- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Opto-magnetic Selection and Isolation of Single Cells
Published: Vol 9, Iss 22, Nov 20, 2019 DOI: 10.21769/BioProtoc.3428 Views: 4597
Reviewed by: Samantha E. R. DundonLinlin SunAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Apr 2019
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Capturing single cells from large heterogenous populations based solely on observable traits is necessary for many cell biology applications and remains a major technical challenge. The protocol we present allows the isolation of viable and metabolically active cells selected for their shape, migration speed, contact to other cells, or intracellular protein localization. We previously introduced a method termed Cell Labeling via Photobleaching (CLaP) for the efficient tagging of cells chosen for visual criteria. Here we describe a new protocol for capturing such cells using ferromagnetic beads termed single-cell magneto-optical capture (scMOCa). This technology is especially useful when the number of target cells represents an extremely low fraction of the total population (potentially one single cell), a situation in which conventional sorting techniques like fluorescent or magnetic activated cell sorting (F/MACS) cannot provide satisfactory results in terms of capture efficiency and specificity. scMOCa uses the lasers of a confocal microscope to photobleach and crosslink biotin-4-fluorecein molecules to cell membranes. Streptavidin coated magnetic beads then adhere to biotin moieties and a magnet allows the capture of illuminated cells. By precisely controlling liquid volumes and spacing between the different parts of a simple setup, high cell selectivity and capture efficacy can be achieved. scMOCA allows visual selection and isolation of any number of cells in a microscopy field and captured cells remain viable to generate new colonies of chosen phenotypes for downstream analyses.
Keywords: Cell sortingBackground
Wide-spread cell sorting techniques rely on the use of highly specific biochemical labels, such as antibodies and fusion proteins, which can be functionalized to allow FACS or MACS (Yan et al., 2009; Kuka and Ashwell, 2013). scMOCa represents a novel alternative which is advantageous when cell selection is based on visual traits such as cell migration (Kurosaka and Kashina, 2008), cell morphology (Prasad and Alizadeh, 2019), intracellular protein organization or cell-to-cell contacts (Garcia et al., 2018) which lack specific molecular markers. For example, cell movement allows embryonic and tumor development, wound healing and immune responses. Morphology reflects different stages of cell growth, differentiation and disease. Cell-to-cell contacts or distance to sources of chemical cues are involved in chemokinesis, differentiation, neural function, and immune responses. Finally, fluorescent protein fusions reveal interesting molecular behavior, such as protein relocalization within cells upon various stimuli. Using smMOCa, these cells can easily be isolated for their extensive characterization. Other approaches that allow the identification of cells without knowledge of a specific marker are based on photoactivated fluorescent proteins (Patterson and Lippincott-Schwartz, 2002; Lovatt et al., 2014). Once tagged, these cells may still need to be isolated, which can be done using cytometry approaches. Unfortunately, FACS or MACS cannot sort these cells since they are sensitive to the total signal intensity rather than its localization. Some rare label free techniques are based on size selection but have very limited applications (Khojah et al., 2017; Zhao et al., 2017). New technologies based on microfluidics and droplets encapsulation reach extremely low capture rates (less than 10%) (Salomon et al., 2019). A microfluidic chip can only capture a given number of cells that depends on the size of the chip (for instance 96 capture sites, or 384…) which is necessarily smaller than the total number of cells in the sample (which may reach millions). Because most cells are lost, chances to extract a cell population that is already rare in the original population is low.
Finally, some situations involve cells for which there is a marker, but that are too rare to be efficiently extracted with FACS or MACS (Leary, 2000). For instance, rare mutated metastatic or transfected cells. scMOCA is optimized for low cell numbers and as such is an ideal technique in these situations.
Our protocol allows manual selection of single cells one by one based on imaging, regardless of their biochemistry. A sample can be imaged or filmed with a microscope to identify fast migrating cells, or cells with different expression patterns (such as the localization of a 53bp1-GFP [Binan et al., 2019]). Chosen cells are then tagged by crosslinking biotin on their membrane using photobleaching: a laser is used to photobleach biotin-4-fluorescein in the close proximity of the membrane of the cell of interest. Upon photobleaching, a reactive free radical is created, which will bind to the cell membrane, hence biotinylating the cell of interest (Binan et al., 2016; Binan et al., 2019). Because this reaction does not depend on the surface chemistry of the cell, any cell type can be labeled. Using scMOCA, as few as one single cell can be extracted as shown in Figure 1 to generate highly pure samples.
