- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Super-resolution Microscopy at Cryogenic Temperatures Using Solid Immersion Lenses

Published: Vol 9, Iss 22, Nov 20, 2019 DOI: 10.21769/BioProtoc.3426 Views: 6966
Reviewed by: Gal HaimovichAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jul 2018
Advertisement





Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics








Abstract
Our mechanistic understanding of cell function depends on imaging biological processes in cells with molecular resolution. Super-resolution fluorescence microscopy plays a crucial role by reporting cellular ultrastructure with 20-30 nm resolution. However, this resolution is insufficient to image macro-molecular machinery at work. A path to improve resolution is to image under cryogenic conditions, which substantially increases the brightness of most fluorophores and preserves native ultrastructure much better than chemical fixatives. Cryogenic conditions are, however, underutilized because of the lack of compatible high numerical aperture (NA) objectives. Here we describe a protocol for the use of super-hemispherical solid immersion lenses (superSILs) to achieve super-resolution imaging at cryogenic temperatures with an effective NA of 2.17 and resolution of ~10 nm.
Keywords: Super-resolution microscopyBackground
Fluorescence microscopy has for many years been one of the most important tools for understanding how biological systems are organized and how they function. Over the last decade, fluorescence microscopy has been revolutionized by the development of “super-resolution” methods that extend the limit of optical microscopy beyond the diffraction limit, providing unprecedented levels of information on the organization of molecular networks in cells. These methods include Structured Illumination Microscopy (SIM) (Gustafsson, 2000), Stimulated Emission Depletion Microscopy (STED) (Hell and Wichmann, 1994), and techniques based on the localization of individual fluorescent molecules with much better precision than the diffraction limit. The latter includes Photo-activated Localization Microscopy (PALM) (Betzig et al., 2006) and Stochastic Optical Reconstruction Microscopy (STORM) (Rust et al., 2006), referred to collectively in this protocol as Single Molecule Localization Microscopy (SMLM) techniques. Because of a requirement to stop motion within the cell when aiming for high resolution, super-resolution methods usually involve the use of chemical fixation, which has the potential to introduce artefacts that can be visible at the resolutions achieved (Whelan and Bell, 2015). This problem has long been recognized in the field of electron microscopy, where a different method is now routinely used to fix cells and preserve their structure: vitrification at cryogenic temperatures. In this method, samples are frozen using a cryogen, cooling rapidly to temperatures of -150 °C or below, resulting in the formation of amorphous ice, with the high rate of cooling preventing the formation of ice crystals (Dubochet and McDowall, 1981). SMLM techniques in particular should benefit from the use of cryogenic conditions. This is because the localization precision of fluorescent molecules is dependent on the number of photons emitted by the fluorophores (Thompson et al., 2002), and at cryogenic temperatures significantly more photons can be collected. Conventional widefield fluorescence microscopy at cryogenic temperatures is already used in Correlative Light and Electron Microscopy (CLEM) (de Boer et al., 2015) workflows, but the mismatch in resolution between optical microscopy and EM limits the value of the fluorescence data, which is usually restricted to locating general areas of interest. Hence, enabling cryogenic super-resolution fluorescence microscopy would be a significant advance for cryogenic CLEM workflows.
Given the advantages of vitrification over chemical fixation, it would be advantageous to apply it to super-resolution optical microscopy methods. However, its use has been severely limited for one major reason: super-resolution methods require the use of high numerical aperture (NA) objective lenses (NA > 1), and achieving this NA typically requires the use of oil immersion objectives, incompatible with cryogenic microscopy. A number of approaches have been taken to avoid this problem, including using non-immersion objectives with extremely bright fluorophores (Liu et al., 2015) and using special cryogenic immersion fluids and custom sample stages (Nahmani et al., 2017). However, these approaches have not been generally applied to the imaging of biological samples.
We have developed a low-cost, generally applicable method for super-resolution cryo-fluorescence microscopy that uses commercially available optical components and cryo-stage (Wang et al., 2019). For this, we use super-hemispherical solid-immersion lenses (superSILs). These lenses are made of high refractive index materials, and take the form of truncated spheres. In our application, their role is to take the place of immersion oil, and fill the gap between the sample and a conventional non-immersion objective. The combination of superSIL and air objective gives a high numerical aperture, dependent on the refractive index of the lens material (Terris et al., 1994; Zhang et al., 2007; Chen et al., 2013).
We have characterized the performance of superSILs for cryo-SMLM, showing that we are able to achieve a point spread function (PSF) size of around 150 nm, compared with around 330 nm for a cryogenic microscope with a conventional low NA air objective. This is because the combination of superSIL and objective achieves a NA of 2.17, much higher than the maximum NA of ~1.4 of oil-immersion objectives. The superSIL microscope achieves single molecule localization precisions of < 10 nm, and a lateral resolution of 12 nm (Wang et al., 2019). A major advantage of the superSIL method is that it can be achieved using readily available off-the-shelf components, with the possibility of easily adding the cryostage to microscopes commonly found in cell biology research laboratories. Moreover, superSIL assemblies can be cleaned and reused multiple times, making them an inexpensive resource. In this protocol, we focus on use of the superSIL system for super-resolution microscopy of bacteria, but also indicate how it might be applied to imaging of mammalian cells. A similar protocol could also be used to image other samples, such as purified protein complexes or vesicles. Used as described, the protocol provides a straightforward method for super-resolution imaging of biological samples at cryogenic temperatures, with the advantage of avoiding potential fixation artefacts. The resolution and localization precision achieved are significantly better than those routinely obtained in conventional room temperature SMLM microscopes. These advantages alone should make superSIL SMLM microscopy a valuable addition to the toolbox of cell biology researchers.
