- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Detection of in vivo Protein Interactions in All Bacterial Compartments by Förster Resonance Energy Transfer with the Superfolder mTurquoise2 ox-mNeongreen FRET Pair

Published: Vol 9, Iss 23, Dec 5, 2019 DOI: 10.21769/BioProtoc.3448 Views: 7096
Reviewed by: Gal Haimovich
Original research article
The authors used this protocol in:

Apr 2019

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
This protocol was developed to qualitatively and quantitatively detect protein-protein interactions in all compartments of Escherichia coli by Förster Resonance Energy Transfer (FRET) using the Superfolder mTurquoise2 ox-mNeonGreen FRET pair (sfTq2ox-mNG). This FRET pair has more than twice the detection range for FRET interaction studies in the cytoplasm or periplasm of E. coli compared to other pairs to date. These protein-interaction studies can be performed in vivo because fluorescent proteins can be genetically encoded as fusions to proteins of interest and expressed in the cell. sfTq2ox and mNG fluorescent protein fusions are co-expressed in bacterial cells and the fluorescence emission spectra are measured. By also measuring reference spectra for the background, sfTq2ox-only and mNG-only samples, expected emission spectra can be calculated. Sensitized emission for mNG above the expected spectrum can be attributed to FRET and quantified by spectral unmixing. This bio-protocol discusses the sfTq2ox-mNG FRET pair and provides a practical guide in preparing the protein fusions, setting up and running the FRET experiments, measuring fluorescence spectra and gives the tools to analyze the collected data.
Keywords: BacteriaBackground
Determining how and which proteins interact to sustain life is at the core of molecular biology research. Many in vitro methods exist but may result in false positives as the interactions are taken out of their biological context. An in vivo method is crucial for the verification of in vitro obtained results. Moreover, in vivo methods allow for the observation of the often, dynamic processes that take place in the cell. Fluorescent proteins (FP)s are ideal for in vivo experiments as they can be fused to proteins of interest, making it possible to microscopically track the localization and dynamics of the fused proteins in living cells. Energy can be transferred between FPs by Förster Resonance Energy Transfer (FRET) provided they have compatible physical properties. The emission spectrum of a donor FP must overlap with the excitation spectrum of an acceptor FP and the physical distance between the chromophores within the folded FPs should be less than 10 nm. The larger the spectral overlap and the smaller the distance between the donor and acceptor FP the more FRET can occur. The stringent distance dependence for FRET is ideal to detect direct protein-protein interactions as they also occur in the nanometer range, whereas indirect protein interactions are usually on a larger distance scale and not detectable by FRET. For this reason, it gives very few false positives but due to the physical requirements of the involved fluorophores, negative results could be false negatives. The advantage of using FPs for FRET measurements is that the chromophore is enclosed in a protein barrel and therefore much better protected from the environment than chemical chromophores. The fluorescence of the latter is extremely sensitive to environmental changes such as pH, salt, and solvents. Consequently, they often cannot be used reliably to measure FRET in a complicated environment like the living cell. A drawback of the 3-4 nm in diameter protein-barrel that encloses the chromophore in FPs is that it reduces the detectable distance between proteins to 1-6 nm. Since 5-7 nm is the typical size of proteins, it usually does not pose a problem, but can again result in false negatives. For a recent perspective on FRET as a biomolecular research tool see Alger et al. (2019). We have used a spectroscopy based FRET method to analyze the interaction between proteins in the cytoplasm and in the periplasm of Escherichia coli (Alexeeva et al., 2010; Fraipont et al., 2011; van der Ploeg et al., 2013 and 2015; Meiresonne et al., 2017 and 2019) and showed that the technique is suitable for mode of action studies of antibiotics (van der Ploeg et al., 2015) and medium throughput screening (Meiresonne et al., 2017 and 2019) using various FP FRET pairs. Building on our experience with bacterial FRET and fluorescent proteins in challenging compartments, we preferentially use the FRET-pair that gives the highest rates of energy transfer in the cytoplasm as well as in the periplasm.
This bio-protocol describes how to perform our sfTq2ox-mNG FRET assay in bacteria to determine protein-protein interactions by spectrally unmixing fluorescence signal into its individual components. Although this protocol is written for E. coli, the method of spectral unmixing is suitable for any organism, provided fusion proteins can be expressed in the species and fluorescence spectra can be collected. The protocol describes how to perform FRET experiments by 2 approaches; 1) Measurements on fixed cell using a fluorometer, the initial determination of protein interactions. 2) Measurements on fixed cells in the plate reader for the faster detection of already confirmed protein interactions that provide clear fluorescence signals.
Materials and Reagents
- Sterile straight neck culturing flasks (DWK Life Sciences, DURAN, catalog number: 2177124) with aluminum anodized metal blue cap (DWK Life Sciences, DURAN, catalog number: 2901324)
- 50 ml sterile Falcon tubes (SARSTEDT, catalog number: 62.657.254)
- 2 ml sterile Eppendorf tubes (Greiner Bio One International, catalog number: 623201)