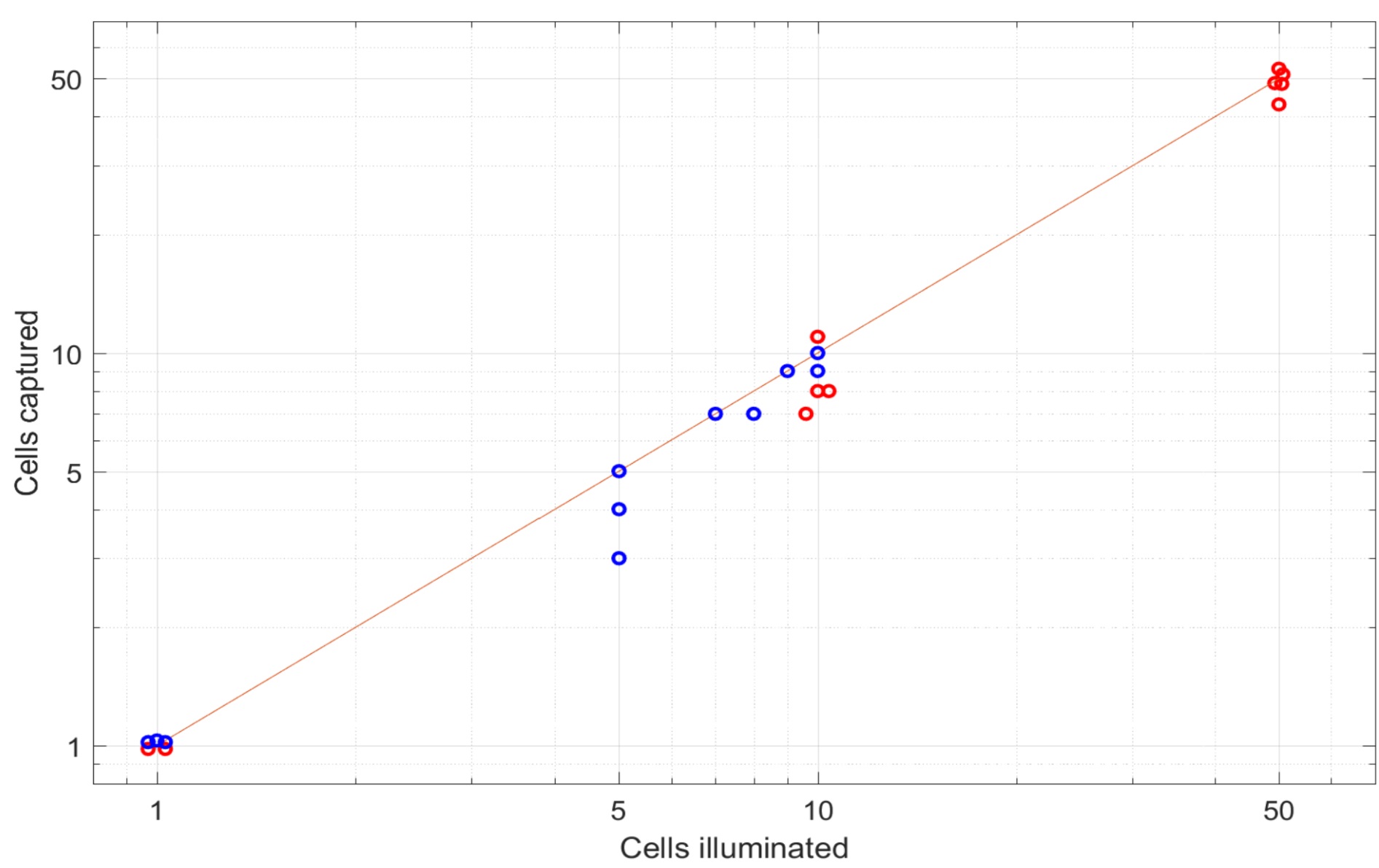
Figure 1. Number of cells captured as a function of the number of cells tagged. scMOCA allows high capture rates, even when low numbers of cells are tagged. Red dots represent experiments done on glass, blue dots represent experiments done on plastic substrates. (Originally published in Binan et al., [2019]. Creative Commons Attribution License).
