and thereby blocks the amplification of wild-type alleles and enriches mutant alleles followed by a Pyrosequencing assay for a sequence based read-out at single nucleotide resolution (How-Kit and Tost, 2015). For DNA methylation analysis, LNA blocker probes are designed to block unmethylated CpG sites on bisulfite-converted DNA enriching thus all other DNA methylation patterns except for completely unmethylated molecules (Mauger et al., 2018).
Therefore, the design of E-ice-COLD-PCR assays is fundamentally different from the more widely used MSP assays as it impedes the amplification of the normal, unmethylated state, but does not make any requirements on the degree or the patterns of DNA methylation in the amplified target region. All molecules different from the blocked pattern will be amplified and the subsequent sequencing-based analysis gives detailed information on the molecules that have been enriched. E-ice-COLD-PCR reaction can be easily multiplexed and the level of mutations or DNA methylation can be quantified using standard curves for the analysis of very low input material such as ccfDNA (Mauger et al., 2016 and 2018; Sefrioui et al., 2017).
The current protocol describes the development and implementation of an E-ice-COLD-PCR assay for the enrichment of low-abundant methylated DNA from a limited quantity of input sample and thus applicable to non-invasive blood-based tests. After bisulfite conversion, the E-ice-COLD-PCR reaction is performed followed by Pyrosequencing analysis. The optimization of E-ice-COLD-PCR includes assay design, optimization of the reaction and analysis of the amplification product.
While the presented protocol focusses on the technically more demanding detection of hypermethylation, blocker probes could also be used to block methylated DNA and enable the enrichment of unmethylated DNA in the presence of an excess of methylated DNA.
Materials and Reagents
- DNA LoBind and PCR Tubes 2 ml (Eppendorf, catalog number: 022431048) or similar
- LightCycler® 480 Mutiwell Plate 96, white (Roche, catalog number: 04729692001)
- LightCycler® 480 Sealing Foil (Roche, catalog number: 04729757001)
- Pyrosequencing plate (PyroMark® Gold Q96 Plate, Qiagen, catalog number: 979101)
- PyroMark® Q96 HS Capillary Tips (Qiagen, catalog number: 979104)
- PyroMark® Q96 HS Reagents Tips (Qiagen, catalog number: 979102)
- PCR Plate 96-well (Thermo Fisher, catalog number: AB0800) or similar
- Adhesive PCR Plate (Thermo Fisher, catalog number: AB0558) or similar
- Polyethylene adhesive PCR Plate (Corning, catalog number: 6524) or similar
- Aluminium Adhesive PCR Plate (Corning, catalog number: 6569) or similar
- 10/20 µl filter tips (Rainin, catalog number: 17002429) or similar
- 200/250 µl filter tips (Rainin, catalog number: 17002428) or similar
- 1,000 µl filter tips (Rainin, catalog number: 17002426) or similar
- 10 µl tips (Rainin, catalog number: 17005091) or similar
- 250 µl tips (Rainin, catalog number: 17005093) or similar
- 1,000 µl tips (Rainin, catalog number:17005089) or similar
- EpiTect control DNA methylated (Qiagen, catalog number: 59655)
- Unmethylated and methylated DNA standards (Zymo Research, catalog number: D5014)
- Qubit dsDNA HS Assay (Thermo Fisher, catalog number: Q33226)
- Epitect Fast DNA bisulfite kit (Qiagen, catalog number:59826)
- EZ DNA Methylation-Gold kit (Zymo Research, catalog number: D5005) or similar
- Primers, blocker probes and Pyrosequencing primers, HPLC-purified grade (TIBMOLBIOL)
- Nuclease Free water (Invitrogen, catalog number: AM9937)
- HotStar Taq Polymerase (provided with the respective HotStar Taq buffer, Qiagen, catalog number: 203203)
- UltraPureTM Agarose (Thermo Fisher, catalog number 16500500)
- dNTP Mix (Thermo Fisher, catalog number: R0192)
- SYTOTM 9 Green Fluorescent Nucleic Acid Stain (Thermo Fisher, catalog number: S34854)
- PyroMark® Gold SQA Q96 Reagents (Qiagen, catalog number: 972812)
- Streptavidin Sepharose beads (GE Healthcare, catalog number: 17-5113-01)
- Denaturing Buffer (0.2 M NaOH)
- 70% Ethanol
- MgCl2
- Tris-HCl
- Tris base
- NaCl
- EDTA
- Tween-20
- Acetic acid
- Binding Buffer (see Recipes)
- Annealing Buffer (see Recipes)
- Wash Buffer (see Recipes)
Equipment
- A pre-PCR room with Laminar flow cabinets and a post-PCR room
- 20, 200 and 1,000 µl and multichannel pipettes (Rainin) or similar and filter tips in pre-PCR room
- Vortex for tubes
- Centrifuge for plates and microtubes (Eppendorf) or similar
- Refrigerant block for 96 plates and ice bucket
- Qubit Fluorometer (Thermo Fisher) or similar
- QIAcube (Qiagen) or similar
- LightCycler® 480 Instrument (Roche LifeScience) or similar
- LightCycler® 96 Instrument (Roche LifeScience) or similar
- Thermocycler (Eppendorf) or similar
- Thermomixer (Eppendorf) or similar
- PyroMark Q96 Vacuum Workstation (Qiagen, catalog number: 9001529)
- Pyrosequencer (PyroMark Q96 MD System, Qiagen) or similar
Software
- Design of PCR primers using MethPrimer (http://www.urogene.org/methprimer/) (Li and Dahiya, 2002)
- Nucleic Acid Sequence Massager (http://www.attotron.com/cybertory/analysis/ seqMassager.htm)
- Design of Pyrosequencing primers can be performed manually or using the commercial PyroMark assay design software (Qiagen)
- Manual design of LNA blocker probe and calculation of Tc using the LNA oligo Tm prediction tool (http://www.exiqon.com/ls/homeoflna/Oligo-tools/tm-prediction-tool.htm)
- LightCycler 480 Software (Roche LifeScience) or similar
- Gradient LightCycler 96 Software (Roche LifeScience) or similar
- PyroMark® CpG Software (Qiagen) or similar
- Statistical or graphical software such as Excel® or similar
Procedure
The development of an E-ice-COLD-PCR assay includes the design of the assay, the optimization of the E-ice-COLD-PCR reaction, the analysis of the samples of interest as well as the data analysis for the E-ice-COLD-PCR assay (Figure 1).