- 1.5 ml sterile Eppendorf tubes (Greiner Bio One International, catalog number: 616201)
- Disposable pipet tips 200 µl (Ultratip, Greiner Bio One International, catalog number: 739290)
- Disposable pipet tips 1,000 µl (Ultratip, Greiner Bio One International, catalog number: 686290)
- Wash bottles (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 2401-0500)
- Glass-bottomed black walled 96 wells plate (Porvair Sciences, catalog number: 324002)
- Black walled clear bottomed 96 wells plastic plate (Greiner Bio One International, catalog number: 675096)
- Stirred Quartz cuvettes (Hellma, catalog number: 119004F-10-40)
- Lens cleaning tissue (Fischer Scientific, FisherbrandTM, catalog number: 10605955)
Note: This product has been discontinued. - Filtropur S 0.2 µm filter (SARSTEDT, catalog number: 83.1826.001)
- Plasmids containing cytosolic and periplasmic controls for the sfTq2ox-mNG FRET pair as well as additional information are available on: https://www.addgene.org/browse/article/28196682/
- Ethanol 70% in lab wash bottle (VWR, Technisolv®, catalog number: 83801.360)
- Ethanol 96% in lab wash bottle (VWR, Technisolv®, catalog number: 83804.360)
- H2O in lab wash bottle
- 1 M β-D-1-thiogalactopyranoside (IPTG) (Duchefa Biochemie, catalog number: I1401)
- Hellmanex III (Hellma, catalog number: 9-307-011-4-507)
- Bacto tryptone (Duchefa Biochemie, catalog number: T1332)
- Yeast extract (Duchefa Biochemie, catalog number: Y1333)
- Sodium chloride (NaCl) (Merck, catalog number: 106404)
- Glucose (Duchefa Biochemie, catalog number: G0802)
- Thiamine hydrochloride (Sigma-Aldrich, catalog number: T4625-25G)
- L-Lysine monohydrochloride (Sigma-Aldrich, catalog number: L8662)
- Ampicillin stock solution 100 mg/ml (Sigma-Aldrich, catalog number: A9518) in 50% ethanol
- Chloramphenicol stock solution 25 mg/ml (Duchefa Biochemie, catalog number: C0113) in 50% ethanol
- di-Potassium hydrogen phosphate (K2HPO4·3H2O) (VWR, catalog number: 26932.290)
- Potassium dihydrogen phosphate (KH2PO4) (Merck, catalog number: 104873)
- Ammonium sulfate, (NH4)2SO4 (Sigma-Aldrich, catalog number: A4418)
- Magnesium sulfate heptahydrate (MgSO4·7H2O) (Carl Roth, catalog number: T888.1)
- Iron(II) sulfate heptahydrate (FeSO4·7H2O) (Sigma-Aldrich, catalog number: 215422)
- Calcium nitrate tetrahydrate, Ca(NO3)2·4H2O (Sigma-Aldrich, catalog number: C1396)
- L-Arginine (Sigma-Aldrich, catalog number: A8094)
- L-Glutamine (Sigma-Aldrich, catalog number: G8540)
- Thymidine (Sigma-Aldrich, catalog number: T1895)
- Uracil (Sigma-Aldrich, catalog number: U1128)
- Potassium chloride (KCl) (VWR, catalog number: 71003-522)
- di-Sodium hydrogen phosphate dihydrate (Na2HPO4·2H2O) (Merck, catalog number: 106580)
- Glutaraldehyde 25% (Merck, catalog number: 1042390250)
- Formaldehyde ≥ 36.0% (Sigma-Aldrich, catalog number: 47608)
- Lysine (Sigma-Aldrich, catalog number: L8662)
- PBS buffer (pH 7.2)
- TY medium (see Recipes)
- 20% glucose solution (see Recipes)
- GB1 medium (see Recipes)
- Stock solution (see Recipes)
- MM1-10x
- MM2-10x
- MM3-100x
- MM4-100x
- Vitamin B1 stock solution (4 mg/ml)
- Lysine stock solution (20 mg/ml)
- Phosphate buffered saline (PBS 1x) (see Recipes)
- Formaldehyde and glutaraldehyde (FAGA) (see Recipes)
Equipment
- P20, P200 & P1,000 Pipetman pipettes (Gilson, catalog number: F167300)
- Gyrotory water bath shaker Model G76 (Eppendorf, New Brunswick Scientific, model: Model G76 or GFL-Gesellschaft für Labortechnik, catalog number: 1092) with holders for culturing flasks
- Photospectrometer (Biochrom, model: Libra S22)
- Compressed air source
- Water purification system (Purelab flex with ELGA LC197-Biofilter from ELGA LabWater, Laner End, HP14 3BY, UK)
Note: All H2O in the protocol indicates water of at least 18.0 MΩ from Water purification system. - Centrifuge for 50 ml Falcon tubes (Eppendorf, model: 5804 R)
- Centrifuge for 1.5 and 2.0 ml Eppendorf tubes (Eppendorf, model: 5424 R)
- Spectrofluorometer Quantamaster 2000-4 (Photon Technology International, NJ) with a red-optimized set-up: R928P PMT tube (185-900 nm), 500 nm blaze (1,200 lines/mm) both excitation and emission gratings
Note: A Photomultiplier tube (PMT) that will reliably detect fluorescence spectra up until 700 nm will be suitable for the described protocol. - Optical filters
- 500/10 nm BrightLine® single-band bandpass filter (Semrock, catalog number: FF01-500/10)
- ET510LP (Chroma, catalog number: ET510lp)
- 435/40 nm BrightLine® single-band bandpass filter (Semrock, catalog number: FF02-435/40)
- 458LP EdgeBasic™ best-value long-pass edge filter (Semrock, catalog number: BLP01-458R)
- Fluorescence scanning plate reader (BioTek Instruments, model: SynergyTM Mx, catalog number: SMATBC)
- Autoclave (Sanyo, catalog number: MLS-3780)
Software
- Fluorescence spectrophotometer software (PTI acquisition software FeliX32 version 1.2 Build 56)
- Microplate reader software Gen5 version 2.00 (Biotek)
- Excel (Microsoft)
Procedure
The FRET method from beginning to end encompasses several steps, which are discussed per subject. The procedure and sections are outlined in Figure 1. This protocol describes our FRET assay with sfTq2ox and mNG, for a full description of sections A and B we refer to https://bio-protocol.org/e2697.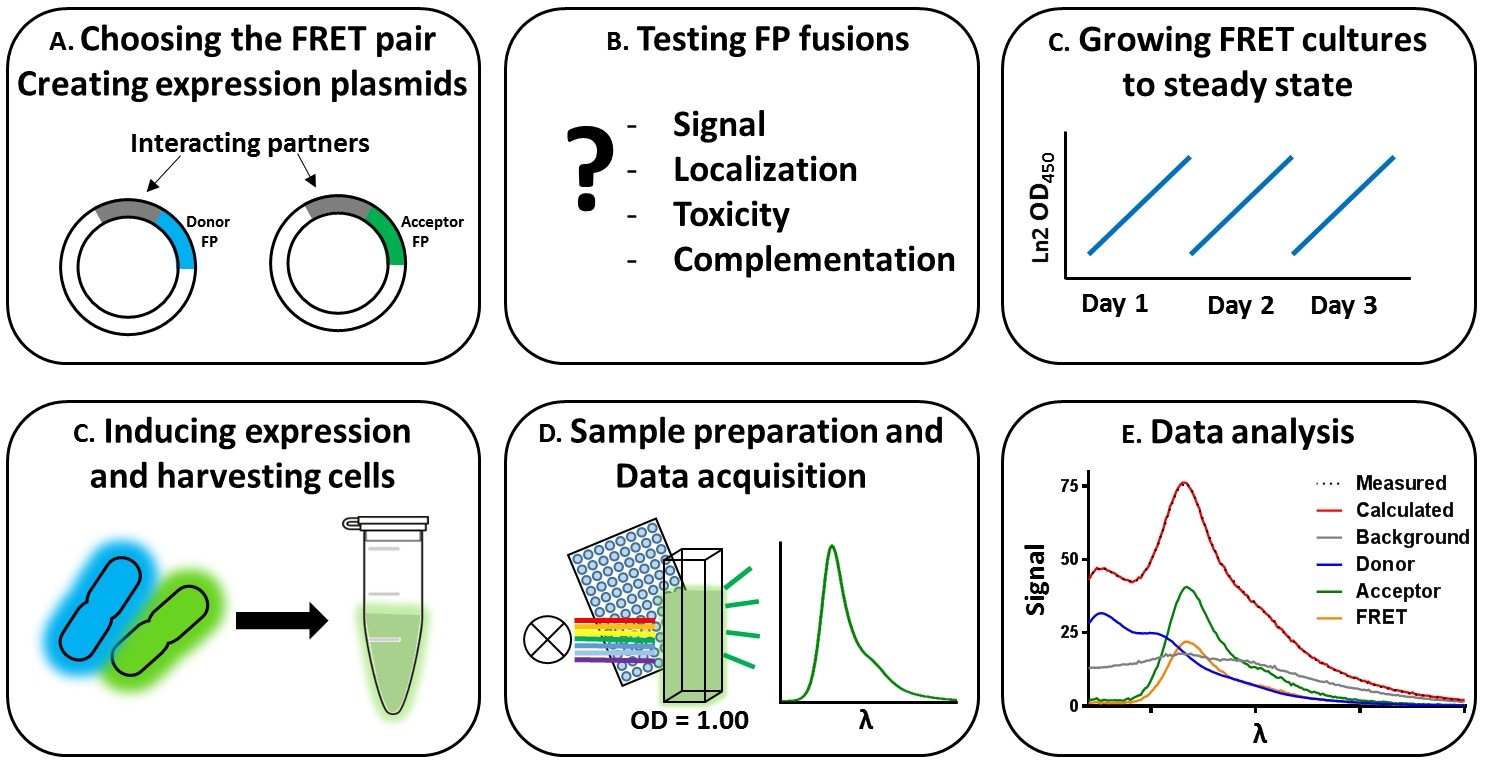
Figure 1. Workflow of the bacterial spectral-based FRET method. The labeling of the panels corresponds to the section describing the specific protocols. A. Choosing the suitable FRET-pair and creating expression plasmids. B. Testing the constructed plasmids for functionality. C. Growing cultures steady state, inducing expression of the FRET constructs and harvesting cells. D. Sample preparation and data acquisition. E. Analysis of the data.