Materials and Reagents
- N35 Neodymium Disc Magnet 3/8’x1/16” Rare Earth Disc Magnets, CMS magnetics, store away from hard drives and cell phones as it may damage them
- Nails from a hardware store. Nails should be made of iron (not steel) and have a 1 mm diameter head. This nail will channel magnetic field lines to the center of the collection chamber
- Coverslip N1 (VWR, catalog number: 4804-455 on US website)
- Pipette tips (10, 200 and 1,000 µl)
- 50 ml conical-bottom centrifuge tubes with Flat cap (VWR, catalog number: 89401-562 on US website)
- Petri dishes
- Disposable biopsy punches, 5 mm (IntegraTM miltex®, catalog number: 21909-142)
- Soft-Jet® 3-Part Dispo sable Syringes, Air-Tite (VWR, catalog number: 89215-238)
- Millex-GS Syringe Filter Unit, 0.22 µm (Sigma-Aldrich, catalog number: SLGSV255F)
- polystyrene (Sigma-Aldrich, catalog number: P5606-400EA)
- Biotin-4-fluorescein (b4f) (Sigma-Aldrich, catalog number: B9431-5 mg), store at 4 °C
- DynabeadsTM M270 streptavidin (Thermo Fisher scientific, catalog number: 65305), store at 4 °C
- Trypsin-EDTA (0.25%), phenol red (ThermoFisher scientific, catalog number: 25200114), store at -20 °C
- Polydimethyl siloxane (PDMS), Dow SYLGARDTM 184 Silicone Encapsulant Clear 0.5 kg Kit, Ellsworth adhesive, 184 SIL ELAST KIT 0.5KG, store at room temperature
- Gelatin from porcine skin (Sigma-Aldrich, catalog number: G2500-100G) or Poly-L-Ornithine Solution (0.01%) (Sigma-Aldrich, catalog number: A-004-C) or Collagen Type I, rat tail (Sigma-Aldrich, catalog number: 08-115)
- Phosphate Buffered Saline, purchased or homemade, pH 7.4
- Phosphate Buffered Saline (containing Ca2+ and Mg2+), purchased or homemade, pH 7.4
- Conditioned medium (see Recipes)
Equipment
- Lego bricks:
- 2x 4211428
- 8x 4211393
- 2x 4211406
- 1x 324526
- 2x 4210963
- 1x 4211394
- 2x 4211452
- Vacuum chamber
- Cell culture hood with UV light
- Confocal microscope
A confocal microscope (inverted), or a similar set-up in which visible lasers can be pointed at specific locations in a microscopy field of view.
Procedure
- Preparing culture chamber
Preparing small chambers takes 2 days, therefore cell culture can be started at the same time in 10 cm dishes. These cells will then be used to fill the chambers, and the sorting can be done 24 h later.- In a 50 ml tube, mix SYLGARD base and curing agent (1/10) as per manufacturer instructions (see number 13 in Materials and Reagents section) and vortex.
- Pour 10 ml of the solution in a 10 cm dish.
- Place the dish to degas overnight in a vacuum chamber.
- Wait for two days for the polymer to be well cured.
- Using a biopsy punch, cut a 5 mm diameter chamber in a block of polymer of approximately 10 mm x 10 mm. Because the polymer is soft, it is easier to first cut the 5 mm chamber, then make a cut around it to separate it from the block of polymer.
- Place chamber on coverslip and press it to generate good adhesion. PDMS naturally adheres to glass or plastic.
- On the day the cells are plated, coat the chamber substrate with a solution of choice for improved cell adhesion (gelatin, poly-ornithine or collagen). Usually, a 1/10 dilution of the coating agent in PBS containing Ca2+ and Mg2+ for an hour at 37 °C, followed by 3 rinses in PBS works well.
Note: We recommend testing different coating solutions for each new cell type used and selecting the one that provides the best outcome in terms of sample purity and sorting efficacy. When working with highly adherent cells, coating may not be necessary. Sensitive cells often require a collagen coating. Nevertheless, this option generates collagen fibers in which both negative and positive cells may be trapped after cell suspension, hence reducing the selectivity of the technique. For this reason, we found that gelatin and poly-ornithine provide good intermediate solutions in most cases. As an example, we used a gelatin coating for sorting embryonic stem cells, but a collagen coating for HUVECS and MDA-MB-231 and no coating at all for U2OS cells. - Place under UV in cell culture hood for sterilization.