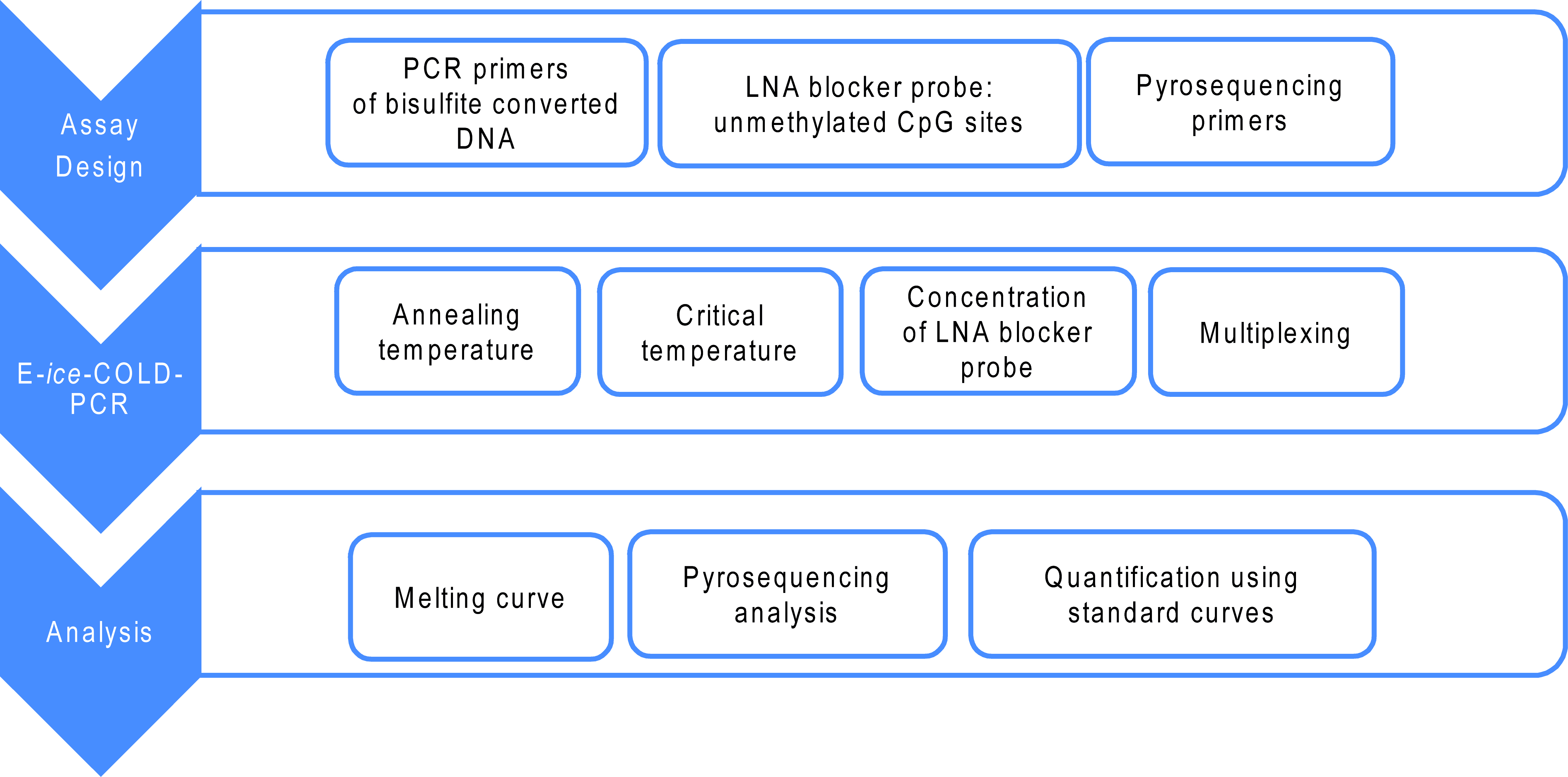
Figure 1. Overview of the development of an E-ice-COLD-PCR assay composed of three main steps. 1) Assay design on bisulfite converted DNA, 2) optimization of the E-ice-COLD-PCR conditions and 3) analysis of E-ice-COLD-PCR to quantify the presence and level of DNA methylation in the samples of interest.
- Assay design
The assay design including the design of PCR and Pyrosequencing primers for the targeted methylated sequence as well as the design of the LNA blocker probe for the optimal enrichment of methylated CpGs, which strongly depends on the frequency and spacing of CpG sites present in the targeted region, is probably the most critical step of the presented protocol.
Design of PCR primers- We recommend designing at least 2 PCR primer sets.
- Design PCR primers for the targeted region on in-silico bisulfite converted DNA using the MethPrimer software. Optimal PCR primers size should be around 20 bases with a melting temperature (Tm) around 60 °C. The size of the amplification product for the E-ice-COLD-PCR amplicons should be kept relatively small (100-180 base pairs maximum).
- Place PCR primers in a region containing four or more cytosines that have been converted to ensure complementary to bisulfite-converted DNA. They should preferentially not contain CpG sites or limited to one CpG site. If CpG sites cannot be completely avoided, they should not be present in the last five bases of the 3' end to avoid preferential amplification of a specific DNA methylation pattern.
- PCR primers should not contain palindromes, complementary sequences, degenerated bases or inosine.
- Check for the presence of single nucleotide polymorphisms (SNPs) within the amplification product using sequence alignment tools such as BLAST or BLAT and associated dbSNP resources. Genetic variants in the sequences targeted by the amplification primers that have a significant frequency in the population of interest must be avoided and amplification primers should be redesigned.
- If no primers are found on the forward strand, create the reverse complement strand using Nucleic Acid Sequence Massager and repeat the design process.
- One PCR primer needs to be biotinylated on the 5' end for the Pyrosequencing reaction later on and the primer to be biotinylated depends on the sense of the Pyrosequencing primer designed in the next step.
Design of Pyrosequencing primers- Pyrosequencing primers can be designed manually or using the commercial PyroMark Assay Design Software. We recommend designing at least 2 Pyrosequencing primers to ensure optimal assay performance.