- Choosing a suitable FRET pair for your experiment and creating FP fusions
- Choosing a suitable FRET pair for your experiment
Before starting FRET experiments, the most suitable FRET-pair should be selected. To date the FRET-pair that yields the highest rates of energy transfer in the cytoplasm and the periplasm of E. coli is super folder mTurquoise2 ox (sfTq2ox) (Meiresonne et al., 2019)–mNeonGreen (mNG) (Shaner et al., 2013). The high FRET efficiency of the sfTq2ox-mNG pair is related to the substantial overlap of their respective emission and excitation spectra, as well as their considerable quantum yield and extinction coefficient (Figure 2, Table 1). It is conceivable that a FRET pair is required with lower intrinsic background fluorescence. For this, the monomeric Kusabira-Orange (mKO) (Karasawa et al., 2004)–mCherry (mCh) (Shaner et al., 2004) could be attempted. The mKO-mCh FRET protocol as well as other alternative FRET pairs is described in https://bio-protocol.org/e2697.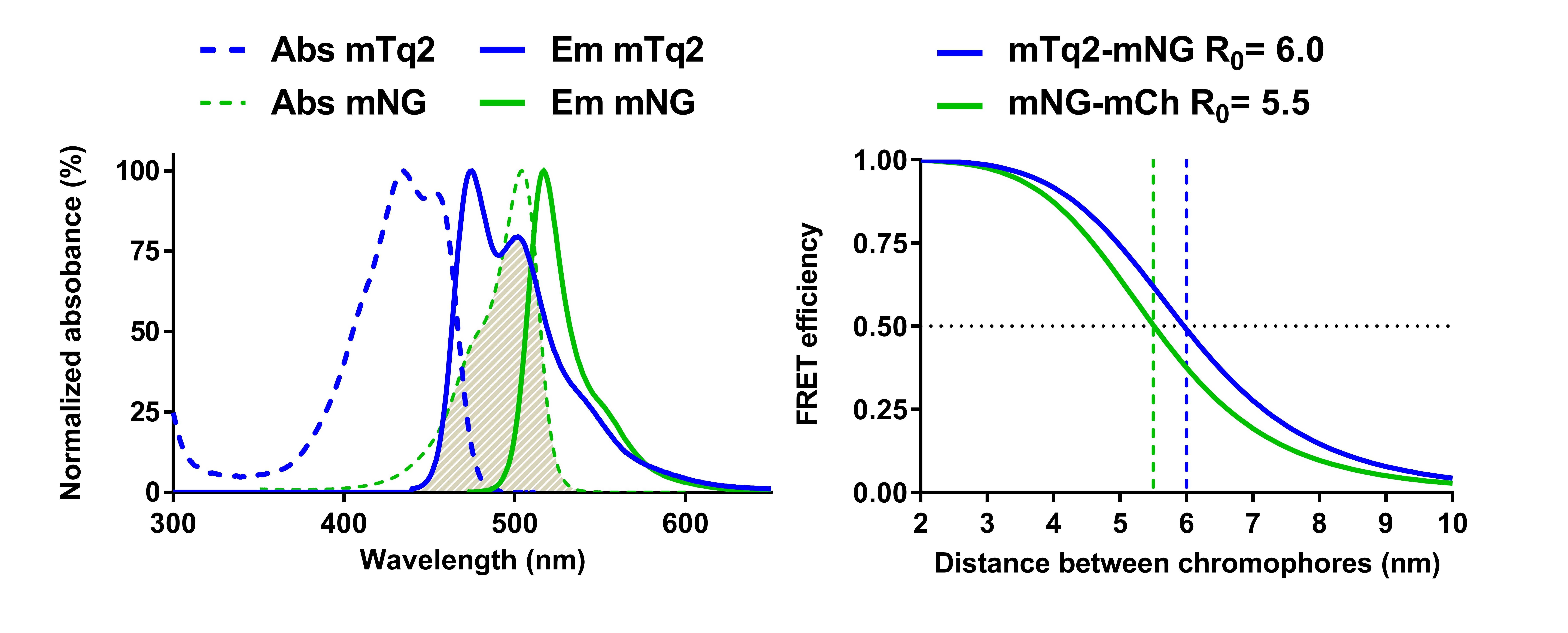
Figure 2. Spectral overlap and Forster distance of the mTq2-mNG FRET pair. On the left, spectral overlap between emission spectrum of mTq2 and absorbance spectrum of mNG. SfTq2ox has to date not been isolated and therefore its spectrum is not available. However, since the spectral properties of mTq2 and sfTq2ox in cells are identical, the shown mTq2 spectrum is identical to that of sfTq2ox. On the right, plotted theoretical FRET efficiencies as a function of chromophore distance for the mTq2-mNG, FRET pairs using the spectral properties in Table 1, the mNG-mCh FRET pair is shown for comparison. R0 is the distance between the fluorophores that will yield 50% energy transfer.
Table 1. Information on the FRET pairs described in this bio-protocol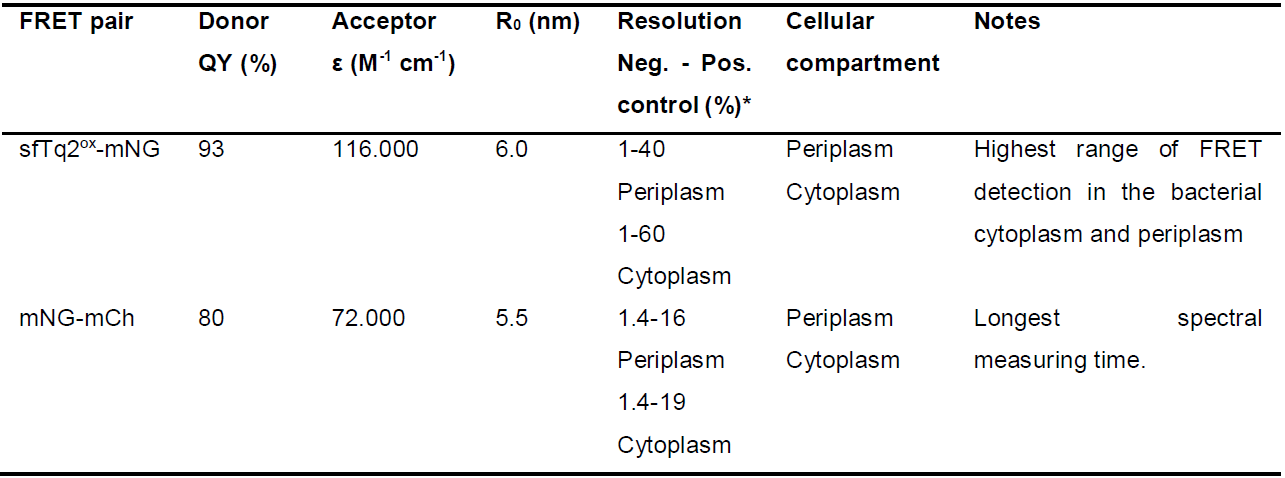
*As described in the original papers (Meiresonne et al., 2017 and 2019).
Notes:- For the R0 values presented in Table 1, the originally published extinction coefficient of 72,000 M-1 cm-1 of mCh was used (Shaner et al., 2004). Variation occurs in the description of FP extinction coefficients based on detection accuracy and sample conditions. The ever-improving field of FP research shows that the extinction coefficient of mCh is more in the range of 85,000 M-1 cm-1(Cranfill et al., 2016; Bindels et al., 2017).
- The quantum yield of sfTq2ox (and sfTq2) was approximated by fluorescence lifetime to not differ from mTq2 (Goedhart et al., 2012; Meiresonne et al., 2019).
- Different FP pairs, have been successfully used for bacterial FRET. For more information, for reference see https://bio-protocol.org/e2697.
- Creating FP fusions
Bacterial genes encoding a protein of interest are fused to a gene encoding one of the described fluorescent proteins and expressed from an inducible expression vector. Any plasmid expression system can be used, provided that the plasmids are compatible (different origins of replication and different antibiotic resistance markers) and the expression level is low (typically 2,000 copies of the protein) to avoid nonspecific interactions. To give the FP and the protein of interest some freedom of movement and reduce steric hindrance, it can be useful to add a linker between the FP and the protein. GGS repeats are often used but we also have good results with ARNNNN and other amino acid sequences. Another consideration is the choice for an N- or C-terminal fusion. For instance, N-terminal fusions of proteins that encode signal sequences for downstream transport or processing should be avoided or circumvented. Additionally, the N- or C-terminus of a protein may be essential for its normal localization or functionality. Procedure B discusses testing the created fusions. The sequence of the plasmid should be confirmed before further experiments. A description of cloning and transforming E. coli with plasmids is beyond the scope of this protocol but other bio-protocols like ‘[BIO101] Standard cloning’ and ‘[BIO101] The Inoue Method for Preparation and Transformation of Competent E. coli: “Ultra Competent” Cells’ (He, 2011; Im, 2011) may be useful.
- Choosing a suitable FRET pair for your experiment
- Testing the FP fusions
Ideally, the FP-fusions should be fully functional and expressed at levels close to their native levels while still resulting in measurable fluorescence intensities. Once the fusions are created on a plasmid, the expressed products should be tested for their effects on phenotype, fluorescence and correct cellular localization. If no fluorescence is observed, degradation of the FP may be assessed by Western blot analysis (anti-GFP for sfTq2ox, Invitrogen A6455 and anti-mNG, Chromogen 32F6). Perhaps the fusion disrupts normal localization, functionality and interactions of the protein of interest. This may be amended by modifying the length of the linker or switching the position of the FP. Some proteins may neither be functional as N nor as C-terminal fusion but will work as a sandwich fusion (van der Ploeg et al., 2015). An FP-fusion should complement the loss of function from a deletion or depletion strain of that same protein. Complementation testing will depend on whether the loss of the gene results in a phenotype. If no complementation experiments are possible, FRET experiments can be done in either a wild-type or deletion background but it should be kept in mind that the fusion protein may not be functional and results could be misinterpreted.
Note: Plasmid-based FP-fusions are a great way of quickly testing localization or the interaction of proteins of interest. However, plasmid-based complementation is not always possible due to stoichiometry imbalance. In such cases, the FP-fusion could be inserted in the chromosome at the original locus. In fact, chromosomal insertions of well-established FRET partners would be preferred for easy assays. However, this would also require chromosomal references in an isogenic background because bacteria can respond differently to plasmid and chromosomal based protein expression with respect to growth differences in and possible autofluorescence due to plasmid maintenance.
Preparation for the FRET experiment
When the FP fusion proteins are ready for use, competent cells can be transformed with the donor and acceptor expressing plasmids. Any E. coli strain should be suitable. Because we are interested in proteins that affect morphology, we prefer to use wild-type strains like MC4100 or BW25113 that are morphogenetically homogenous.