It is also possible to consider other potential advantages of the superSIL microscopy protocol. Firstly, because of the ultra-high NA, the resolution that can be achieved even at room temperature is significantly better than that obtained in conventional SMLM microscopes. The superSIL protocol therefore provides a simple low-cost method by which a basic fluorescence microscope could be converted to a high-performance super-resolution system. Our protocol describes use of the superSIL in an upright microscope, but the method could also be used in an inverted microscope by using a modified version of the superSIL assembly.
We believe that superSIL microscopy could have an important role to play in CLEM, helping to bridge the resolution gap between cryo-fluorescence microscopy and EM. We do not describe any CLEM approaches in this protocol, but possible methodologies could involve the transferring and imaging of thin lamellae produced by focused ion beam (FIB) milling under both superSIL and transmission EM modes, or the correlation of superSIL SMLM images with serial block-face scanning EM. Given the small size of the lens assembly, it may also be possible to incorporate a superSIL into the stage of an electron microscope, potentially permitting simultaneous or near-simultaneous imaging of the same sample with both light and electrons.
Materials and Reagents
- superSIL assembly
- Hyper Hemispheric Ball lens, Cubic Zirconia, 0.89 mm diameter x 0.73 mm thickness, λ\4 flatness (Knight Optical, LBB2018-C)
- Platinum Foil (Goodfellow, catalog number: PT000264 876-861-73)
- Cryo-compatible adhesive (Loctite Stycast, 2850 FT)
- Preparation of bacterial and fiducial solutions
- Flat cap centrifuge tubes, 50 ml (Fisherbrand, catalog number: 05-539-13)
- Eppendorf tubes, 2 ml (Eppendorf, catalog number: 0030 123.344)
- Tetraspeck 0.1 μm blue/green/orange/dark red beads (Molecular probes, Thermo Fisher Scientific, catalog number: T7279)
- Agar plate(s) with colonies of E.Coli expressing plasmid of interest
- LB medium granular (e.g., Melford, L24400-5000.0)
- 1 M HCl solution in water for pH adjustment (made up from HCl 37% ACS grade, 320331, Sigma-Aldrich)
- Selection antibiotics stock solutions in water (e.g., Kanamycin 25 mg/ml from powder-Sigma-Aldrich, K1377; Carbenicillin 100 mg/ml ready-made solution, Sigma-Aldrich, C1613)
- IPTG 1 M stock solution in water (made up from powder e.g., Fluorochem, M02726)
- PBS 1x pH 7.4 (Gibco, catalog number: 10010-015)
- Ultrapure deionized water (MilliQ or equivalent)
- LB medium (see Recipes)
- Cleaning of superSIL assemblies
- Clean glass slides (e.g., Fischer Scientific 16 x 38 mm microscope slides 244879)
- Filter paper (e.g., qualitative filter paper grade A, SLS 2036)
- Clean glass scintillation vials (e.g., Thermo Chromacol 20 ml vials 20-EPSVCA)
- superSIL assemblies (produced as described in Procedure A), cubic zirconia crystal NA 2.17
- Sulphuric acid ≥ 95% pure (e.g., Fischer Scientific S/9231/PB15)
- 30% Hydrogen Peroxide with inhibitor, ACS grade (e.g., Sigma-Aldrich, catalog number: 216763)
- At least 100 ml of MilliQ (or equivalent) water
- Piranha Solution (see Recipes)
- Plunge-freezing
- Ethane gas bottle
- Cleaned and glow discharged superSILs (prepared as described in Procedure D)
- Bacterial suspension (prepared as described in Procedure B)
- Fiducial suspension (prepared as described in Procedure C)
- Liquid nitrogen (at least a full 4 L dewar)
- 70% ethanol
- MilliQ water
Equipment
- superSIL assembly and microscope
- Ceramic hot plate (VWR, catalog number: 444-0624)
- Coordinate measuring machine (OGP, SmartScope ZIP 250)
- Cerna Microscope Body with Epi-Illumination Arm (Thorlabs, catalog number: CEA1350)
- Nikon D-FL Epi-Fluorescence Illuminator (Thorlabs, catalog number: CSE1000)
- Microscope cube assembly for Nikon TE2000 (Thorlabs, catalog number: TLV-TE2000)
- Single objective arm (Thorlabs, catalog number: CSA1100)
- Objective focusing module (Thorlabs, catalog number: ZFM2020)
- 3-axis controller and knob box for 1" Cerna stages (Thorlabs, catalog number: MCM3001)
- TIRF module comprising of X/Y translator and right-angle mirror mount (Thorlabs, ST1XY-D and KCB1/M)
- Translation stage (Prior, H117P2IX/G)
- Translation stage controller (Prior, ProScan III H31XYZEF)
- Image splitting module (Cairn, Twincam)
The following components are used in the microscope schematic illustrated in Figure 1. In principle, most popular commercial microscope stands could be modified to create a similar configuration since various adapter plates for the cryo-stage are available from Linkam Scientific Ltd and the Cairn Twincam can be purchased with c-mount threads on the input and output ports to suit most types of scientific camera.