- Pyrosequencing primers should not overlap with potentially variable positions and must not produce primer dimers or a hairpin structure.
- Position the Pyrosequencing primer at least five bases from the end of the amplification product. The last consecutive 4-5 bases at the 3'-end of the Pyrosequencing primer should be complementary to a unique sequence in the amplification product to avoid background signals due to non-specific annealing during the sequencing-by-synthesis reaction.
- Design the Pyrosequencing primer in a non-polymorphic region next to the variable region of interest. A number of nucleotide dispensations can be used to approach the region of interest to ensure specific annealing if necessary.
- Indicate polymorphic positions in the dispensation order with degenerated bases.
Blocker probe design
The blocker probe for E-ice-COLD-PCR contains Locked Nucleic Acid (LNA) nucleobases in order to anneal very strongly to the complementary sequence. For the detection and analysis of hypermethylated molecules, the LNA bases will be complementary to unmethylated CpGs, i.e., to the thymines generated through PCR amplification after bisulfite conversion. Methylated CpGs (cytosines following bisulfite conversion and PCR amplification) will generate mismatches (heteroduplexes) and the blocker probe will dissociate from the target strand at the critical temperature allowing the amplification and preferential enrichment of methylated bisulfite-converted CpGs while inhibiting amplification of unmethylated bisulfite-converted CpGs.- We recommend designing at least 3 LNA blocker probes for evaluation for each target region.
- LNA blocker probes should contain more than two closely neighbored LNA bases at the unmethylated CpG sites in the center of the probe and a phosphate group at the 3' end to avoid any elongation of the blocker probe during the E-ice-COLD-PCR reaction.
Note: LNA blocker probe could also be designed to block methylated bisulfite-converted DNA to enrich specifically unmethylated bisulfite-converted DNA molecules. The enrichment of unmethylated DNA is thermodynamically favored as unmethylated DNA has a lower melting temperature compared to methylated DNA after bisulfite conversion. Therefore, a greater flexibility on the assay design is possible for the enrichment of unmethylated DNA. - The LNA blocker probe should have a length of between 40 and 65 bases and overlap five to six nucleotides at the 3' end with one of the amplification primers.
- If it is not possible to cover all CpG sites present in the amplification product, the LNA blocker probe should cover a maximum of consecutive CpG sites representing at least 45-50% of the total CpG sites of the amplification product. The blocker probes should contain LNA bases at each unmethylated bisulfite-converted CpG sites underlying the probe sequence.
Note: The blocker probe sequence should be complementary to the DNA methylation patterns which are present in the non-disease relevant cell populations (e.g., white blood cells). If specific CpG sites are highly methylated in samples of control individuals, this should be incorporated in the design to avoid enrichment of uninformative methylated molecules. - Do not incorporate LNA bases in the last six nucleotides from the 5' or the 3' ends of the blocker sequence.
- Make sure that LNA blocker probe and PCR primers cannot generate primer dimers.
- Verify the prediction of the Tm of the blocker probe using the LNA oligo Tm prediction tool. The Tm should be above 70 °C. The Tm of the LNA blocker probe can be modulated by increasing the length and/or the number of LNA bases. If necessary, incorporate LNA bases at both bases of an unmethylated bisulfite-converted CpG site (i.e., the A, which is complementary to the T of an unmethylated cytosine after bisulfite treatment, and the C complementary to the G of the CpG dinucleotide). This strategy might prove especially useful if there are less than three CpGs sites underlying the LNA blocker probe.
Note: The calculated Tm is only an estimation of temperature, which might differ from its actual behavior during E-ice-COLD-PCR experiments. If it is not possible to increase the length of the blocker probe or the number of LNA bases within the blocker probe, a predicted temperature between 68 °C and 70 °C can be used.
- Bisulfite conversion
Following the design, the next step of the protocol is the bisulfite conversion of the samples to be analyzed.- Work in a pre-PCR room with filter tips.
- Quantify the DNA content of the samples to be analyzed using a Qubit Fluorometer according to the manufacturer’s instructions.
Note: This method is currently used in our laboratory and allows the quantification of circulating cell-free DNA (Mauger et al., 2015). Several commercially available methods could also be used to quantify DNA as well as qPCR-based assays, which will also yield information on the fragment size of the cell-free DNA (Mauger et al., 2015). - Use unmethylated and methylated DNA standards to control for the efficiency of the bisulfite conversion (see Note 1).
- If at least 200 ng of DNA quantified by fluorescence are available, perform bisulfite conversion using the EpiTect Fast DNA Bisulfite Kit on a QIAcube automated system (Qiagen) according to the manufacturer’s instructions in an elution volume of 40 µl.
- If only a few ng of circulating cell-free DNA is available, perform bisulfite conversion using the EZ DNA Methylation-Gold Kit according to the manufacturer’s instructions with an elution volume of 30 µl (see Note 1).
- Prepare serial dilutions of known bisulfite-converted methylated and unmethylated standard DNA: 100%, 75%, 50%, 20%, 10%, 5%, 2.5%, 1%, 0.5%, 0.25%, 0.1% and 0%.
Note: Commercially available standard bisulfite-converted methylated and unmethylated DNA should be used for the optimization of E-ice-COLD-PCR and to establish standard curves for the enrichment during E-ice-COLD-PCR.
- E-ice-COLD-PCR reaction
The optimization of the E-ice-COLD-PCR reaction consists of three main steps: the determination of the annealing temperature (Ta) of the PCR primer set, the determination of the critical temperature (Tc) for the LNA blocker probe and a concentration gradient of the designed LNA blocker probes (Figure 1, Figure 2 and Figure 3).- Evaluate at least three different LNA blocker probes as the enrichment of E-ice-COLD-PCR depends on the optimal combination of an amplification primer set, the critical temperature and concentration of LNA blocker probe.
- Order all PCR primers, LNA blocker probe and Pyrosequencing primers with HPLC purification grade. We recommend using the same lot of synthesized primers for the analysis of all samples in the project. For a new synthesis of LNA blocker probe, verify the enrichment by using a concentration gradient of the LNA blocker probe. New syntheses might show a slightly different enrichment efficiency.
- Work in a dedicated pre-PCR room with Laminar flow cabinets and filter tips.