- Building Chamber holder
Build chamber holder using Lego bricks. This structure will be used to maintain the collection chamber and the magnets at appropriate distances from the donor chamber.- We provide building instructions in Figure 2 and the setup is visible in Video 1.

Figure 2. Instructions to build the structure with Lego bricks (Originally published in Binan et al., [2019]. Creative Commons Attribution License) Video 1. Assembly and use of the setup
Video 1. Assembly and use of the setup - If the setup is built without Lego bricks, important parameters that must be kept constant are the distance between chambers (6 mm from the bottom of the donor chamber to the bottom of the collection chamber) and the ability to maintain 10 magnets close to a nail which head is centered in the collection chamber as shown Video 1.
- The coverslip with the collection chamber will be inserted just above the long bricks from Step 7 in Figure 2. The long transversal bars from Step 8 in Figure 2 will be used to pinch it in position.
- The double height, black brick in Step 3 in Figure 2 (number 16.d from the Material and Reagents list above) is hollow and two magnets are inserted inside. They allow holding the rest of the pile of magnets on the side of the brick, as visible in Figure 3 and Video 1.

Figure 3. Position of the chambers in the setup (Originally published in Binan et al., [2019]. Creative Commons Attribution License)
- We provide building instructions in Figure 2 and the setup is visible in Video 1.
- Conditioned medium preparation
When preparing chambers, the day prior to the experiment, keep medium from the culture and filter it through a 0.22 µm filter to remove debris (with syringe and syringe filters). This medium is rich in secreted factors and helps grow cells at very low density, improving sorted cell survival. - Laser alignment
In order to precisely tag single cells, it is key to locate the exact location at which the laser points. If the experiment is performed on a microscope that allows pointing at a given location on the image and to bleach cells, this step is not necessary and can be skipped. On the other hand, on home-made optical systems without this functionality, or if cells of interest are identified with bright field or epifluorescence imaging, precise recording of the laser position is required.- Prepare a fluorescent coverslip: place a drop of fluorescein (or b4f) stock solution between two coverslips and let it dry.
- Once dried, observe this fluorescent slide and adjust focus using an inverted microscope.
- Immobilize the blue laser from the confocal microscope and illuminate the slide for a few seconds.
- Without moving the slide/stage, observe again in fluorescence and record the position of the bleached spot (we used a simple pen mark on the computer screen, but one must then be careful not to move the position of the windows on the screen afterwards). This dark photobleached area is the precise location that will be illuminated in the sample.
- Laser-assisted membrane biotinylation and bead attachment
- Seed cells 24 h in advance in culture chambers prepared in Procedure A.
- On the day of the experiment, add biotin-4-fluorescein to medium to reach 0.04 mg/ml.
- Observe sample and choose the cells to capture. It is easier (but not necessary) to use the same objective to select cells of interest and to label them by photobleaching. We generally used a 10x, 0.4NA objective, unless the observation of the features of interest required higher magnification. In this particular case, a 60x can be used to identify cells that will be captured. Then the objective can be replaced by a 10x, 0.4NA to label the cells with the usual power settings. Using a relatively low NA increases the chances of successfully tagging the cells by reducing the importance of adjusting the focus for each cell of interest.
- Depending on the setup used, either point the laser to the cell of choice and illuminate for 2 s, or move the sample such that one of the chosen cells is positioned in the laser illumination mark obtained in Sorting Procedure D and open the shutter to irradiate the cell for 2 s. Laser power needs to be optimized depending on cell type and experimental setup, with ideal power varying from 30 µW to 400 µW using a 0.4NA objective. The ideal power is the lowest power that allows efficient labeling of the cells of interest. It is only dependent on the setup used and should not vary from a cell type to another. Repeat this step for each cell that needs to be captured.
- Rinse thoroughly in PBS.
The medium has to be well rinsed as remaining free biotin-4-fluorescein will block binding sites on the beads. It is key that the chamber never dries, which can happen very fast due to its extremely small total volume. In our hands, dipping the culture chamber in a PBS beaker several times provides excellent results whereas pipetting tends to detach some cells, and the tip may scratch the bottom of these small chambers. - Replace liquid in the chamber with medium containing 3 µl of beads washed in PBS as per manufacturer instructions, without further dilution.