Note: Check the emission spectra of your fluorophore or dye of choice at cryogenic temperature and select dichroic and emission filters to suit. The chromatic aberration between channels can be compensated for by changing the strength of lens l6 and axial translation of sCMOS camera 2.- l1: f = 30 mm achromatic doublet lens (Thorlabs, catalog number: AC254-030-A-ML)
- l2: f = 150 mm achromatic doublet lens (Thorlabs, catalog number: AC254-150-A-ML)
- l3: f = 100 mm achromatic doublet lens (Thorlabs, catalog number: AC254-100-A-ML)
- l4: f = 150 mm achromatic doublet lens (Thorlabs, catalog number: AC254-150-A-ML)
- l5: f = 200 mm 1x camera tube lens (Thorlabs, catalog number: WFA4100)
- l6: f = 500 mm B-BK7 plano-convex lens (Thorlabs, catalog number: LA1908)
- l7: 50x, 0.75 NA Microscope objective lens (Mitutoyo, M Plan Apo HR 378-814-4)
- p1: 30 μm pinhole (Thorlabs, P30D)
- t1: Top-hat beam shaping optic (TOPAG Lasertechnik GmbH, GTH-5-250-4-VIS)
- d1: Dichroic beam splitter (Semrock, Di-R405/488/561/635)
- d2: Dichroic beam splitter (Chroma, t565lpxr)
- f1: Quad-band emission filter (Semrock, FF01-446/523/600/677-25)
- f2: Green channel band-pass filter (Semrock, catalog number: FF03-525/50-25)
- f3: Red channel band-pass filter (Semrock, catalog number: FF01-593/40-25)
- q1: Achromatic quarter wave-plate (Thorlabs, AHWP10M-600)
- m1: Broadband dielectric mirror (Thorlabs, BB1-E02)
- Omicron LightHUB-6 containing four laser modules
- 405 nm 60 mW Phoxx diode laser
- 488 nm 200 mW Phoxx diode laser
- 561 nm 150 mW Cobolt Jive DPSS laser
- 642 nm 140 mW Phoxx diode laser
- Two back-illuminated sCMOS cameras with 18.7 mm chip sensor size (Teledyne Photometrics Ltd, 18.7 mm Prime 95B)
- Cryo-stage with autofill Dewar (Linkam, CMS196M and AUTOFILL)
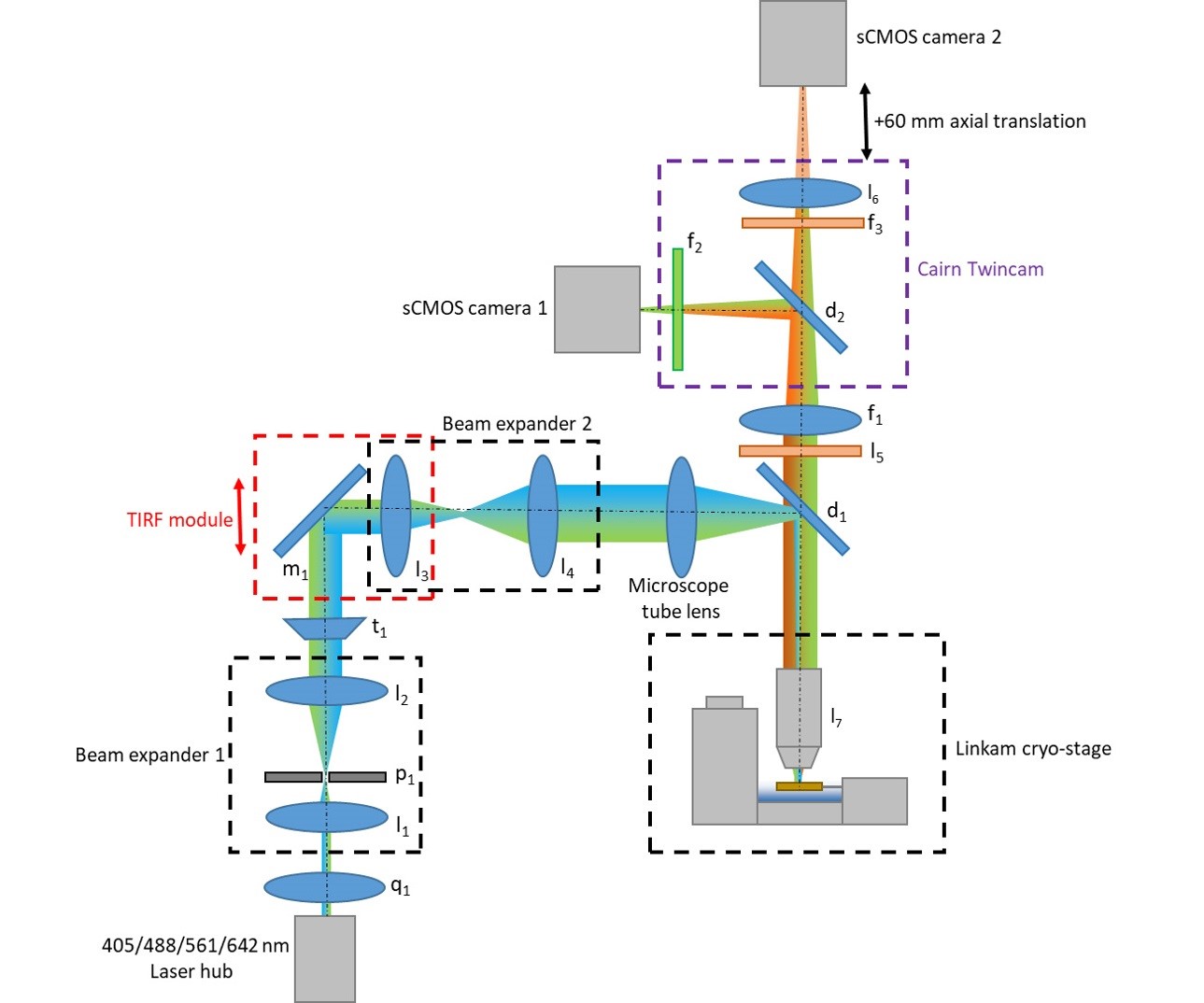
Figure 1. Microscope schematic detailing optics layout
- Preparation of bacterial and fiducial solutions
- Swinging bucket centrifuge (e.g., Beckman Coulter Allegra X-15R centrifuge)
- P1000, P100 and P10 pipettes and tips
- Autoclave (e.g., Prestige Medical Classic tabletop autoclave)
- Spectrophotometer (e.g., Amersham Biosciences Ultrospec 10 cell density meter)
- MSC cabinet (e.g., Scanlaf Mars Safety Class 2)
- Temperature-controlled, agitating incubator (e.g., New Brunswick Scientific Innova 44R)
- Vortex (e.g., IKA MS2 minishaker)
- Immersion probe pH meter (e.g., Hanna PH21)
- Cleaning of superSIL assemblies
- Chemical fume hood
- P1000 pipette and tips
- Glow discharger (e.g., Quorum GloQube)
- Inverted tweezers (e.g., Dumont N1)
- Timer
- Plunge-freezing
- FEI Vitrobot or equivalent plunge-freezer
- Flammable gas regulator (e.g., Air Liquide HBD-240-4-2.S)
- Gas leak detector (e.g., Everbuild Plumber’s P18 Gas Leak Detector)
- Inverted tweezers (e.g., Dumont N1)
- Curved tweezers (e.g., Dumont 7, 0130-7-PO)
- Blotting paper (e.g., Agar round filter paper for Vitrobot, 47000-100)
- P10 pipette and tips
- 1 L Thermos flask
- Funnel
- Tissue paper
- Grid boxes (e.g., SwissCI cryo-EM grid box MD16-104, Molecular Dimensions Limited)
- A 50 ml centrifuge tube attached to a piece of synthetic, non-absorbent twine
- Transport cryodewar (e.g., Statebourne Cryogenics OD-1)
- Handheld liquid nitrogen dewar (e.g., Worthington LD4)
- Storage cryodewar (e.g., Worthington VHC35)
- Hot plate or hair dryer
- Adequate personal protective equipment (PPE) for work with cryogenics (refer to local health and safety regulations at your facility)
- Optional: PFTE thread seal tape (e.g., Buffalo 12 m x 12 mm x 0.075 mm BS7786: 2006 Grade L)
- Optional: Local Exhaust Ventilation (LEV) inlet
- Optional: orientable tabletop lamp