Note: If deletion strains are used the references and controls should be in the wild-type variant. The FRET assay described, requires at least three reference samples that are expressed in cells of the same genetic background as the assessed interaction. Strongly advised is the use of positive and negative controls consisting of a plasmid expressing sfTq2ox fused to mNG and two plasmids expressing the sfTq2ox or mNG fused to unrelated proteins that are known not to interact, respectively. The best controls are expressed in the same compartment as where the protein-protein interaction takes place to be able to assess molecular crowding effects. It is worthwhile to also switch donor and acceptor fusions (i.e., protein 1 is fused to sfTq2ox or mNG and protein 2 has the opposite FP). This is to rule out biased results due to differences of expression due to plasmid copy number etc. and the found FRET values should be similar. A list of samples for a minimal sfTq2ox-mNG FRET experiment are shown in Table 2.
Table 2. Samples and plasmids required for a minimal FRET experiment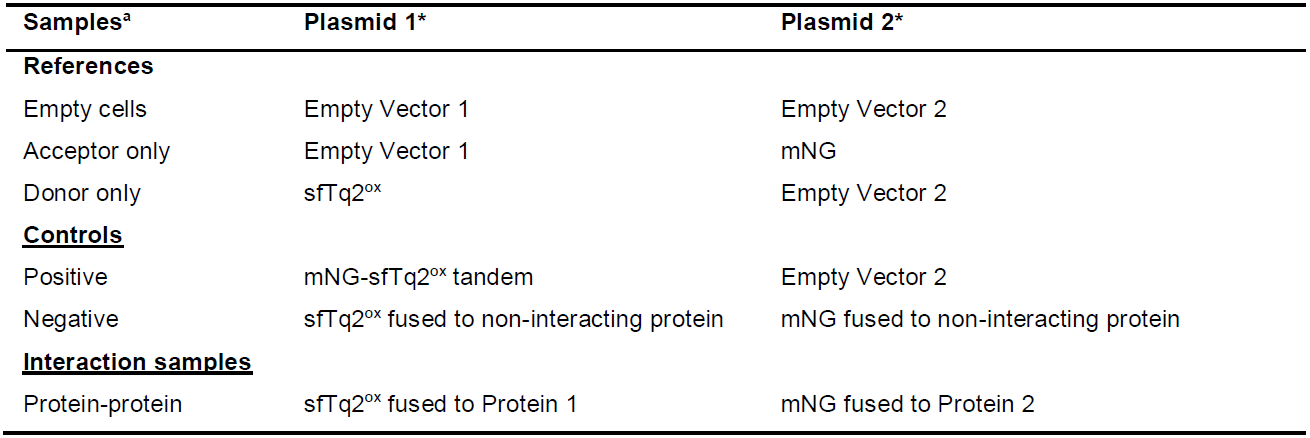
aTo detect the interaction between two proteins of interest at least three references are required. Positive and negative controls are advised. The described protocol is for two compatible plasmids that have a p15 origin of replication combined with chloramphenicol resistance and a ColE1 origin of replication combined with ampicillin resistance. Both plasmids have a weakened trc99A promoter and two terminators.
*Several positive control plasmids for cytoplasmic and periplasmic expression are available on https://www.addgene.org/browse/article/28196682/.
- Steady state growth, induction and harvesting cells
Bacterial cells that are grown in rich medium often contain a high concentration of autofluorescing molecules that decrease the signal to noise ratio. For that reason, the cells are grown in minimal medium and FRET experiments performed in rich medium are discouraged. To allow comparison of the various bacterial cultures expressing FRET constructs the cells are grown to steady state in about 20 generations before the expression of the proteins is induced (Figure 3). This assures identical autofluorescence backgrounds in all samples. After induction of expression for at least 2 mass doublings the cells are fixed and harvested.- Prepare starter cultures of all strains required to run your FRET experiment in 5 ml TY + ampicillin 100 µg/ml + chloramphenicol 25 µg/ml + 0.5% glucose medium at 37 °C while shaking until bacterial growth becomes visible (Day 0).
Note: Glucose is added to the medium to repress the expression of the lacIq controlled gene from the plasmids. - Use the starter cultures to inoculate 25 ml (1:250) of Gb1 medium + ampicillin + chloramphenicol and continue overnight growth at 28 °C while shaking.
- Day 1. Dilute the overnight cultures 1:250 in fresh Gb1 medium + ampicillin + chloramphenicol and continue growth at 28 °C while shaking.
Note: From now on periodically measure and note OD450 values of the cultures and do not let them reach above 0.2. When the cultures approach this value dilute with fresh pre-warmed medium. - Calculate the doubling time (TD) for all growing cultures using the provided Excel sheet ‘Growth calculation.xlsx’ and use it to calculate the dilution factor to reach OD450 values of 0.1 the next morning when the experiment is continued.
- Day 2. Repeat the growing in fresh Gb1 medium + ampicillin + chloramphenicol while keeping the OD450 values below 0.2 and dilute for the next morning like in Step C4.
- Day 3. Repeat the growing in fresh Gb1 medium + ampicillin and chloramphenicol while keeping the OD450 values below 0.2 and dilute for the next morning like in Step C4 but this time aim for an OD450 value of 0.025 in 35 ml fresh Gb1 + ampicillin + chloramphenicol.
- Day 4. Measure OD450 values of the cultures and if they are around 0.03-0.04 induce expression from the plasmids by adding IPTG at an appropriate concentration (e.g., 10-20 µM) and continue growth.
- Before the cultures approach OD450 values of ~0.2, prepare a sufficient amount of FAGA fixative (Recipe 5).
Note: The total volume of FAGA needed differs depending on the total volume of the to-be-fixed cultures, 1 ml of FAGA is required to fix 12.5 ml of culture. - When the cells reach OD450 values of 0.2, fix the cells for 10 min in the shaking water bath at 28 °C with FAGA, which amount depends on how much of the culture’s volume is left.
- Decant the fixed cultures in 50 ml Falcon tubes and pellet the cells by centrifugation for 10 min at 6,300 x g at room temperature (Eppendorf 5804 R).
- Discard the supernatant and wash 3 x with 1 ml PBS in a 1.5 ml Eppendorf tube centrifuging for 5 min at 4,600 x g at room temperature in a tabletop centrifuge (Eppendorf 5424 R).
- Resuspend the last pellet in 1 ml of PBS and store the samples overnight at 4 °C and measure the next day.
Note: sfTq2ox and mNG fold fast and their chromophore matures quickly so direct measurements could be considered, but it is more practical to do the spectral measurements a day after.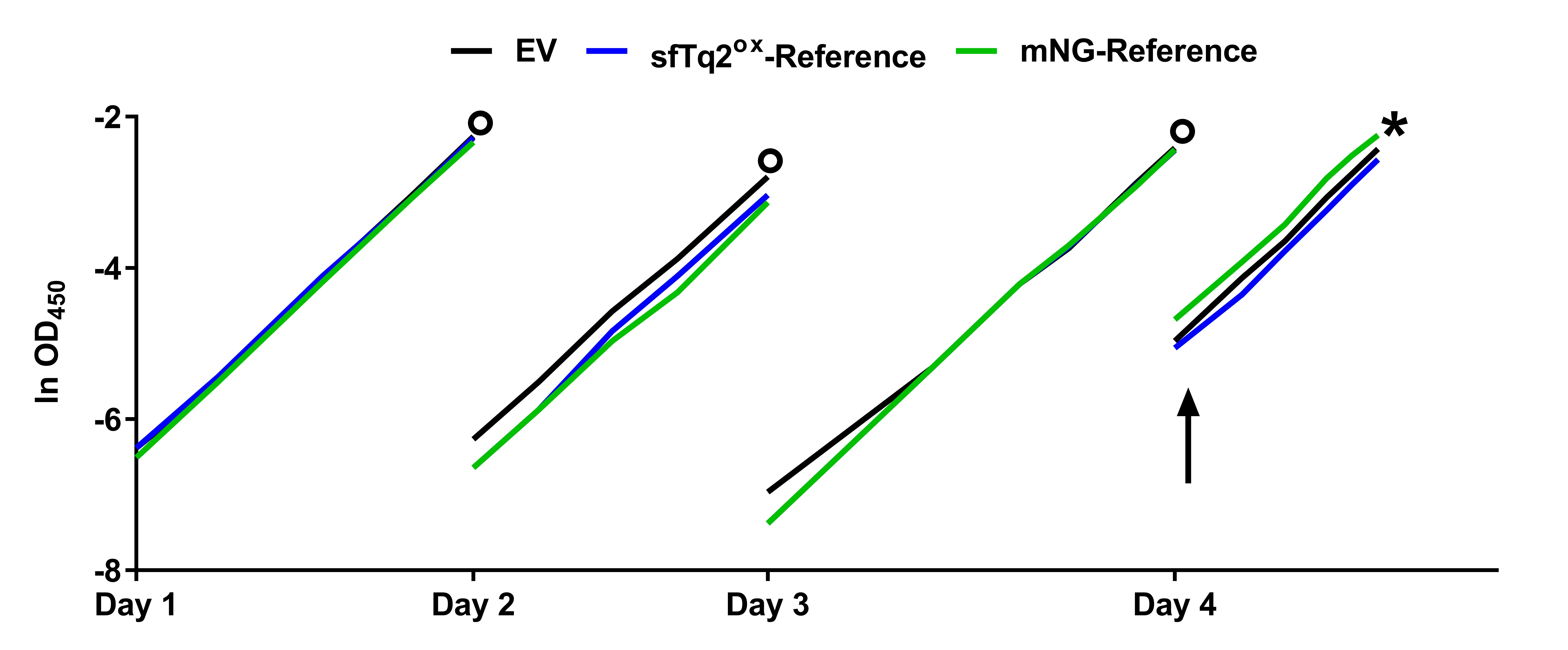
Figure 3. Example of steady state growth. Flask culture growth curves of MC4100 based E. coli cultures were grown to steady state in Gb1 at 28 °C expressing FP-fusions. The circles represent the time points at which overnight dilutions were made. At Day 4 the cultures were induced with IPTG (arrow) to induce production of FP-fusions. The EV (Empty Vector) culture does not express any FP and serves as a control. The X-axis is given in OD450 to signify exponential growth. Cells were fixed and harvested (asterisk) after at least two mass doublings after induction. This figure was adapted from (Meiresonne et al., 2017).