- Use a magnet to pull beads down towards the cells.
When strongly adherent cells are used, the magnet can be used to pull beads across the whole area to ensure maximal attachment of the beads. When sensitive cells are manipulated, beads can be pulled down with the magnet, then resuspended either by pipetting the solution up and down a few times or by placing the magnets above the sample, then pulled back down with the magnets three times. The ideal strategy has to be determined for each different cell type and culture substrate as it is dependent on adhesion strength. - Rinse again in PBS. Three repeats of a 30 s immersion in a beaker of PBS is still the ideal solution here.
- Observe under a bright field microscope to verify that beads decorate the cells of choice. Expected result is shown in Figure 4.
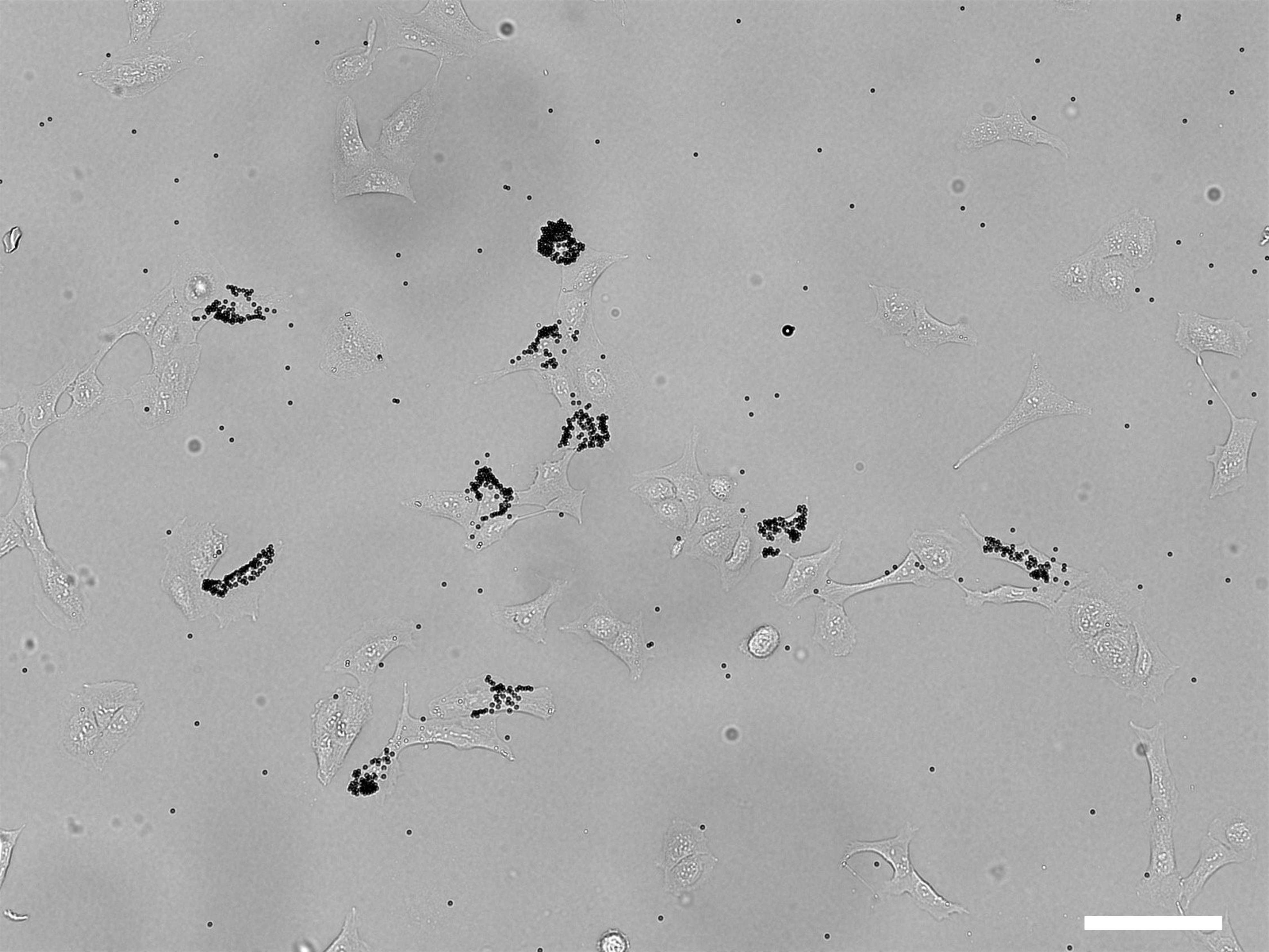
Figure 4. Bright field image of 10 tagged U2Os cells. Scale bar: 100 µm.