- FEI Vitrobot or equivalent humidity controller
Software
- Micro-Manager v1.4.23 20161114 (Edelstein et al., 2014)
- Omicron Control Centre v2.1.4
- ImageJ version v1.52h (Schindelin et al., 2015)
- ThunderSTORM v1.3-2014-11-08 plugin (Ovesný et al., 2014)
Procedure
- Manufacturing superSIL assemblies
Note: This step assumes the use of a mechanical workshop equipped with a laser cutter and drying oven.
Figure 2. superSIL assembly manufacturing process. A. A 3 mm diameter platinum disc with a 1 mm diameter hole laser-cut in the center. B. the assembled disc with superSIL glued in place. Scale bar: 1 mm.- Cut a 3 mm diameter circle from the platinum foil using the laser cutter. Drill a 1 mm diameter hole in the center (as shown in Figure 2A).
- Glue the superSIL into the hole using the cryo-compatible adhesive (as shown in Figure 2B).
- Cure the assembly in the oven for 24 h at 45 °C.
- Measure the angle between the flat surface of the superSIL and the flat surface of the platinum disk using the coordinate measuring machine. A flatness of < 1° is essential to minimize imaging aberrations.
- Preparation of bacterial suspensions
Note: Preserve sterility of your cultures by performing all operations until Step B6 under an MSC or next to a Bunsen burner.
The procedure assumes that the selected bacterial strain is expressing a suitable fluorescent protein for SMLM. We have imaged successfully using Enhanced GFP (EGFP) (Wang et al., 2019) and there have been reports of cryo-SMLM using monomeric EGFP and mVenus (Kaufmann et al., 2014).- Inoculate one colony of the bacterial strain of interest in 10 ml of LB agar.
- Add selecting antibiotics depending on selection markers present on plasmid of interest.
- Grow at 37 °C 220 rpm for 3 h, then check O.D. 660 on a spectrophotometer, using LB medium (Recipe 1) as a blank.
- If cultures have reached O.D. ~0.6, induce with IPTG 0.2-1 mM (exact amounts will have to be optimized in a case-by-case basis) and culture for 18 h at 25 °C.
- Keeping the culture sterile, take 2 ml of culture and transfer them to a 2 ml Eppendorf.
- Centrifuge at 3,000 x g 10 min, discard supernatant.
- Add 2 ml PBS, resuspend and centrifuge at 3,000 x g 10 min, then discard supernatant.
- Resuspend in 100 μl PBS 1x, keep on ice.
- Preparation of fiducial suspensions
- Vortex Tetraspeck vial at maximum setting for at least 10 s.
- Dilute Tetraspeck 1:10 in MilliQ (or equivalent purity) water.
- Vortex diluted bead suspension at maximum setting for at least 10 s.
- Keep on ice.
- Cleaning of superSIL assemblies
This procedure should be used to clean superSILs after each use, before applying a new sample. This makes superSIL assemblies re-usable for a few cycles. Cleaning degrades the glue that attaches the lens to the platinum disk. When the superSIL becomes loose it can be re-attached as described in Procedure A.
Important safety advice- Piranha solution is highly acidic, oxidizing and exothermic. Do not make more than you need, do not cap the vial where you are making it, and do not store. Always use appropriate PPE. Prepare and use Piranha exclusively under a fume hood.
- DO NOT mix with organic solvents as this is likely to cause an explosion.
- Do not use hydrogen peroxide at concentrations higher than 30% or this may cause an explosion.
- After use, dilute leftovers with plenty of MilliQ water and leave it to outgas and degrade in a fume hood overnight, and then dispose down the drain with plenty of cold water.
- This procedure is visually demonstrated in Video 1.
 Video 1. SuperSIL cleaning process
Video 1. SuperSIL cleaning process
- Prepare Piranha solution (Recipe 2) as described in Recipes.