- Prepare starter cultures of all strains required to run your FRET experiment in 5 ml TY + ampicillin 100 µg/ml + chloramphenicol 25 µg/ml + 0.5% glucose medium at 37 °C while shaking until bacterial growth becomes visible (Day 0).
- Sample preparation and data acquisition
- Pellet the cells as described in Step C10 and resuspend in 1 ml PBS.
- Prepare at least 1,300 µl of samples of with OD450 values of 1.00 ± 0.005 by diluting the cells with fresh PBS and add 1,200 µl to the stirred quartz cuvette.
Notes:- The dilution can be made inside the photospectrometer; the more equal the OD450 values are; the more reliable will be the results.
- Do not forget to add the magnetic stirrer to the cuvette before measuring.
- In theory plastic cuvettes could be used for measurements of spectra at visible wavelengths, however, that was not attempted for the experiments described in this protocol and should be tested first. The cuvettes should be identical for each sample to avoid differences in background.
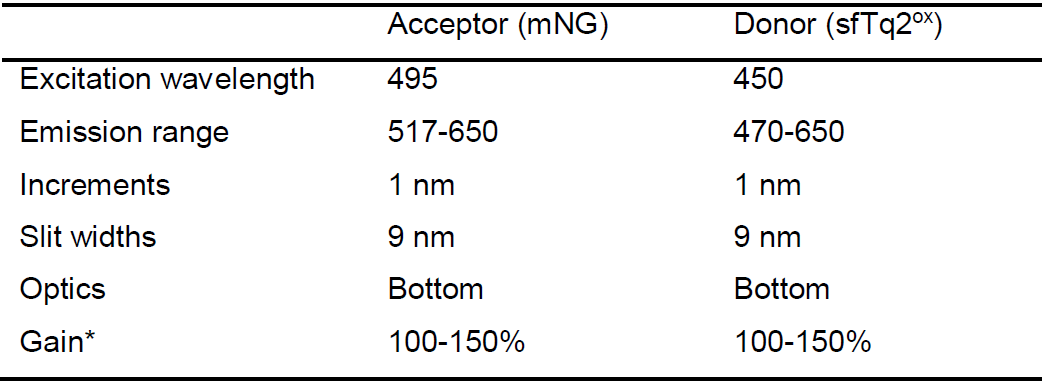
- Measure the mNG emission spectrum using the settings and optical filters indicated in Table 3 and described in ‘Measuring spectra using a fluorometer–Fluorometer settings and practical setup’.
Table 3. Filter setup for fluorometer spectral measurements
aFilters with similar transmission properties can be used. - Measure the donor emission spectrum using the settings and optical filters indicated in Table 3 and described in ‘Measuring spectra using a fluorometer–Fluorometer settings and practical setup’.
Note: It is advisable to measure the mNG and sfTq2ox spectra of each sample in the same order to prevent possible differences in FP bleaching. - Repeat Steps D2-D4 for each sample and also measure a blank PBS only sample at the start of the measurement and the end of all measurements. This allows you to verify any change in the instrument response.
- Save the data in any convenient file format that can be exported to Microsoft Excel.
Notes:- Before loading a new sample in the quartz cuvette, it needs to be thoroughly cleaned by performing the steps outlined in the cleaning protocol below.
- By saving the OD450 = 1.00 samples they can directly be reused for spectral measurements with the plate reader described in ‘Measuring spectra with the plate reader’.
Measuring spectra using a fluorometer–Fluorometer settings and practical setup
Emission spectra are measured using a fluorometer. The fluorometer should excite a sample with a narrow band wavelength, which then emits fluorescence that is measured. A schematic overview of a fluorometer is shown in Figure 4. The light source emits white light, which passes a slit to the excitation monochromator where it is separated into its wavelength components. The light passes a slit before passing the excitation filter and hits the sample suspension in the quartz cuvette. The cells in the sample will be excited and emit light in every direction but excitation light also scatters from them. At an angle of 90°, the scattered light is filtered by an emission filter and the fluorescence emission passes a slit entering the emission monochromator. Here, the emission wavelengths are scanned and pass the last slit before entering a photomultiplier where quanta of photons are being transformed into a measurable electrical current. The resulting emission spectra are the raw data needed to calculate FRET. The filter and measurement settings are shown in Table 3. Slit widths are usually set to 6 nm and generally give a good result if the fluorescent proteins are expressed well. Smaller slit widths result in more specific measurements but will also let less light pass to be detected so slits should only be more closed when fluorescent signals are very high. If the fluorescent signals are low, the slits could be widened but this will result in less specific signals. When background fluorescence is an issue, this will increase in a proportional manner.
Notes:- The white light source has to heat up before the light output will be stable. Thirty minutes after turning it on, check whether the power of the lamp is 75 W and if not adjust. Other fluorometers will have different requirements.
- The use of other white light sources or equipment may require different preparations.
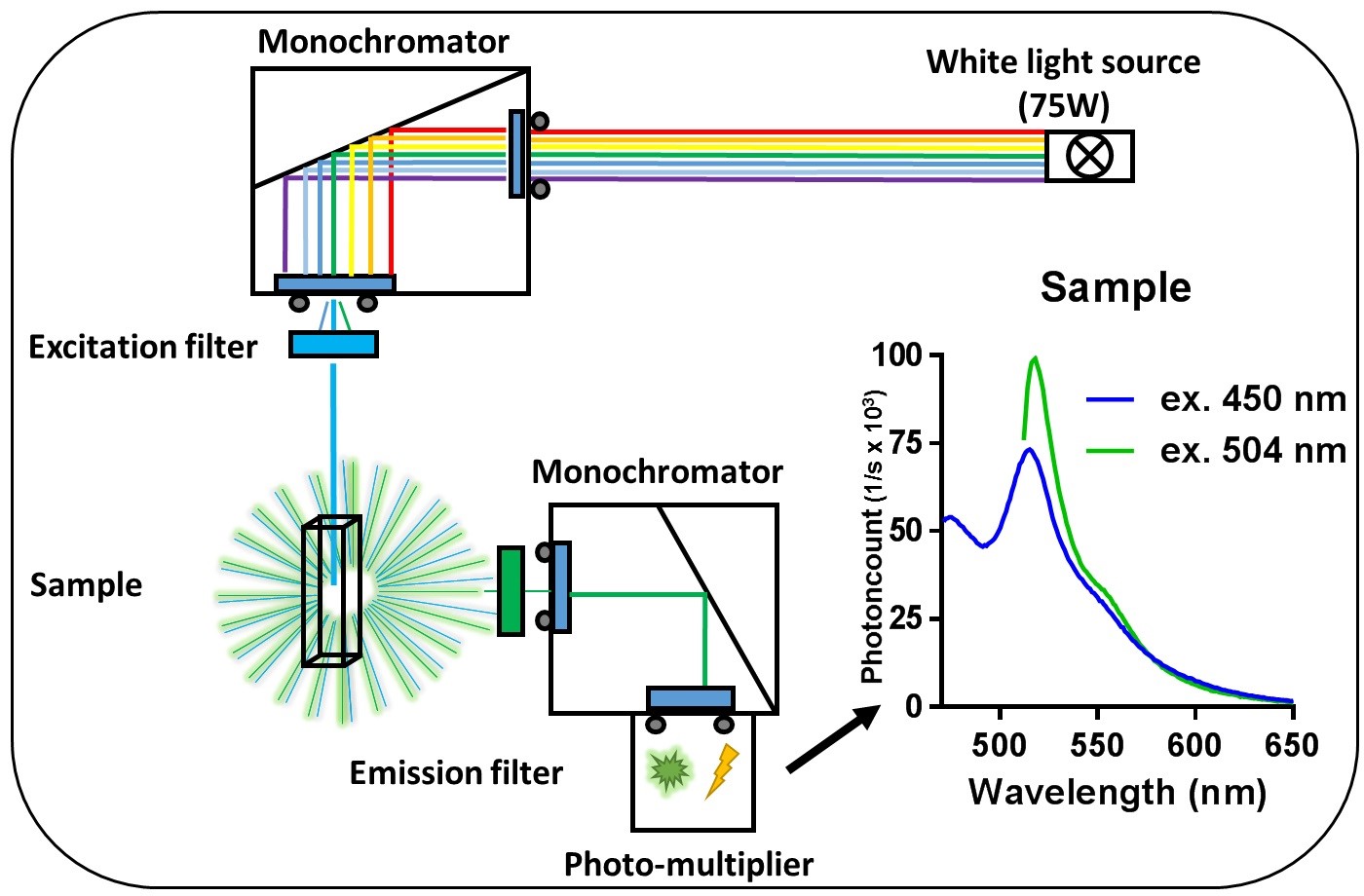
Figure 4. Schematic overview of measuring a sample in the fluorometer. The double black and gray circles at the monochromators represent adjustable slits.