- E-ice-COLD-PCR is optimized for low input of samples starting with 2 ng of bisulfite-converted DNA (corresponding to ~700 DNA molecules). For higher input of DNA (≤ 25 ng), we recommend reducing the number of cycles of the reaction and/or the quantity of the PCR primers.
- Typical E-ice-COLD-PCR reaction conditions are 1x HotStar Taq buffer (Qiagen) supplemented with 1.6 mM MgCl2, 200 µM of each dNTP, 2.0 U of HotStar Taq polymerase, 200 nM of forward and reverse primers 2 µM of SYTO9, 0-100 nM of blocker probe in a 25 µl volume.
Note: HotStar Taq Polymerase is currently used in our laboratory for both E-ice-COLD-PCR analysis and standard bisulfite Pyrosequencing analysis. Other commercially available enzymes for DNA methylation could be used, but the efficiency of the enzyme for the amplification of the E-ice-COLD-PCR product as well as its compatibility with bisulfite converted DNA should be verified. - We recommend analyzing each sample in triplicate and using negative and positive controls.
- The E-ice-COLD-PCR cycling conditions include an initial denaturation step for 10 min at 95 °C, followed by 6 cycles of 30 s at 95 °C, 20 s at the Ta °C (see “Determination of annealing temperature” in Procedure C) and 10 s at 72 °C, followed by 52 cycles of 20 s at 95 °C, 30 s at 70 °C, 20 s at Tc °C (see “Determination of the critical temperature” in Procedure C), 20 s at Ta °C and 10 s at 72 °C, followed by a melting curve analysis with 20 acquisition per degree from 65 to 95 °C and a final cooling step to 40 °C (Figure 2).
Note: Determination of the required number of cycles of the E-ice-COLD-PCR reaction should be performed especially for higher quantity of input sample. - We recommend verifying the E-ice-COLD-PCR reaction using a melting curve analysis prior to proceeding to the Pyrosequencing analysis (Figure 4A).

Figure 2. Schematic overview of the different steps of an E-ice-COLD-PCR assay composed of three major steps: Initial denaturation, standard PCR amplification and E-ice-COLD-PCR amplification
Determination of annealing temperature
The optimal annealing temperature should be determined in a temperature gradient thermocycler using bisulfite-converted DNA controls of 100%, 50%, 0% of DNA methylation as well as a negative control.- The PCR reaction conditions are the same as used for E-ice-COLD-PCR without adding the LNA blocker probe.
- The gradient PCR program consists of a denaturing step of 15 min at 95 °C, followed by 52 cycles of 30 s at 95 °C, 20 s at a gradient Ta and 10 s at 72 °C, with a final extension at 72 °C.
- In a post-PCR room, deposit 10 µl of the PCR product as well as a positive and negative control on a 2% agarose gel.
- The optimal annealing temperature is selected taking the expected size and the intensity of the PCR product band as well as specificity of the reaction into account. Although not absolutely necessary for E-ice-COLD-PCR, the absence of a strong amplification bias during PCR reaction should be verified by Pyrosequencing.
Note: Due to the difference in GC content between completely unmethylated and methylated molecules following bisulfite conversion, a preferential amplification in most cases of unmethylated molecules is commonly observed. This preferential amplification might be attenuated using different primer pairs and/or enzymes for the PCR amplification, but persists in many cases (Warnecke et al., 1997). - Analyze the PCR product by Pyrosequencing to verify the correct sequence has been amplified and the Pyrosequencing primer is annealing uniquely and does not create any background noise. The optimal Pyrosequencing primer should yield signals (Pyrosequencing peaks) of expected height of the bisulfite-converted DNA standards and no observable signals on negative controls (no template, genomic DNA).
Determination of the critical temperature (Tc)- Identify the critical temperature using a temperature gradient quantitative PCR between 70 °C and 90 °C.
Note: The use of a gradient-enabled qPCR cycler allows for the rapid determination of the critical temperature and associated blocker probe concentration. If no gradient qPCR thermocycler is available, several independent reactions using different temperatures have to be performed. Alternatively, optimization can be performed using a standard thermocycler and analyzing PCR products on an agarose gel followed by a sequencing reaction for reaction conditions yielding a PCR product. - We recommend for the first test using concentrations of 0, 25 and 50 nM of LNA blocker probe.
- Control the reaction by using serial dilutions of bisulfite-converted methylated DNA controls into bisulfite converted unmethylated DNA yielding a DNA methylation degree of 0%, 10%, 25%, 50% and 100% and a negative control.
- We recommend analyzing samples in triplicates especially if quantification of the DNA methylation level is performed as some variation between individual amplifications exists.
- Quantify the enrichment of DNA methylation using Pyrosequencing by comparing the E-ice-COLD-PCR reaction to a standard qPCR reaction without adding the LNA blocker probe (Figure 4C).
- For some combinations (LNA blocker probes, concentration of LNA blocker probe and/or Tc of one LNA blocker probe) no enrichment, no PCR product, or presence of primer dimers might be encountered. In this case, decrease the concentration of blocker probe for Tc determination.
- The enrichment of DNA methylation should clearly be obtained for all replicates, different % of methylated DNA as input (except for unmethylated DNA) and for several concentrations of the same LNA blocker probe. At the optimal Tc a high enrichment should be obtained but yield a high quality Pyrogram (without dispensation artefacts and sufficient intensity of peaks) and it should avoid non-specific amplification of the E-ice-COLD-PCR reaction (see Note 2).
Note: It is important to avoid a complete blocking of unmethylated DNA, as in this case an amplification failure due to low quality samples will be indistinguishable from a completely unmethylated sample. For all samples, a PCR amplification product should be obtained, with an enrichment of methylated molecules in those which contain methylated molecules.
Determination of concentration of LNA blocker probe- Use a concentration gradient of the blocker probe at 0, 20, 40, 60, 80 and 100 nM.
- Use a serial dilution of standard bisulfite-converted methylated DNA of 0%, 0.5%, 1%, 5%, 10%, 20 % and 100% into unmethylated DNA as well as a negative control.
- Analyze samples in triplicates.
- Analyze the melting curve of each reaction for E-ice-COLD-PCR, amplification and enrichment of unmethylated and/or methylated DNA, primer dimer formation and reproducibility of replicates (Figure 4A).