- Spot ~100 μl of Piranha solution of a clean glass slide. Prepare a spot for each superSIL to be cleaned.
- Using a pair of inverted tweezers, place each superSIL on a spot of Piranha solution, flat face down.
- Incubate for 7 min at RT.
- Using a pair of inverted tweezers, rinse each superSIL thoroughly by dunking it in a vial of MilliQ water and blotting it on clean filter paper. Repeat the dunking and blotting at least three times.
- Repeat Step D5 once more using a clean vial of MilliQ water.
- Leave superSILs to dry on a piece of clean filter paper at least 1 h.
- Glow discharge superSILs flat face up for 120 s at 40 mA in a glow discharger.
- Plunge-freezing
Important safety notice- This step of the protocol entails works with cryogenics and with flammable gases, which can cause cryogenic burns, explosions and suffocation by oxygen displacement. Always wear appropriate PPE and make sure you are working in an area with adequate ventilation and oxygen depletion alarms. Always refer to the health and safety guidance on working with cryogenics and with flammable gases available at your facility. Obtain adequate training and/or supervision before commencing work.
- Procedures will be described assuming the use of a Vitrobot plunge freezer (FEI). Please refer to manufacturers’ instructions for how to translate these for use with equivalent equipment from other manufacturers.

Figure 3. The cryo-bath of the Vitrobot plunge-freezer
- Turn on the Vitrobot at the main switch and install the humidity controller.
- Set humidity at 70% and temperature at 22 °C. Wait until they equilibrate (usually 25-30 min).
- In the options menu, set blot number to zero and allow the use of footpedal.
Note: Automated blotting needs to be disabled to preserve the structural integrity of the superSILs. - Fill the outer chamber of the cryo-bath with approximately 1 L of liquid nitrogen. Make sure the “spider” is in place (as shown in Figure 3).
Note: Do not fill straight from the handheld dewar, rather use it to fill a smaller thermos flask. Use the funnel and tissue paper to filter out macroscopic ice crystals that might contaminate your sample. - Wait 10-15 min for the inner chamber to cool down and place grid boxes in the dedicated slots (max 4 boxes, so max 16 samples at any one time).
- Fit the regulator on the ethane bottle and check for leaks using the leak detector spray.
Note: If leaks are detected, close all valves and restart. Use PVDF tape to wrap regulator thread if necessary. Abide by local safety regulations regarding the use of flammable gases. Video 2. SuperSIL plunge freezing process - Slowly dispense the ethane into the inner cup and wait until it is almost full to remove the spider. Ethane should be in liquid state for plunge freezing. If necessary, melt solidified ethane by inserting a warm metal instrument into the ethane inner cup.
Notes:- Orienting a local exhaust ventilation (LEV) head close to the cryo-bath is quite helpful in clearing cold ethane vapor and gauging levels more accurately.
- he spider needs to be removed for the following passages. Failing to do so might cause significant damage to the machine.
- Take up a superSIL with the Vitrobot-compatible tweezers, making sure that the flat face is facing the side of your dominant/favorite hand and load it on the shaft.
Note: Make sure the tweezers are not proud of the shaft on either side as this can cause serious damage to the machine as the shaft is retracted. - Press pedal to retract tweezers into waiting position.
- Place cryo-bath on the pedestal and press pedal to lift.
- Once cryo-bath has finished moving, press pedal to lower the tweezers into sample loading position.
- Apply 2.5 μl of bacterial suspension to the flat side of the superSIL through the side port.
- Open the front door and blot away most liquid using a piece of filter paper.
- Repeat sample application and blotting.
- Apply 2.5 μl of fiducial solution to the flat side of the superSIL through the side port.
- Repeat Step E13.
- Press pedal to start plunging sequence.
- Once the shaft and the cryo-bath have reached the end of the course, top up liquid nitrogen in the outer chamber, unload the tweezers from the shaft and move the superSIL from the ethane cup into the nitrogen bath.
Notes:- Extra light from an orientable tabletop lamp is quite helpful at this stage.
- Take the tweezers out of the ethane slowly to prevent liquid ethane from sticking to the superSIL surface, then transfer quickly in liquid nitrogen.
- Carefully release the catch on the tweezers and depose the superSIL in one of the slots of the grid box.
- Warm and dry all instruments using the heated plate or the hair dryer.
Note: After each sample, wash the Vitrobot tweezers with 70% ethanol and let dry to prevent cross-contamination of the superSILs. - Once all samples have been plunged, fill the transport dewar with liquid nitrogen and allow to settle.
- Cool down the tube on a string inside the transport dewar.
- Using cold tweezers, quickly transfer the grid boxes from the outer nitrogen bath of the Vitrobot cryo-bath into the tube.
- Store samples in a storage dewar filled with liquid nitrogen until ready to image (and no longer than a month).
- Sample transfer to the cryo-stage

Figure 4. Linkam cryo-stage layout with lid removed- Switch on the Linkam CMS196M cryo-stage and perform a bake-out cycle.
- Fill the Linkam Autofill Dewar with liquid nitrogen and couple to the CMS196M stage. Initiate cooling of the cryo-stage.
- Once the temperature inside the cryo-chamber has equilibrated to -196 °C, insert the Linkam sample transfer puck into one of the empty receptacles inside the cryo-chamber and allow to cool. Insert a blank sample cassette (as shown in Figure 4).
- Transfer a storage puck containing your superSIL samples into the remaining receptacle inside the cryo-chamber.
- Transfer the superSIL assemblies from the storage puck to the sample cassette using tweezers, paying close attention to the orientation; the curved surface of the superSIL needs to point towards the microscope objective once mounted.
- Once the superSILs have been mounted, close the cassette and transfer it to the brass bridge using the magnetic transfer tool. Remove the storage and transfer pucks from the cryo-chamber to minimize vibration during imaging.