Protocol cleaning quartz cuvettes- Profusely rinse the inside and outside of the cuvette with H2O.
- Carefully shake off the excess H2O.
- Profusely rinse the inside and outside with 70% ethanol.
- Carefully shake off the excess 70% ethanol.
- Repeat steps a-d (Protocol cleaning quartz cuvettes), three times.
- Profusely rinse the inside and outside with 96% ethanol.
- Carefully shake off the excess ethanol 96%.
- Blow dry the cuvette with compressed air.
- Polish the cuvette with lens tissue to get rid of accidental smudges like for instance fingerprints. Now the cuvettes are properly cleaned and can hold a sample for measurement.
Notes:- The magnetic stirrers beans are washed by the same rinsing protocol in the palm of your hand.
- After the last measurement, wash all cuvettes and stirrer beans one more time and then submerge them in 2% Hellmanex III for storage. If cuvettes are not needed for longer time periods, store them clean and dry.
- Quartz cuvettes should be cared for meticulously as they are fragile and expensive. Only touch the cuvettes with your fingers close to the opening to prevent finger prints on the important part of the quartz. Carry the cuvette between your index finger and thumb while supporting the bottom with your pinky or ring finger as shown in Figure 5.
Figure 5. Cartoon of how to hold quartz cuvettes properly (i.e., between thumb and index finger supported by the little finger)
Measuring spectra with the plate reader
A convenient way of measuring the emission spectra of your samples is by using a fluorescence plate reader. A word of caution is in order as the sensitivity of most fluorescence plate readers is lower than that of a dedicated fluorometer and provide weaker fluorescence signals. If fluorescence is low, the fluorometer should be used. This bio-protocol describes the use of the BioTek synergy MX plate reader, which has excitation and emission monochromators without the possibility for additional optical filtering. In addition, this plate reader has slits with a minimal opening of 9 nm. The, on average, lower signal to noise and less specific signals from a plate reader may therefore lead to unmixing problems. It is thus recommended to first firmly establish FRET interactions using a fluorometer and then confirming them using the plate reader. If results are reproducible, experiments can be done with the plate reader only.Table 4. Settings for spectral measurements using the plate reader
Protocol plate reader FRET for fixed cells- Prepare samples with OD450 values of 1.00 ± 0.005 like described in Steps D1-D2.
- Dispense 200 µl replicates of the samples in a glass bottomed black walled 96-well plate. If eight replicates are made, there is space for 11 prepared samples and 1 PBS measurement in a 96-well plate.
- Measure the donor and acceptor emission spectra of each well using the settings shown in Table 4.
- Export the emission data in an Excel compatible format and plot the replicates to check for obvious outlying data.
- Exclude the possible outliers based on large and obvious deviations from the other replicates and average the acceptor and donor emission spectra. These data will be used for spectral unmixing and FRET calculations.
*Optimal gain may differ between experiments. It is better to have slightly lower signals than to oversaturate the detector.
Note: Measuring the OD450 values in the plate reader before the spectral measurements will give an indication how similar the samples are to each other. - Data analysis
When the acceptor and donor emission spectra of all samples are measured either by the fluorometer or by the plate reader, they can be unmixed. Spectral unmixing dissects the emission data into its components based on the references that were provided. The sfTq2ox channel is composed of background emission, sfTq2ox-only emission, directly excited mNG emission and possible FRET mNG emission (Figure 6). The mNG channel is composed of background emission and mNG-only emission. From this spectrum, the amount of mNG in the sample can be calculated using the background and reference spectra by unmixing. Knowing the amount of mNG fluorescence, it can be calculated how much fluorescence is expected from the mNG due to cross-excitation of the donor and the acceptor in the sample that contains both. Using the reference spectra and the knowledge on the amount of mNG in the sample, the emission spectrum for a sample can be dissected into the background spectrum, the sfTq2ox spectrum and the mNG spectrum. Remaining is a spectrum of identical shape as the mNG spectrum that cannot be accounted for if no sensitized emission (energy transfer) had occurred. This extra mNG signal corresponds to the amount of energy transfer. The supplementary data of (Alexeeva et al., 2010) gives a detailed description of the calculations based on biophysical work beyond the scope of a protocol (Clegg, 1992; Clegg et al., 1992; Wlodarczyk et al., 2008; Gadella, 2009). In the supplementary information, ready-to-use Excel sheets are provided to calculate FRET efficiencies from sfTq2ox-mNG experiments measured with the fluorometer or plate reader ‘sfTq2ox-mNG.xlsx’ ‘sfTq2ox-mNG platereader.xlsx’.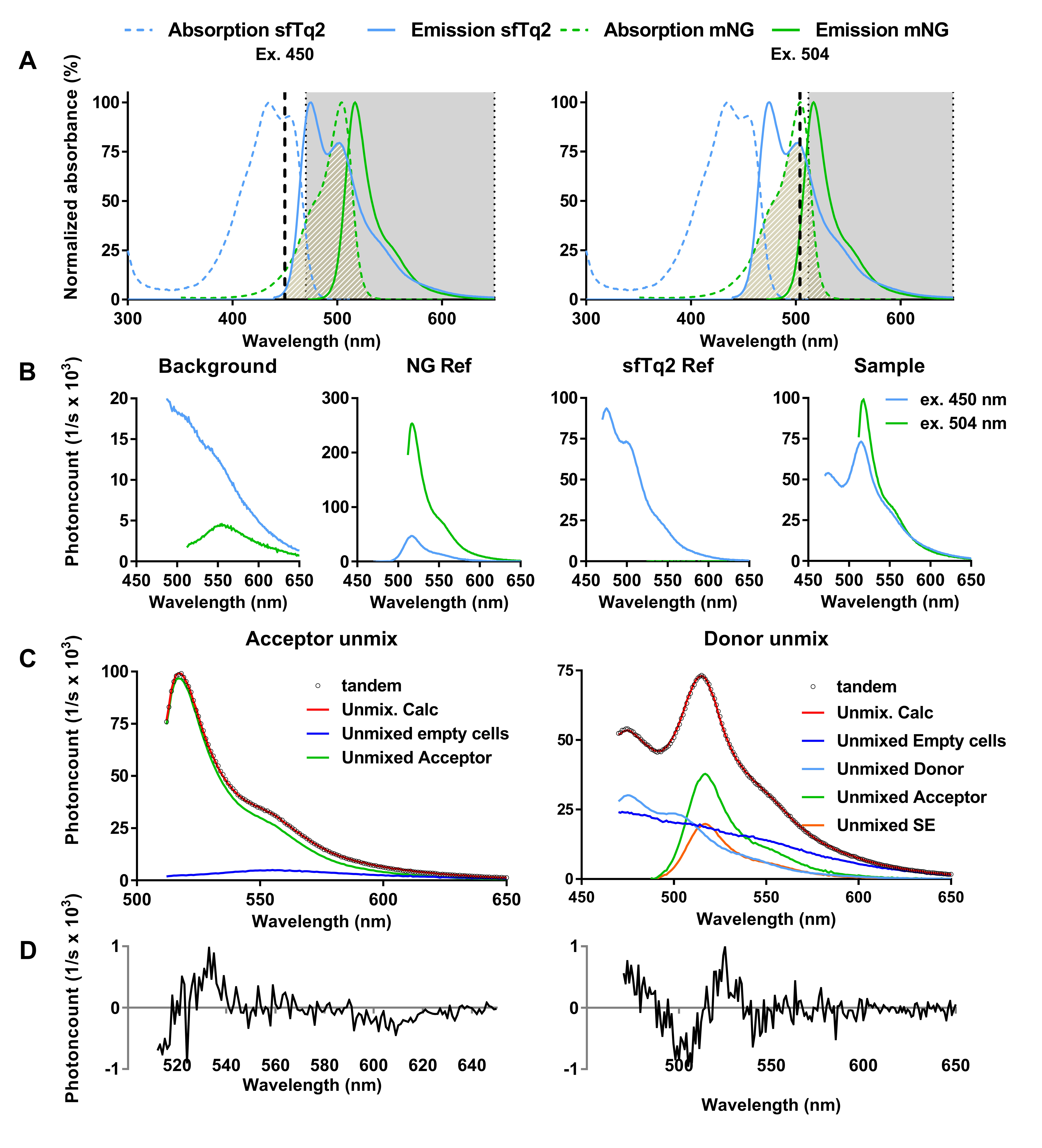