- The optimal concentration of the blocker probe should be obtained by analyzing the Pyrograms selecting the concentration with the highest enrichment without any non-specific reactions.
- Another gradient of concentration should be tested with a narrower concentration range around the pre-defined concentration to find the optimal concentration of LNA blocker probe.
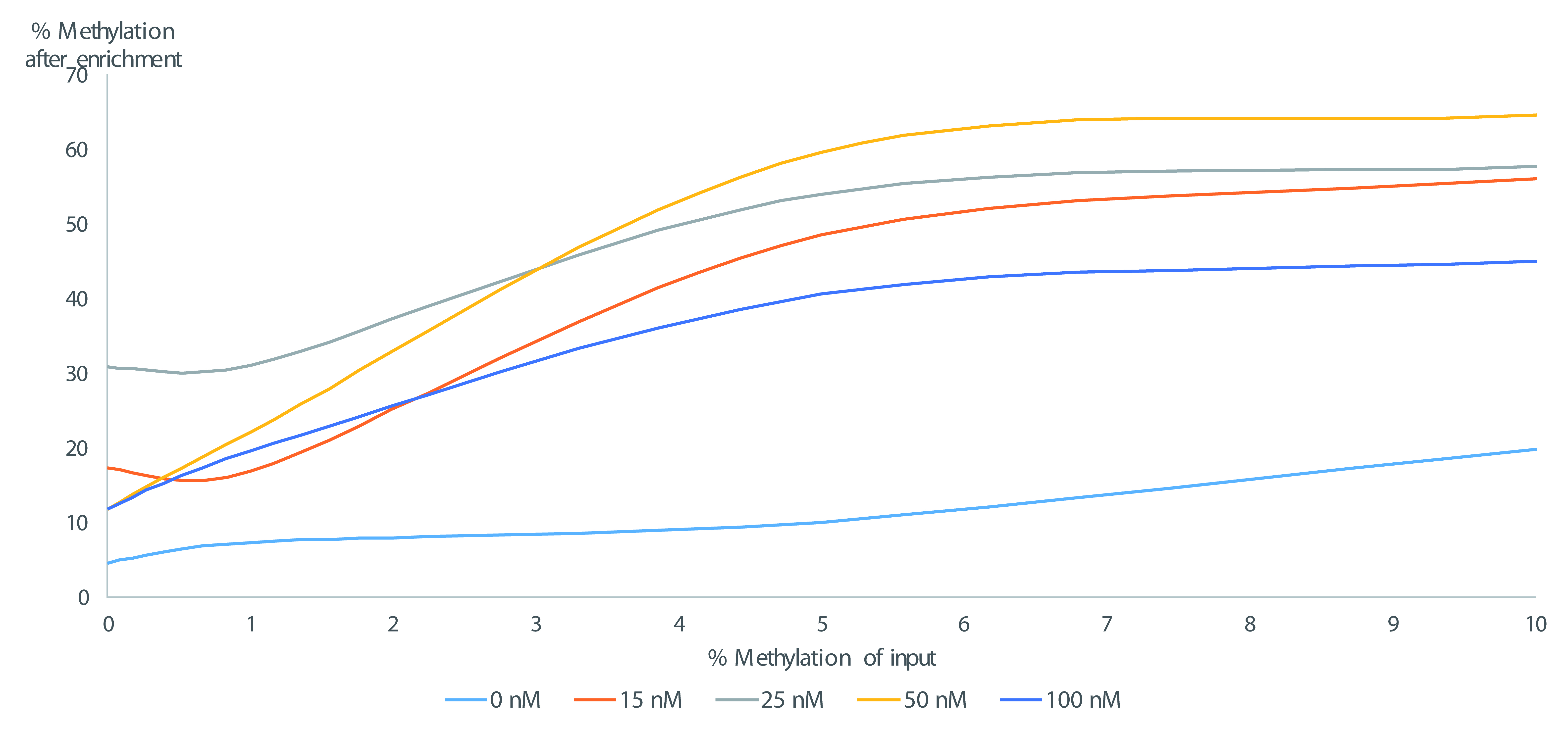
Figure 3. Enrichment of DNA methylation levels by E-ice-COLD-PCR detected by pyrosequencing in function of the input of methylated DNA for various LNA blocker probe concentrations. The example shows enrichment of DNA methylation at the AIM2 gene promoter (Mauger et al., 2018) and demonstrates that the enrichment depends on the concentration of LNA blocker probe.
Quantification of DNA methylation by E-ice-COLD-PCR standard curve
E-ice-COLD-PCR is an enrichment method for the detection of low-abundant methylated DNA and thus yields mainly a qualitative result on the presence of DNA methylation, but it can also be used to quantify the level of DNA methylation using standard curves.- Control optimal conditions of E-ice-COLD-PCR by comparing standard curves of CpG sites to those obtained using standard qPCR without the blocker probe.
- Analyze serial dilutions of standards with a known degree of methylation (100%, 75%, 50%, 20%, 10%, 5%, 2.5%, 1%, 0.5%, 0.25%, 0.1% and 0%) in sextuplicates (see Note 2).
- Analyze samples in triplicates.
Multiplex E-ice-COLD-PCR
If the amount of sample available is limited such as for the analysis of circulating cell-free DNA, E-ice-COLD-PCR reaction can be multiplexed analyzing several target regions in parallel (Sefrioui et al., 2017; Mauger et al., 2018).- E-ice-COLD PCR conditions for multiplex assays should be the same as for the simplex assay except that the concentration of each PCR primer should be decreased (example: half the quantity for a duplex) (Mauger et al., 2018).
- Multiplex E-ice-COLD-PCR reactions using the same Ta and Tc.
Note: The critical temperature of the different assays might not be identical, in that case a critical temperature should be chosen, that allows the best enrichment of all targets included in the multiplex assay (Mauger et al., 2016). - Optimize the multiplex PCR by comparing the enrichment obtained with the simplex assay and standard qPCR varying the blocker concentration and/or critical temperature slightly.
- Analyze the multiplexing experiment using a melting curve analysis. Amplifications should not yield primer dimers.
- Pyrosequencing assay
E-ice-COLD-PCR reaction is analyzed by Pyrosequencing (Tost and Gut, 2007; Busato et al., 2018). Prior to the Pyrosequencing reaction on the PyroMark Q96 MD System, a sample preparation step is performed which includes the purification and the denaturation of E-ice-COLD-PCR product to obtain single-stranded product followed by hybridization of the Pyrosequencing primer.