- Finding focus
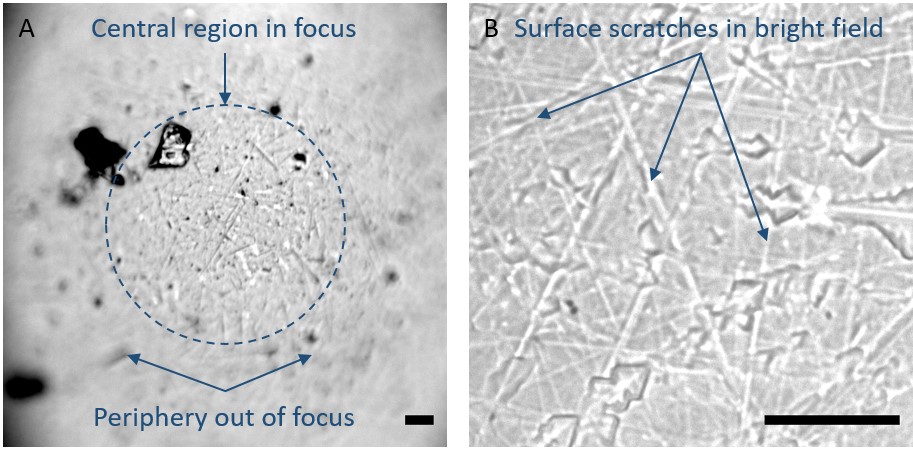
Figure 5. Finding focus under bright field illumination. The top of the superSIL curved surface is shown focused in (A), viewed under the 50x, 0.75 NA objective lens. Viewing through the superSIL, characteristic scratches on the flat surface should become visible under bright field illumination, shown in (B), indicating that the sample plane is in focus. Scale bar: 10 µm.- Start Micro-Manager and bring up a live view of the camera(s).
- Switch on the bright field condenser LED built-in to the Linkam CMS196M.
- Lower the microscope objective in to the Linkam chamber towards the superSIL. Monitor the green channel camera output. Pay close attention to the distance between the objective and superSIL; a collision will likely destroy the Linkam brass bridge leading to large repair costs and delays.
Note: Light from the built-in LED condenser is unlikely to appear in the red channel. - As the objective approaches the superSIL, you will notice the brightness of the green channel increase. Adjust the microscope stage laterally to ensure the brightest area remains in the center of the camera’s field of view as you lower the objective further.
- The curved surface of the superSIL will appear in focus. Translate the microscope stage laterally so the very top of the curved surface is in the center of the camera’s field of view (Figure 5A) and the periphery blurs out of focus.
- Now lower the objective approximately 1 mm to find the virtual imaging plane that is located below the flat surface of the superSIL. You will notice inherent scratches in the flat surface of the superSIL appearing into focus first (Figure 5B). If the ice that has been produced in the plunge-freezing procedure is relatively thin, the plane at which the sample is located should be very close to this flat surface of the superSIL.
- Imaging
Note: You have the option to store the collected images in memory or to stream them straight to hard disk. sCMOS cameras produce large quantities of data so make sure you have enough available space before continuing otherwise the system may become unresponsive and crash. A typical dual-color STORM experiment comprising of 10,000 full-frames per channel consumes approximately 10 GB of data. Computer memory equal to or greater than 32 GB and a solid-state hard drive of capacity equal to or greater than 1 TB is recommended as a minimum requirement.- Take an optional bright field image snapshot. Switch off the LED condenser.
- Switch on the required lasers once they have warmed up sufficiently.
- Open the laser shutter on the microscope to illuminate your sample.
- Fine-tune the objective height until the fiducial markers are in focus.
- Optional: Adjust the micrometer on the TIRF module to increase the contrast from your sample.
- Start with a low laser power density (~50 W/cm2). Wait for the sample to photo-bleach sufficiently until you see clear well-separated single molecules blinking. Increase the laser power density if necessary until you observe the desired blinking characteristics for your fluorophore of choice. The time that the molecule spends in its emitting bright state should be as low as possible compared to the time spent in the dark state.
- Adjust the camera exposure time and acquire an appropriate number of frames for your experiment based on your fluorophore of choice and sample conditions. For E. Coli containing eGFP, we chose to acquire 10,000 frames with a 50 ms exposure time.
- Save the acquired frames as 16-bit unsigned integer TIF stacks.
- Reverse the “Finding Focus” steps to safely remove your samples from the cryo-microscope stage.
Data analysis
- Single molecule localization
- Open ImageJ and load a TIF stack of interest.
- Run the ThunderSTORM plugin. Verify your camera settings are correct under “Camera setup” and you have entered appropriate fitting and sub-pixel localization settings based on your theoretical point spread function size (Figure 6).
- Select a desired method for visualizing the reconstructed image.
- Click “Preview” to see how the analysis handles the current frame of data and verify that it detects all of the visible single molecules.
- Click “OK” to run the analysis for the entire dataset.
- Once the analysis has completed, you will notice a table of results appears on the screen alongside a reconstructed image. Save the table of results as a .csv file using the “Export” feature as this can be used to re-generate the reconstructed image at a later time.