- Cell sorting
- Replace liquid with trypsin-EDTA (0.25%) in the donor chamber. Wait for the cells to become round and detach. Perform a few ups-and-downs with a pipette to carefully detach all cells.
- Place a collection chamber (an identical cell culture chamber, without cells) in the sorting setup, fill it with a drop of trypsin/PBS/medium (see details below) and place it above the donor chamber in a symmetric position, to bring both liquid drops to merge as shown in Figure 3 and Video 1. Chamber dimensions are designed to allow surface tension forces to maintain the drop of liquid inside the chamber without falling, even when the chamber is upside down.
- Wait 4 min for positive cells to gather in the collection chamber while negative cells settle in the donor chamber.
- Flip the setup, remove collection chamber and verify with a microscope that cells were captured.
- Gently pipette the solution up and down a few times to resuspend the cells and repeat the procedure up to 4 times to reach a pure sample. The number of repeats depends on how sticky the cells are, and on the density of the original cell culture. These sorting steps are depicted in Figure 5. When working with resistant cells, all repeats may be performed in trypsin. When working with more sensitive cells, they may be done in PBS. One must be careful to not let the cells re-adhere in any of the intermediate chambers by thoroughly pipetting the solution up-and-down to efficiently resuspend them between each step. For each intermediate sorting, the collection chamber should not be coated but rather made of bare glass to avoid undesired cell adhesion to the substrate. The final step should be performed in a coated chamber, filled with medium. Because collected cells usually end up being at very low density in the collection chamber, their viability may be reduced. To solve this issue, the medium used for the last sorting step, and for following medium changes in this chamber, should be conditioned medium (see Procedure C).
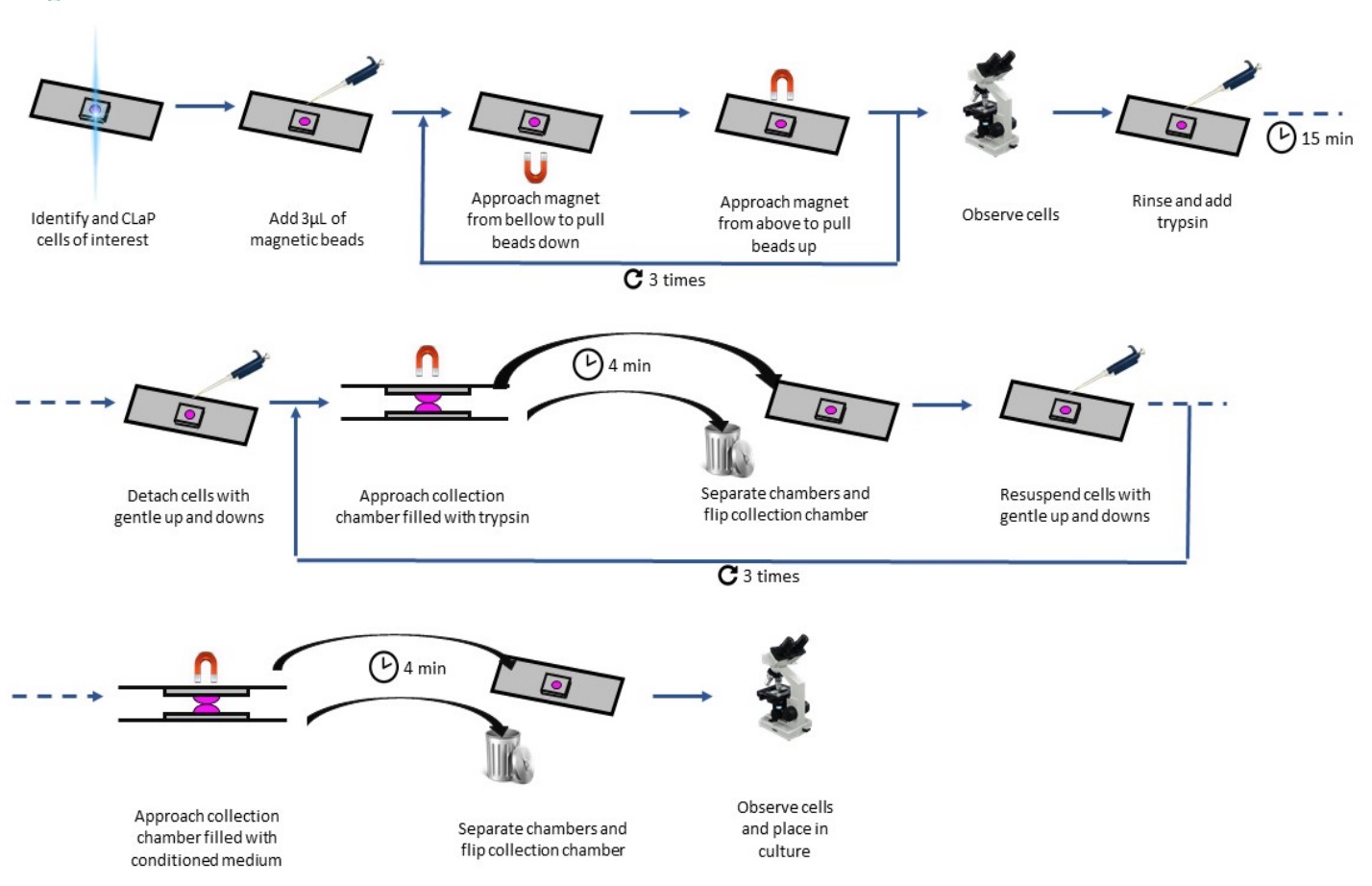
Figure 5. Detailed schematic of the sorting procedure (Originally published in Binan et al., [2019]. Creative Commons Attribution License)
Notes
- Before its first use on any setup and with any new biological system, the procedure’s parameters should be optimized. The adequate laser power that permits efficient cell labeling without killing has to be determined for each experiment and is highly dependent on the alignment of the laser with the microscope objective but should not vary with the cell type. The ideal laser power is the lowest power that allows efficient cell labeling on the setup used. We usually use around 50 µW.