Notes:
- E-ice-COLD-PCR reaction is currently analyzed by Pyrosequencing in our laboratory as it allows for short-turnaround time, but the amplification products could be analyzed by Sanger or NGS sequencing or if no detailed information on individual CpG positions is required–simply by the included high-resolution melting analysis.
- This protocol uses the PyroMark Q96 MD System, but the Pyrosequencing analysis can also be performed on the more recent PyroMark Q24 Advanced System according to the manufacturer’s instructions.
Sample preparation- We recommend working in a dedicated post-PCR room.
- Prepare the PCR plate by transferring 10 µl of E-ice-COLD-PCR reaction, adding 40 µl of binding buffer and 2 µl of sepharose beads and completing to 80 µl with water in a new PCR plate.
Note: The different buffers used for Pyrosequencing sample preparation are also commercially available from Qiagen. We commonly prepare them ourselves. - Seal the PCR plate and incubate the reaction for 10 min at room temperature under agitation (1,400 rpm) allowing the sepharose beads to capture the biotinylated amplification products.
- During the incubation, prepare the Pyrosequencing plate by diluting 4 pmol of the Pyrosequencing primer into 12 µl of annealing buffer per well. Several Pyrosequencing primers can be used in the same plate.
Note: For multiplexing experiments, it might be necessary to re-amplify each enriched PCR product separately in a standard PCR reaction as Pyrosequencing is performed at low temperature due to the thermal instability of the enzyme mix and sequencing primers might anneal to several of the amplified regions simultaneously leading to a high background signal. - Prepare the vacuum workstation by adding approximately 180 mI of 70% Ethanol, 180 ml of denaturing solution, 200 ml of wash buffer and 200 ml of water.
- Turn on the vacuum of the workstation to create a vacuum in the aspiration device.
- Clean the tips of the filters by immersion in water for several seconds.
- Remove the PCR plate from the plate shaker and the seal from the PCR plate. Aspirate the remaining binding buffer from the PCR plate. The beads remain on the filters of the tips.
- Immerse the tips of the filters in successive baths of 70% Ethanol for 5 s, denaturing solution for 5 s and wash buffer for 10 s. Turn over the tool and release the vacuum.
- Immerse the tips of the filters in the annealing mix of the Pyrosequencing plate and shake gently to release the beads into the wells.
- Incubate the Pyrosequencing plate at 80 °C for 2 min on a thermoplate placed on a heating device.
- Let the Pyrosequencing plate cool down at room temperature to allow annealing of the sequencing primer.
Pyrosequencing reaction- During the cooling of the Pyrosequencing plate, program the Pyrosequencing run on the Pyrosequencer.
- Calculate the amount of reagents for the Pyrosequencing run using the software. Dispense the reagent in the appropriate tips and place the tips in the cartridge avoiding bubble formation.
- Place the reagent’s cartridge and the sealed Pyrosequencing test plate into the Pyrosequencer. Perform the dispensation test to be sure that the dispensing tips are working properly. Droplets should be clearly visible and homogenous.
- Deposit the Pyrosequencing plate into the Pyrosequencer and start the Pyrosequencing run. The length of a run is proportional to the number of dispensations (1 per min).
- In some cases, the signals corresponding to part of the sequence may be missing. Run a dispension test to check if some tips are blocked.
- After the end of the Pyrosequencing run, remove the cartridge and clean the tips by rinsing with pure water.
Data analysis
- Analyze E-ice-COLD-PCR reactions using the LightCycler 480 Software.
- Compare the melting curves of each sample with serial dilutions of bisulfite-converted unmethylated and methylated DNA standards and a negative control to verify the amplification by E-ice-COLD-PCR and to check for the absence of primer dimers.
- In multiplex E-ice-COLD-PCR reactions, the peak of unmethylated and/or methylated DNA can be obtained for each reaction (Figure 4A).
- Analyze the Pyrosequencing data using the PyroMark MD Software.
- Export the results to be treated with statistical or graphical software such as Excel®.
- Calculate the % of DNA methylation for each CpG site using the intensity of the peak of cytosine and thymine of the CpG site for each sample (Figure 4B).
- Establish standard curves for each CpG site using serial dilutions of standard bisulfite-converted unmethylated and methylated DNA. Calculate the equation and the standard deviation of each standard curve (see Note 3) (Figure 4C).
- Quantify the % of DNA methylation of each sample using the standard curves and the % of DNA methylation obtained by Pyrosequencing (Figures 4B and 4C).
- Calculate, for each sample, the average % of DNA methylation of all CpG sites analyzed.
- Calculate the standard deviation of the % of the DNA methylation using triplicates for each sample.