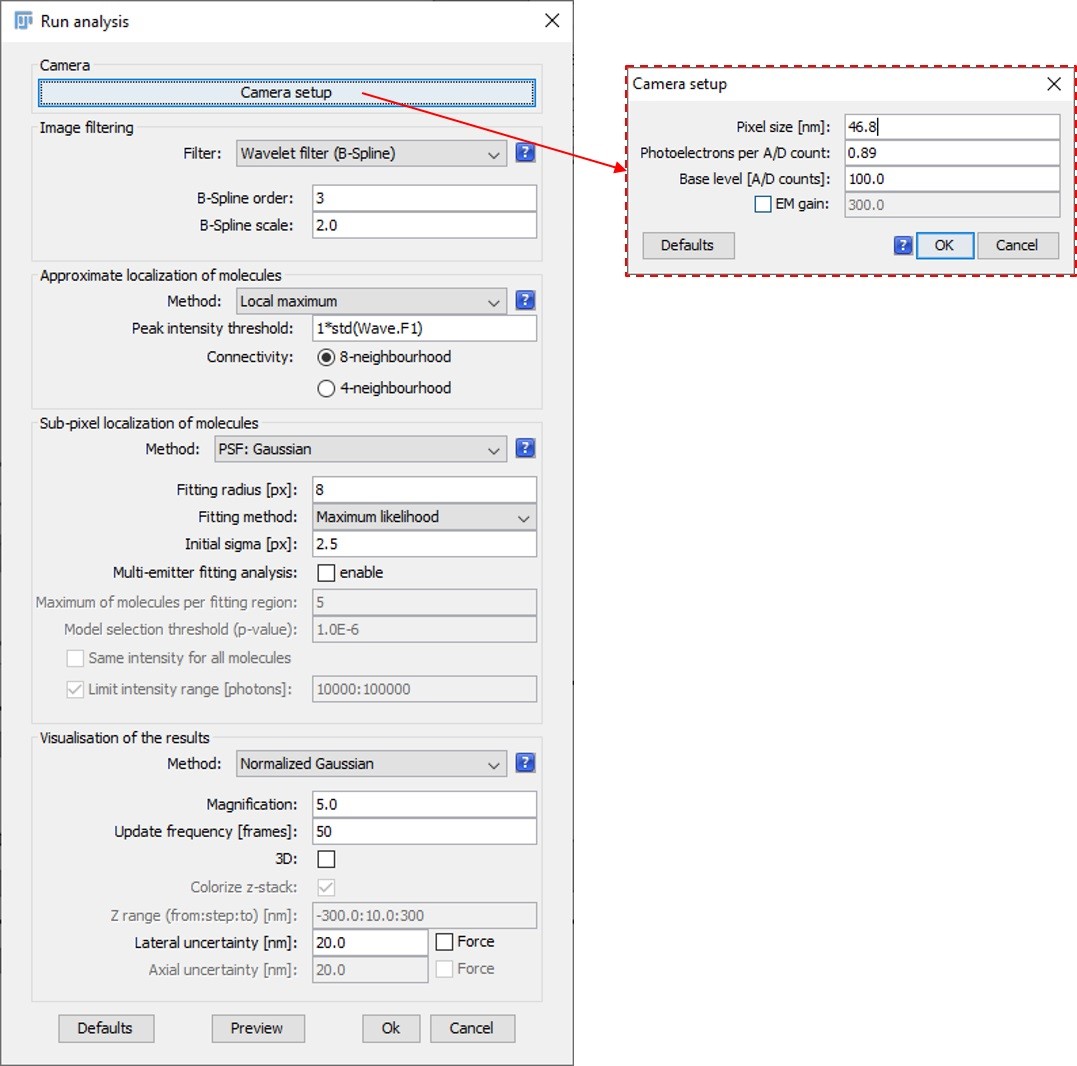
Figure 6. Typical ThunderSTORM settings based on the effective point spread function and pixel size at the imaging plane of the superSIL
- Drift correction
- You can apply drift correction to your dataset if fiducial markers are present and in focus. Click the “Drift Correction” tab and load the sub-menu by clicking on the double arrow icon. We have found that setting a maximum distance of 500 nm, minimum marker ratio of 0.9 and trajectory smoothing factor of 0.001 under the drift correction options produce satisfactory results given the nature of the drift in our particular cryo-stage.
- Once the drift correction has been calculated and applied, a new reconstruction appears reflecting the changes. Zoom in to inspect the fiducial markers using the magnifier tool from the ImageJ toolbar; if there is still a visible drift then you may need to adjust the variables discussed in the previous step.
- Save the revised results table as a .csv file using the “Export” function.
- Filtering
Note: The fitting algorithms within ThunderSTORM can be used to remove unwanted localization artifacts arising from sources such as ice crystals, aggregated fiducials, sample auto-fluorescence or scatter.- On the “Filter” tab, you can define your desired filters using Boolean algebra expressions.
- For example, if your system has a theoretical point spread function sigma of 125 nm, you could choose to include only those localizations with a sigma in the range 100-150 nm with the expression “sigma > 100 & sigma < 150”.
- Your sample of interest may have an intensity range of 100-1000 ADU counts with the fiducial markers having an intensity over 6000 ADU counts. In this scenario, you can exclude the fiducial markers by using expression “intensity < 6000”.
- Visualization and measurements
- It is possible to apply color look up tables (LUTs) to your reconstructed image by selecting “File > Import > LUT…”.
- The pixel size needs to be correctly defined in order to make measurements in your reconstructed image. This can be verified by selecting the reconstructed image window and navigating to “Image > Properties”. The pixel size should be set to that of the camera divided by the final magnification used in the visualization settings of ThunderSTORM. For instance, if you have a pixel size of 46.8 nm and chose to have a 5x final magnification, then your reconstructed pixel size is 9.36 nm.
- You can now measure approximate distances between features using the ruler tool located on the main ImageJ toolbar.
- Advanced data analysis
The .csv data table generated by ThunderSTORM can be imported into other software packages for advanced data analysis, including single-molecule clustering, stoichiometry and co-localization between channels.