- The appropriate surface preparation for the culture chambers depends on the cell type. Most sensitive cell types may require collagen coating, but this option makes following steps more difficult. When cells are more resistant, one may prefer to choose gelatin or poly-ornithine, or even bare glass when working with resistant cells.
- A frequent cause of failure is inefficient rinsing. The concentration of biotin-4-fluorescein (b4f) is extremely high and therefore the three conventional rinses in PBS are often not sufficient to remove all traces of free b4f. This is detrimental to the binding of magnetic beads to tagged cells because any free floating b4f will block binding sites. Observation of the sample after incubation with beads allows troubleshooting this issue: any beads remaining in the chamber should not be fluorescent in the fluorescein channel. Beads only become fluorescent if free b4f binds to streptavidin.
- Because losing as few as one cell may be very detrimental to this experiment, rinsing should be well optimized. In our hands, the best solution was obtained by immersing the culture chamber in large volumes (10 to 30 ml) of PBS.
- A few parameters are key to successful captures and should not be modified: the chamber must be 5 mm in diameter. Thus, surface tension prevents the liquid drop from falling when the chamber is upside down and reduces the distance from the edge to the center of the chamber, where the cells will be attracted by the magnets. The distance between the two chambers substrates should be kept at 6 mm, which is close enough for the magnets to efficiently pull up positive cells, but also far enough to maintain negative cells far from the collection chamber.
Recipes
- Conditioned medium
When passaging cells, the day prior to the experiment, keep old medium and filter it through 0.22 µm filter to remove debris (with syringe and syringe filters). This medium is rich in secreted factors and helps culture cells at very low density.
Acknowledgments
This work was supported by grants from the Natural Science and Engineering Research Council of Canada, Genome Canada/Génome Québec and Canadian Cancer Society, Fonds de Recherche du Québec-Nature et Technologies. S.C. holds salary awards from the Fonds de Recherche du Québec-Santé. This protocol is derived from the paper Opto-magnetic capture of individual cells based on their phenotype (Binan et al., 2019).