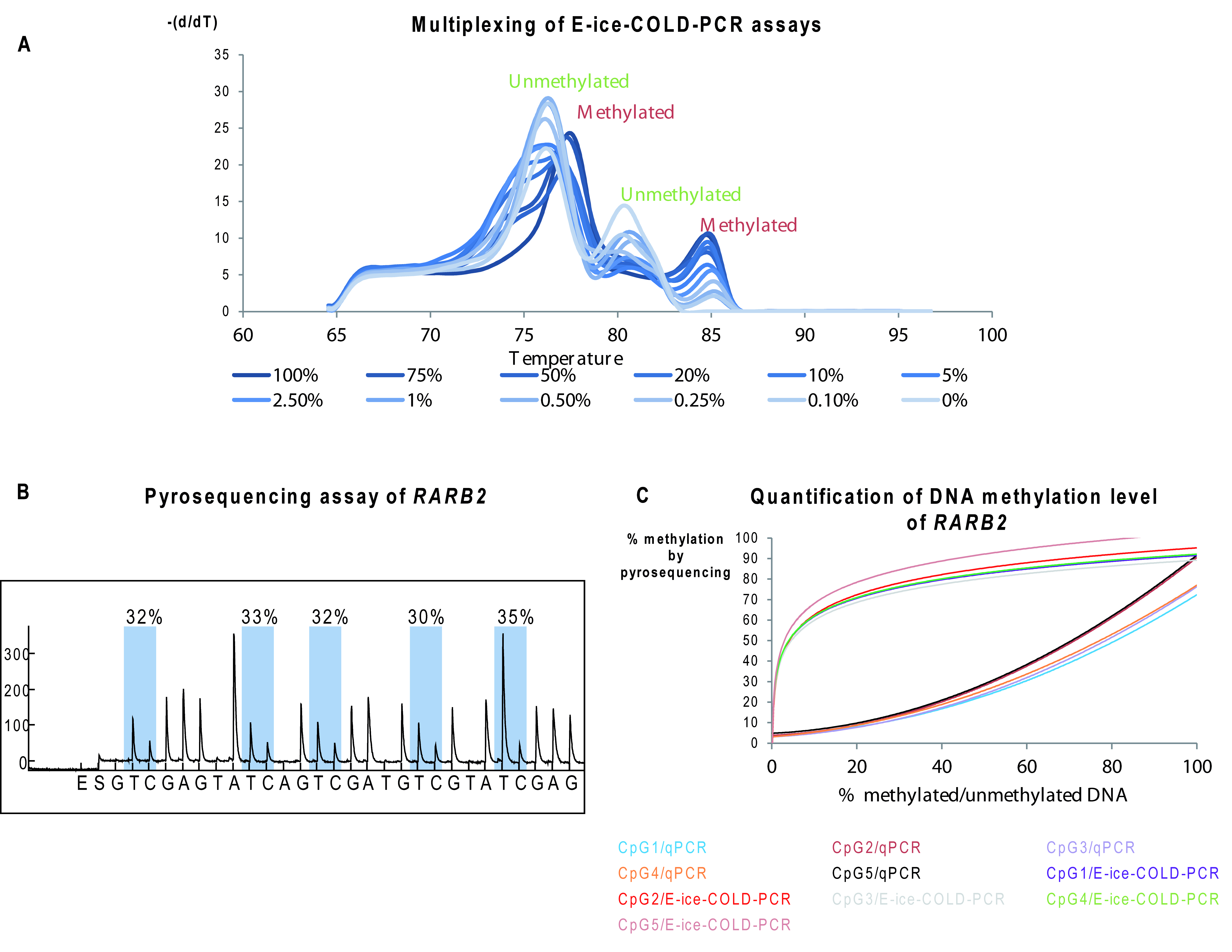
Figure 4. Analysis of DNA methylation by E-ice-COLD-PCR. A. Example of a melting curve of duplex E-ice-COLD-PCR reaction analyzing methylation in the AIM2 and RARB2 genes that shows unmethylated and methylated peaks for each amplicon. B. Example of a Pyrogram of RARB2 following E-ice-COLD-PCR. C. Example of E-ice-COLD-PCR and qPCR standard curves for 5 CpGs sites in the RARB2 promoter.
Notes
- We currently use the EZ DNA Methylation-Gold (Zymo Research) and the Epitect Fast DNA bisulfite kits (Qiagen) in our laboratory. Several commercially available kits can be used but they could have different efficiencies for different sample types (Worm Ørntoft et al., 2017).
- The standard curve of the qPCR reaction (without blocker) can also be used to estimate the enrichment by E-ice-COLD-PCR (Figure 4C).
- E-ice-COLD PCR standard curves can saturate at high levels of DNA methylation due to the enrichment by E-ice-COLD-PCR reaction. Consequently, the quantification using these standard curves cannot be performed for high DNA methylation level. In this case, qPCR reaction without enrichment can be performed to quantify high levels of DNA methylation (Figure 4C) or another E-ice-COLD-PCR reaction can be performed using a lower concentration of the LNA blocker probe.
Recipes
- Binding Buffer
10 mM Tris-HCl
2 mM NaCl
1 mM EDTA
0.1% Tween-20, pH 7.6 - Annealing Buffer
20 mM Tris-acetate
2 mM Mg-acetate, pH 7.6 adjusted with acetic acid 4 M - Wash Buffer
10 mM Tris-acetate, pH 7.6 adjusted with acetic acid 4 M
Acknowledgments
This work was supported by the institutional budget of the Centre National de Recherche en Génomique Humaine (CNRGH). This protocol was adapted from previous work (How-Kit and Tost et al., 2015; Busato et al., 2018; Mauger et al., 2018).
Competing interests
The authors declare no conflict of interest.
Ethics
Use of breast cancer samples in the framework of the original publication (Mauger et al., 2018) was approved by the Norwegian Regional Committee for Medical Research Ethics (approval numbers 429-04148 and 2014/895), and patients have given written informed consent for the use of material for research purposes. No additional samples were used for the preparation of this manuscript.
References
- Alnaes, G. I., Ronneberg, J. A., Kristensen, V. N. and Tost, J. (2015). Heterogeneous DNA methylation patterns in the GSTP1 promoter lead to discordant results between assay technologies and impede its implementation as epigenetic biomarkers in breast cancer. Genes (Basel) 6(3): 878-900.
- Barlebo Ahlborn, L. and Østrup, O. (2019). Toward liquid biopsies in cancer treatment: application of circulating tumor DNA. APMIS 127(5): 329-336.
- Busato, F., Dejeux, E., El Abdalaoui, H., Gut, I. G. and Tost, J. (2018). Quantitative DNA methylation analysis at single-nucleotide resolution by pyrosequencing®. Methods Mol Biol 1708: 427-445.
- Cabel, L., Proudhon, C., Romano, E., Girard, N., Lantz, O., Stern, M. H., Pierga, J. Y. and Bidard, F. C. (2018). Clinical potential of circulating tumour DNA in patients receiving anticancer immunotherapy. Nat Rev Clin Oncol 15(10): 639-650.
- Campan, M., Weisenberger, D. J., Trinh, B. and Laird, P. W. (2018). MethyLight and digital methyLight. Methods Mol Biol 1708: 497-513.
- Candiloro, I. L., Mikeska, T. and Dobrovic, A. (2011). Closed-tube PCR methods for locus-specific DNA methylation analysis. Methods Mol Biol 791: 55-71.
- Castellanos-Rizaldos, E., Milbury, C. A., Karatza, E., Chen, C. C., Makrigiorgos, G. M. and Merewood, A. (2014). COLD-PCR amplification of bisulfite-converted DNA allows the enrichment and sequencing of rare un-methylated genomic regions. PLoS One 9(4): e94103.