Recipes
- LB medium
- Weigh out 25 g of granules
- Add 1 L of MilliQ water
- Adjust pH to 7.2 using HCl
- Autoclave to sterilize
- Piranha Solution
- Add 900 μl of concentrated sulphuric acid to a glass scintillation vial
- Add 300 μl of 30% hydrogen peroxide to the vial and quickly mix
Note: The vial will heat up considerably. - Use immediately. Do not cap the vial
- Piranha solution is highly acidic, oxidizing and exothermic. Do not make more than you need, do not cap the vial where you are making it, and do not store. Always use appropriate PPE. Prepare and use Piranha exclusively under a fume hood.
- Do not mix with organic solvents as this is likely to cause an explosion.
- Do not use hydrogen peroxide at concentrations higher than 30% or this may cause an explosion.
- After use, dilute leftovers with plenty of MilliQ water and leave it to outgas and degrade in a fume hood overnight, and then dispose down the drain with plenty of cold water.
- This recipe makes enough Piranha solution to clean 20+ superSILs with the method described above. Note that the hazard associated with preparation of Piranha solution increases if larger volumes are made. For volumes larger than 5 ml, the solution should always be prepared on ice to minimize heating.
Acknowledgments
This work has been funded by Medical Research Council grant MR/K015591/1 to MMF. The protocol is based on work published in Wang et al. (2019).
Competing interests
The authors declare no competing interests.
References
- Betzig, E., Patterson, G. H., Sougrat, R., Lindwasser, O. W., Olenych, S., Bonifacino, J. S., Davidson, M. W., Lippincott-Schwartz, J. and Hess, H. F. (2006). Imaging intracellular fluorescent proteins at nanometer resolution. Science 313(5793): 1642-1645.
- Chen, R., Agarwal, K., Sheppard, C. J., Phang, J. C. and Chen, X. (2013). A complete and computationally efficient numerical model of aplanatic solid immersion lens scanning microscope. Opt Express 21(12): 14316-14330.
- de Boer, P., Hoogenboom, J. P. and Giepmans, B. N. (2015). Correlated light and electron microscopy: ultrastructure lights up! Nat Methods 12(6): 503-513.
- Dubochet, J. and McDowall, A. W. (1981). Vitrification of pure water for electron-microscopy. J Microsc-Oxford 124(3): Rp3-Rp4.
- Edelstein, A. D., Tsuchida, M. A., Amodaj, N., Pinkard, H., Vale, R. D. and Stuurman, N. (2014). Advanced methods of microscope control using muManager software. J Biol Methods 1(2).
- Gustafsson, M. G. (2000). Surpassing the lateral resolution limit by a factor of two using structured illumination microscopy. J Microsc 198(Pt 2): 82-87.
- Hell, S. W. and Wichmann, J. (1994). Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy. Opt Lett 19(11): 780-782.
- Kaufmann, R., Schellenberger, P., Seiradake, E., Dobbie, I. M., Jones, E. Y., Davis, I., Hagen, C. and Grünewald, K. (2014). Super-resolution microscopy using standard fluorescent proteins in intact cells under cryo-conditions. Nano Lett 14(7): 4171-4175.
- Liu, B., Xue, Y., Zhao, W., Chen, Y., Fan, C., Gu, L., Zhang, Y., Zhang, X., Sun, L., Huang, X., Ding, W., Sun, F., Ji, W. and Xu, T. (2015). Three-dimensional super-resolution protein localization correlated with vitrified cellular context. Sci Rep 5: 13017.
- Nahmani, M., Lanahan, C., DeRosier, D. and Turrigiano, G. G. (2017). High-numerical-aperture cryogenic light microscopy for increased precision of superresolution reconstructions. Proc Natl Acad Sci U S A 114(15): 3832-3836.
- Ovesný, M., Křížek, P., Borkovec, J., Svindrych, Z. and Hagen, G. M. (2014). ThunderSTORM: a comprehensive ImageJ plug-in for PALM and STORM data analysis and super-resolution imaging. Bioinformatics 30(16): 2389-2390.
- Rust, M. J., Bates, M. and Zhuang, X. (2006). Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat Methods 3(10): 793-795.
- Schindelin, J., Rueden, C. T., Hiner, M. C. and Eliceiri, K. W. (2015). The ImageJ ecosystem: An open platform for biomedical image analysis. Mol Reprod Dev 82(7-8): 518-529.
- Terris, B. D., Mamin, H. J. and Rugar, D. (1994). Near-field optical data storage using a solid immersion lens. Appl Phys Lett 65(4): 388-390.
- Thompson, R. E., Larson, D. R. and Webb, W. W. (2002). Precise nanometer localization analysis for individual fluorescent probes. Biophys J 82(5): 2775-2783.
- Wang, L., Bateman, B., Zanetti-Domingues, L. C., Moores, A. N., Astbury, S., Spindloe, C., Darrow, M. C., Romano, M., Needham, S. R., Beis, K., Rolfe, D. J., Clarke, D. T. and Martin-Fernandez, M. L. (2019). Solid immersion microscopy images cells under cryogenic conditions with 12 nm resolution. Commun Biol 2: 74.
- Whelan, D. R. and Bell, T. D. (2015). Image artifacts in single molecule localization microscopy: why optimization of sample preparation protocols matters. Sci Rep 5: 7924.
- Zhang, J., See, C. W. and Somekh, M. G. (2007). Imaging performance of widefield solid immersion lens microscopy. Appl Opt 46(20): 4202-4208.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Bateman, B. C., Zanetti-Domingues, L. C., Moores, A. N., Needham, S. R., Rolfe, D. J., Wang, L., Clarke, D. T. and Martin-Fernandez, M. L. (2019). Super-resolution Microscopy at Cryogenic Temperatures Using Solid Immersion Lenses. Bio-protocol 9(22): e3426. DOI: 10.21769/BioProtoc.3426.
Category
Biophysics > Microscopy > Cryogenic microscopy
Cell Biology > Cell imaging > Super resolution imaging
Cell Biology > Cell imaging > Fluorescence
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.