Figure 6. Principle of the sfTq2ox-mNG FRET assay. A. Excitation and emission spectra of mTq2 and mNG, indicating the wavelengths used to measure the mNG channel (left) or the sfTq2ox channel (right). The bold dotted line represents the used excitation wavelengths. The hatched blue area represents the spectral overlap between the mTq2 emission spectrum and the mNG excitation spectrum. The gray shaded area represents the wavelengths for which the emission was measured. B. Samples measured to calculate periplasmic FRET. References of background, mNG, and sfTq2ox are needed to calculate their contributions to the FRET within a sample. C. Unmixing of the tandem FRET sample, showing the measured spectrum as black dots (tandem) and the calculated spectrum as a solid red line (Unmix. Calc.). The measured spectrum for the mNG channel is composed of the background fluorescence (Unmixed Empty cells) and the amount of mNG (Unmixed Acceptor) present in the sample excited at 504 nm. The direct excitation of mNG is used to determine the amount of mNG present in the sample. This amount is then used to calculate how much extra mNG (Unmixed SE) is detected in the sfTq2ox excited sample. The measured spectrum for the sfTq2ox channel contains the background (Unmixed Empty cells), sfTq2ox (Unmixed Donor), and mNG (Unmixed Acceptor) fluorescence and sensitized emission (Unmixed SE), which is the extra fluorescence that the unmixing algorithm cannot attribute to direct excitation of mNG. D. The difference between the measured and calculated spectra represents signal that cannot be attributed to the reference components and is a measure of the quality of unmixing.
Protocol using the unmixing sheets
An overview of how the unmixing process would appear on the computer screen is shown in Figure 7. High resolution screenshots are available in the supplementary data.
Note: Be aware that the raw data may have different output formats depending on the equipment used and adjust accordingly.What determines a good measurement?- Copy the spectral data in the appropriate excel sheet 1 ‘Raw data’.
- Subtract the PBS measurement from all samples in sheet 2 ‘Minus PBS’.
- Copy the empty vector, acceptor reference and the donor reference in the indicated places in sheet 3 ‘Data’. This will automatically subtract the background signal from the acceptor and donor reference spectra.
- Copy sample data to the indicated place in sheet 3 ‘Data’ the sheet will unmix these spectra based on the given references.
Note: The reference and sample spectra are plotted at the bottom of sheet 3 for inspection. - Inspect the unmixing overview on sheet 4 ‘Result’ as described further below.
- Copy paste the results to sheet 5 ‘Summaries’.
- Repeat steps 4-6 for the remaining samples.
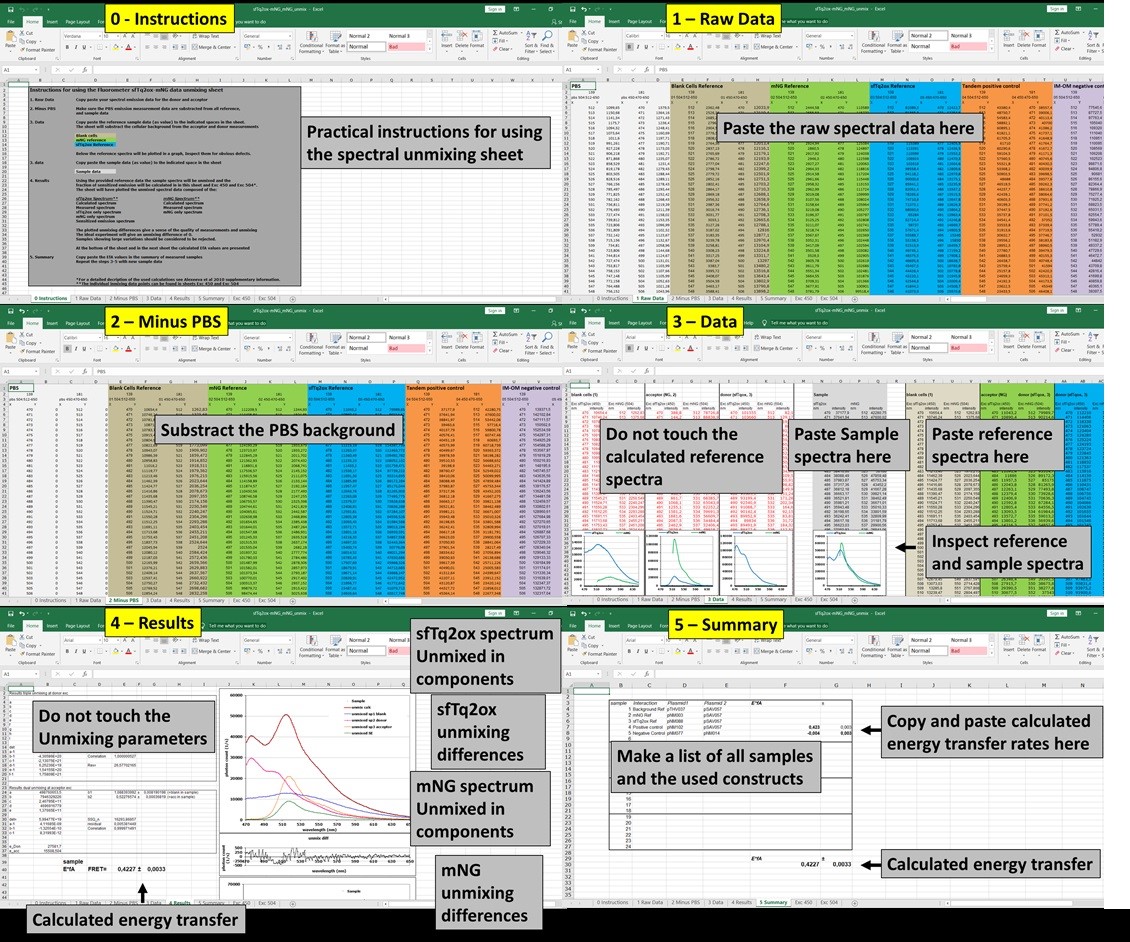
Figure 7. The unmixing process for the calculation of FRET efficiencies as it would appear on the screen. Raw donor and acceptor unmixing calculations that will not be directly used are shown in the high-resolution screenshots in Supplementary file ‘Unmixing screens.pptx’.
Spectral unmixing is highly dependent on clear fluorescent signals that should be measured under identical conditions. The donor and acceptor reference spectra are obtained by subtracting the PBS and empty cells data from their respective emission measurements as described in ‘Protocol using the unmixing sheets’. This should yield a clean emission spectrum of sfTq2ox only or mNG only. Sometimes spectra that do not resemble the measured FP anymore or even negative can be seen. This can be caused by strong levels of autofluorescence in the empty cells sample and low FP signals in the reference samples. If one of the references is not good, the unmixing of the spectra will not give trustworthy results. Alternatively, reference spectra of a previous measurement may be used to still get a rough indication of the FRET values of the samples. It is not advised to use these values as definite and the experiment needs to be repeated to yield trustworthy data. When the experiment results in good references, sample data can be unmixed. Emission signal that could not be attributed to the mNG spectrum is called the unmixing difference. The unmixing difference is a good measure for the quality of the measured FRET spectra and in an ideal experiment it should be zero. More realistically, it should be a small percentage of the measured spectra as shown in Figure 6D.