- Distler, J. (2019). Quantification of methylated DNA by HeavyMethyl duplex PCR. Methods Mol Biol 507: 339-46.
- Frommer, M., McDonald, L. E., Millar, D. S., Collis, C. M., Watt, F., Grigg, G. W., Molloy, P. L. and Paul, C. L. (1992). A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc Natl Acad Sci U S A 89(5): 1827-1831.
- How-Kit, A. and Tost, J. (2015). Pyrosequencing®-based identification of low-frequency mutations enriched through enhanced-ice-COLD-PCR. Methods Mol Biol 1315: 83-101.
- Jung, M., Kristiansen, G. and Dietrich, D. (2018). DNA methylation analysis of free-circulating DNA in body fluids. Methods Mol Biol 1708: 621-641.
- Kumar, M., Choudhury, Y., Ghosh, S. K. and Mondal, R. (2018). Application and optimization of minimally invasive cell-free DNA techniques in oncogenomics. Tumour Biol 40(2): 1010428318760342.
- Lehmann-Werman, R., Neiman, D., Zemmour, H., Moss, J., Magenheim, J., Vaknin-Dembinsky, A., Rubertsson, S., Nellgard, B., Blennow, K., Zetterberg, H., Spalding, K., Haller, M. J., Wasserfall, C. H., Schatz, D. A., Greenbaum, C. J., Dorrell, C., Grompe, M., Zick, A., Hubert, A., Maoz, M., Fendrich, V., Bartsch, D. K., Golan, T., Ben Sasson, S. A., Zamir, G., Razin, A., Cedar, H., Shapiro, A. M., Glaser, B., Shemer, R. and Dor, Y. (2016). Identification of tissue-specific cell death using methylation patterns of circulating DNA. Proc Natl Acad Sci U S A 113(13): E1826-1834.
- Lewis, J. M., Heineck, D. P. and Heller, M. J. (2015). Detecting cancer biomarkers in blood: challenges for new molecular diagnostic and point-of-care tests using cell-free nucleic acids. Expert Rev Mol Diagn 15(9): 1187-1200.
- Li, L. C. and Dahiya, R. (2002). MethPrimer: designing primers for methylation PCRs. Bioinformatics 18(11): 1427-1431.
- Liu, Y., Song, C., Ladas, I., Fitarelli-Kiehl, M. and Makrigiorgos, G. M. (2017). Methylation-sensitive enrichment of minor DNA alleles using a double-strand DNA-specific nuclease. Nucleic Acids Res 45(6): e39.
- Mauger, F., Daunay, A., Deleuze, J. F., Tost, J. and How-Kit, A. (2016). Multiplexing of E-ice-COLD-PCR assays for mutation detection and identification. Clin Chem 62(8): 1155-1158.
- Mauger, F., Dulary, C., Daviaud, C., Deleuze, J. F. and Tost, J. (2015). Comprehensive evaluation of methods to isolate, quantify, and characterize circulating cell-free DNA from small volumes of plasma. Anal Bioanal Chem 407(22): 6873-6878.
- Mauger, F., How-Kit, A. and Tost, J. (2017). COLD-PCR technologies in the area of personalized medicine: methodology and applications. Mol Diagn Ther 21(3): 269-283.
- Mauger, F., Kernaleguen, M., Lallemand, C., Kristensen, V. N., Deleuze, J. F. and Tost, J. (2018). Enrichment of methylated molecules using enhanced-ice-co-amplification at lower denaturation temperature-PCR (E-ice-COLD-PCR) for the sensitive detection of disease-related hypermethylation. Epigenomics 10(5): 525-537.
- Molnár, B., Tóth, K., Barták, B. K. and Tulassay, Z. (2015). Plasma methylated septin 9: a colorectal cancer screening marker. Expert Rev Mol Diagn 15(2): 171-184.
- Sefrioui, D., Mauger, F., Leclere, L., Beaussire, L., Di Fiore, F., Deleuze, J. F., Sarafan-Vasseur, N. and Tost, J. (2017). Comparison of the quantification of KRAS mutations by digital PCR and E-ice-COLD-PCR in circulating-cell-free DNA from metastatic colorectal cancer patients. Clin Chim Acta 465: 1-4.
- Siravegna, G., Marsoni, S., Siena, S. and Bardelli, A. (2017). Integrating liquid biopsies into the management of cancer. Nat Rev Clin Oncol 14(9): 531-548.
- Tost, J. and Gut, I. G. (2017). DNA methylation analysis by pyrosequencing. Nat Protoc 2(9): 2265-75.
- Tost, J. (2016). Current and emerging technologies for the analysis of the genome-wide and locus-specific DNA methylation patterns. Adv Exp Med Biol 945: 343-430.
- Wan, J. C. M., Massie, C., Garcia-Corbacho, J., Mouliere, F., Brenton, J. D., Caldas, C., Pacey, S., Baird, R. and Rosenfeld, N. (2017). Liquid biopsies come of age: towards implementation of circulating tumour DNA. Nat Rev Cancer 17(4): 223-238.
- Warnecke, P. M., Stirzaker, C., Melki, J. R., Millar, D. S., Paul, C. L. and Clark, S. J. (1997). Detection and measurement of PCR bias in quantitative methylation analysis of bisulphite-treated DNA. Nucleic Acids Res 25(21): 4422-4426.
- Warton, K., Mahon, K. L. and Samimi, G. (2016). Methylated circulating tumor DNA in blood: power in cancer prognosis and response. Endocr Relat Cancer 23(3): R157-171.
- Worm Ørntoft, M. B., Jensen, S. Ø., Hansen, T. B., Bramsen, J. B. and Andersen, C. L. (2017). Comparative analysis of 12 different kits for bisulfite conversion of circulating cell-free DNA. Epigenetics 12(8): 626-636.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Mauger, F. and Tost, J. (2019). Enhanced-ice-COLD-PCR for the Sensitive Detection of Rare DNA Methylation Patterns in Liquid Biopsies. Bio-protocol 9(23): e3452. DOI: 10.21769/BioProtoc.3452.
Category
Cancer Biology > Genome instability & mutation > Genetics
Cancer Biology > General technique > Genetics > Gene expression
Molecular Biology > DNA > DNA synthesis
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.