Competing interests
The authors declare no competing financial interests.
References
- Binan, L., Belanger, F., Uriarte, M., Lemay, J. F., Pelletier De Koninck, J. C., Roy, J., Affar, E. B., Drobetsky, E., Wurtele, H. and Costantino, S. (2019). Opto-magnetic capture of individual cells based on visual phenotypes. Elife 8. pii: e45239.
- Binan, L., Mazzaferri, J., Choquet, K., Lorenzo, L. E., Wang, Y. C., Affar, E. B., De Koninck, Y., Ragoussis, J., Kleinman, C. L. and Costantino, S. (2016). Live single-cell laser tag. Nat Commun 7: 11636.
- Garcia, M. A., Nelson, W. J. and Chavez, N. (2018). Cell-Cell Junctions Organize Structural and Signaling Networks. Cold Spring Harb Perspect Biol 10(4).
- Khojah, R., Stoutamore, R. and Di Carlo, D. (2017). Size-tunable microvortex capture of rare cells. Lab Chip 17(15): 2542-2549.
- Kuka, M. and Ashwell, J. D. (2013). A method for high purity sorting of rare cell subsets applied to TDC. J Immunol Methods 400-401: 111-116.
- Kurosaka, S. and Kashina, A. (2008). Cell biology of embryonic migration. Birth Defects Res C Embryo Today 84(2): 102-122.
- Leary, J. F. (2000). Rare-Event detection and sorting of rare cells. In Emerging Tools for Single‐Cell Analysis pp: 49-72.
- Lovatt, D., Ruble, B. K., Lee, J., Dueck, H., Kim, T. K., Fisher, S., Francis, C., Spaethling, J. M., Wolf, J. A., Grady, M. S., Ulyanova, A. V., Yeldell, S. B., Griepenburg, J. C., Buckley, P. T., Kim, J., Sul, J. Y., Dmochowski, I. J. and Eberwine, J. (2014). Transcriptome in vivo analysis (TIVA) of spatially defined single cells in live tissue. Nat Methods 11(2): 190-196.
- Patterson, G. H. and Lippincott-Schwartz, J. (2002). A photoactivatable GFP for selective photolabeling of proteins and cells. Science 297(5588): 1873-1877.
- Prasad, A. and Alizadeh, E. (2019). Cell form and function: Interpreting and controlling the shape of adherent cells. Trends Biotechnol 37(4): 347-357.
- Salomon, R., Kaczorowski, D., Valdes-Mora, F., Nordon, R. E., Neild, A., Farbehi, N., Bartonicek, N. and Gallego-Ortega, D. (2019). Droplet-based single cell RNAseq tools: a practical guide. Lab Chip 19(10): 1706-1727.
- Yan, H., Ding, C. G., Tian, P. X., Ge, G. Q., Jin, Z. K., Jia, L. N., Ding, X. M., Pan, X. M. and Xue, W. J. (2009). Magnetic cell sorting and flow cytometry sorting methods for the isolation and function analysis of mouse CD4+ CD25+ Treg cells. J Zhejiang Univ Sci B 10(12): 928-932.
- Zhao, W., Cheng, R., Lim, S. H., Miller, J. R., Zhang, W., Tang, W., Xie, J. and Mao, L. (2017). Biocompatible and label-free separation of cancer cells from cell culture lines from white blood cells in ferrofluids. Lab Chip 17(13): 2243-2255.
Article Information
Copyright
![]() Binan et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Binan et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Binan, L., Roy, J. and Costantino, S. (2019). Opto-magnetic Selection and Isolation of Single Cells. Bio-protocol 9(22): e3428. DOI: 10.21769/BioProtoc.3428.
- Binan, L., Belanger, F., Uriarte, M., Lemay, J. F., Pelletier De Koninck, J. C., Roy, J., Affar, E. B., Drobetsky, E., Wurtele, H. and Costantino, S. (2019). Opto-magnetic capture of individual cells based on visual phenotypes. Elife 8.
Category
Biophysics > Bioengineering > Cell capture technique
Cell Biology > Cell imaging > Live-cell imaging
Cell Biology > Cell isolation and culture > Cell isolation
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.