Data analysis
This bio-protocol describes how to prepare, perform and analyze in vivo bacterial FRET experiments. At the relevant sections (Procedure E), information is provided on data analysis and references to in-depth technical information are provided. If the protocol is rigorously followed every time, the resulting data will be highly similar. The significance of the obtained FRET values can be calculated using standard confidence interval statistical approaches. The FRET values should be significantly higher than the values obtained from non-interacting samples due to crowding. For this analysis, standard t-tests can be used.
Recipes
- TY medium (1 L)
10 g Bacto tryptone
5 g yeast extract
5 g NaCl
Add H2O up to 1 L and autoclave for 15 min at 121 °C - 20% glucose solution (200 ml)
40 g glucose
Add H2O up to 200 ml and filter sterilize with Filtropur S 0.2 µm filter - Stock solution
- MM1-10x (1 L)
63.3 g K2HPO4·3H2O
29.5 g KH2PO4
Add H2O up to 1 L and autoclave for 15 min at 121 °C - MM2-10x (1 L)
10.5 g (NH4)2SO4
1 g MgSO4·7H2O
Add H2O up to1 L and autoclave for 15 min at 121 °C - MM3-100x (1 L)
28 mg FeSO4·7H2O
710 mg Ca(NO3)2·4H2O
Add H2O up to 1 L and autoclave for 15 min at 121 °C
Note: After sterilization, the MM3 will show a red precipitation that should be allowed to deposit and not be touched when pipetting. - MM4-100x (50 ml)
250 mg L-arginine
250 mg L-glutamine
10 mg thymidine
100 mg uracil
Add H2O up to 50 ml and filter sterilize with Filtropur S 0.2 µm filter - Vitamin B1 stock solution (4 mg/ml) (50 ml)
200 mg thiamine hydrochloride
Add H2O up to 50 ml and filter sterilize with Filtropur S 0.2 µm filter - Lysine stock solution (20 mg/ml) (50 ml)
1 g lysine
Add H2O up to 50 ml and filter sterilize with Filtropur S 0.2 µm filter
- MM1-10x (1 L)
- Gb1 medium (1 L) (using aseptic technique)
Note: MM4-100x has to be added to GB1 medium for BW25113, TB28 and MG1655 based E. coli strains.100 ml MM1-10x 100 ml MM2-10x 10 ml MM3-100x 10 ml MM4-100x
Add sterile H2O up to 1 L20 ml 20% glucose 1 ml 4 mg/ml thiamine hydrochloride 2.5 ml 20 mg/ml L-lysine monohydrochloride 1 ml 100 mg/ml ampicillin 1 ml 25 mg/ml chloramphenicol - Phosphate buffered saline (PBS 1x)
PBS buffer (pH 7.2)
140 mM NaCl
27 mM KCl
10 mM Na2HPO4·2H2O
2 mM KH2PO4 - Formaldehyde and glutaraldehyde (FAGA) fixative
Per 1 ml of formaldehyde add 21 µl of glutaraldehyde
Acknowledgments
This bio-protocol is a sfTq2ox-mNG specific adaptation of https://bio-protocol.org/e2697, which was co-authored by Svetlana Alexeeva and René van der Ploeg. Adaptations of this bio-protocol have been used in the following publications (Alexeeva et al., 2010; Fraipont et al., 2011; van der Ploeg et al., 2013 and 2015; Meiresonne et al., 2017 and 2019). NYM was supported by the NWO, ALW open program (822.02.019) and HFSP Program RGP0034/2013. E.C. and L.M.Y.M. were supported by the European Commission via the International Training Network Train2Target (721484).
References
- Alexeeva, S., Gadella, T. W., Jr., Verheul, J., Verhoeven, G. S. and den Blaauwen, T. (2010). Direct interactions of early and late assembling division proteins in Escherichia coli cells resolved by FRET. Mol Microbiol 77(2): 384-398.
- Algar, W. R., Hildebrandt, N., Vogel, S. S. and Medintz, I. L.(2019) FRET as a biomolecular research tool–understanding its potential while avoiding pitfalls. Nat Methods 16(9): 815–829.
- Bindels, D. S., Haarbosch, L., van Weeren, L., Postma, M., Wiese, K. E., Mastop, M., Aumonier, S., Gotthard, G., Royant, A., Hink, M. A. and Gadella, T. W., Jr. (2017). mScarlet: a bright monomeric red fluorescent protein for cellular imaging. Nat Methods 14(1): 53-56.
- Clegg, R. M. (1992). Fluorescence resonance energy transfer and nucleic acids. Methods Enzymol 211: 353-388.
- Clegg, R. M., Murchie, A. I., Zechel, A., Carlberg, C., Diekmann, S. and Lilley, D. M. (1992). Fluorescence resonance energy transfer analysis of the structure of the four-way DNA junction. Biochemistry 31(20): 4846-4856.
- Cranfill, P. J., Sell, B. R., Baird, M. A., Allen, J. R., Lavagnino, Z., de Gruiter, H. M., Kremers, G. J., Davidson, M. W., Ustione, A. and Piston, D. W. (2016). Quantitative assessment of fluorescent proteins. Nat Methods 13(7): 557-562.
- Fraipont, C., Alexeeva, S., Wolf, B., van der Ploeg, R., Schloesser, M., den Blaauwen, T. and Nguyen-Disteche, M. (2011). The integral membrane FtsW protein and peptidoglycan synthase PBP3 form a subcomplex in Escherichia coli. Microbiology 157(Pt 1): 251-259.
- Gadella T. W. J. (2009). FRET and FLIM techniques. Elsevier.
- Goedhart, J., von Stetten, D., Noirclerc-Savoye, M., Lelimousin, M., Joosen, L., Hink, M. A., van Weeren, L., Gadella, T. W., Jr. and Royant, A. (2012). Structure-guided evolution of cyan fluorescent proteins towards a quantum yield of 93%. Nat Commun 3: 751.
- He, F. (2011). Standard DNA cloning. Bio-101: e52.
- Im, H. (2011). The inoue method for preparation and transformation of competent E. coli: "Ultra competent" cells. Bio-101: e143.
- Karasawa, S., Araki, T., Nagai, T., Mizuno, H. and Miyawaki, A. (2004). Cyan-emitting and orange-emitting fluorescent proteins as a donor/acceptor pair for fluorescence resonance energy transfer. Biochem J 381(Pt 1): 307-312.
- Meiresonne, N. Y., Consoli, E., Mertens, L. M. Y., Chertkova, A. O., Goedhart, J. and den Blaauwen, T. (2019). Superfolder mTurquoise2(ox) optimized for the bacterial periplasm allows high efficiency in vivo FRET of cell division antibiotic targets. Mol Microbiol 111(4): 1025-1038.
- Meiresonne, N. Y., van der Ploeg, R., Hink, M. A. and den Blaauwen, T. (2017). Activity-related conformational changes in d,d-Carboxypeptidases revealed by in vivo periplasmic forster resonance energy transfer assay in Escherichia coli. MBio 8(5).
- Shaner, N. C., Campbell, R. E., Steinbach, P. A., Giepmans, B. N., Palmer, A. E. and Tsien, R. Y. (2004). Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol 22(12): 1567-1572.
- Shaner, N. C., Lambert, G. G., Chammas, A., Ni, Y., Cranfill, P. J., Baird, M. A., Sell, B. R., Allen, J. R., Day, R. N., Israelsson, M., Davidson, M. W. and Wang, J. (2013). A bright monomeric green fluorescent protein derived from Branchiostoma lanceolatum. Nat Methods 10(5): 407-409.
- van der Ploeg, R., Goudelis, S. T. and den Blaauwen, T. (2015). Validation of FRET assay for the screening of growth inhibitors of Escherichia coli reveals elongasome assembly dynamics. Int J Mol Sci 16(8): 17637-17654.
- van der Ploeg, R., Verheul, J., Vischer, N. O., Alexeeva, S., Hoogendoorn, E., Postma, M., Banzhaf, M., Vollmer, W. and den Blaauwen, T. (2013). Colocalization and interaction between elongasome and divisome during a preparative cell division phase in Escherichia coli. Mol Microbiol 87(5): 1074-1087.
- Wlodarczyk, J., Woehler, A., Kobe, F., Ponimaskin, E., Zeug, A. and Neher, E. (2008). Analysis of FRET signals in the presence of free donors and acceptors. Biophys J 94(3): 986-1000.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Meiresonne, N. Y., Consoli, E., Mertens, L. M. and den Blaauwen, T. (2019). Detection of in vivo Protein Interactions in All Bacterial Compartments by Förster Resonance Energy Transfer with the Superfolder mTurquoise2 ox-mNeongreen FRET Pair. Bio-protocol 9(23): e3448. DOI: 10.21769/BioProtoc.3448.
Category
Microbiology > in vivo model > Bacterium
Biochemistry > Protein > Interaction > Protein-protein interaction
Molecular Biology > Protein > Protein-protein interaction
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.