- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

High Throughput Traction Force Microscopy for Multicellular Islands on Combinatorial Microarrays
Published: Vol 9, Iss 21, Nov 5, 2019 DOI: 10.21769/BioProtoc.3418 Views: 6982
Reviewed by: Chiara AmbrogioPia GiovannelliLili Wang
Original research article
The authors used this protocol in:

Dec 2018


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
The composition and mechanical properties of the cellular microenvironment along with the resulting distribution of cellular devolved forces can affect cellular function and behavior. Traction Force Microscopy (TFM) provides a method to measure the forces applied to a surface by adherent cells. Numerous TFM systems have been described in literature. Broadly, these involve culturing cells on a flexible substrate with embedded fluorescent markers which are imaged before and after relaxion of cell forces. From these images, a displacement field is calculated, and from the displacement field, a traction field. Here we describe a TFM system using polyacrylamide substrates and a microarray spotter to fabricate arrays of multicellular islands on various combinations of extra cellular matrix (ECM) proteins or other biomolecules. A microscope with an automated stage is used to image each of the cellular islands before and after lysing cells with a detergent. These images are analyzed in a semi-automated fashion using a series of MATLAB scripts which produce the displacement and traction fields, and summary data. By combining microarrays with a semi-automated implementation of TFM analysis, this protocol enables evaluation of the impact of substrate stiffness, matrix composition, and tissue geometry on cellular mechanical behavior in high throughput.
Keywords: Traction force microscopyBackground
Cellular generated forces are transmitted to the extracellular matrix (ECM) through integrins, and to surrounding cells by cadherins. The signaling associated with the transduction of these forces plays an important role in the regulation of proliferation, differentiation, morphogenesis, and homeostasis. Further, the magnitude and distribution of cellular forces are impacted by the conditions of the microenvironment such as ECM composition, geometry, substrate stiffness, and the presence of other ligands or drugs (Discher et al., 2009). We recently showed how ECM composition and substrate stiffness work together to affect liver progenitor cell differentiation and contractility (Kourouklis et al., 2016), and that cell contractility and traction stress distribution can facilitate spatial patterning of liver progenitor cell differentiation (Kaylan et al., 2018). Investigating the ways that the conditions of microenvironment such as ECM composition, geometry, substrate stiffness, and the presence of other ligands or drugs, mediate biomechanical and biochemical responses requires engineered systems that enable systematic control of the environment along with a way to assay the resulting behavior.
A variety of techniques have merged to measure cell-generated forces (Polacheck and Chen, 2016). Traction force microscopy (TFM) is a well-established method used to measure the forces applied by cells to the surface they are adhered to (Butler et al., 2002). While this concept has been expanded to three-dimensional cell culture, this protocol with only address TFM in two dimensional monolayers. Since its establishment, many authors have implemented TFM systems using various approaches and algorithms (Schwarz and Soine, 2015; Kulkarni et al., 2018). Broadly, in TFM, a flexible cell culture substrate is prepared with embedded fluorescent markers, often polymer beads. This substrate is modified such that cells will adhere, often by attaching a suitable ECM protein. Cells are then cultured on these substrates. Using an inverted fluorescence microscope, an image is taken the substrate surface, and the fluorescent markers, under each cell area of interest. The cells lysed using a detergent or enzyme, resulting in any forces applied to the surface by the cells to be relaxed. The same region is imaged once again. From these images, a displacement field is calculated using specialized software, based on the movement of the fluorescent markers after cell lysing. From the displacement field, a traction stress field is the calculated, using the known mechanical characteristics of the substrate.
In the MATLAB code provided with this protocol, the displacement field is calculated using a publicly available digital image correlation (DIC) algorithm (Landauer et al., 2018). Traction forces calculation is performed using an available Fourier transform traction cytometry (FFTC) algorithm (Sabass et al., 2008; Han et al., 2015). These methods were chosen based on their relatively low computational expense and time and relatively few required user inputs. This supports the high throughput nature of the analysis and goal to allow “off the shelf” use for labs who do not focus on TFM as a core competency. However, the code was intended to enable users to substitute alternative displacement or traction field algorithms based on the needs and computational resources of the user and application.
Performing TFM often involves bulk preparations of adhesive substrates which requires relatively large amounts of adhesive proteins, decreasing the practicality of performing TFM on multiple ECM proteins or surface bound ligands. Here we use contact printed microarrays which allows multiple ECM/ligand combinations, with replicates, to be included on a single substrate with low material usage (Flaim et al., 2005). TFM is often implemented either on single cells, or randomly distributed small colonies. In the microarray system, we can assess the mechanical behavior of multicellular islands where forces are transmit to the substrate as well as neighboring cells, resulting in collective behavior (Mertz et al., 2013). Further, these multi cellular islands are of consistent size and shape. This permits analysis of average mechanical behavior of these islands as a function of the environmental conditions. Island diameter can be tuned by using differently sized microarray pins, adding geometry as an additional parameter to investigate. The MATLAB code provided includes techniques to identify island boundaries and align data from replicate islands to enable analysis of these average behaviors. These cellular micro-arrays can be assayed in parallel using immunocytochemistry, which has been described in more detailed elsewhere (Kaylan et al., 2017), to allow correlation of mechanical and phenotypic behavior.
Our application has focused on the relationship between spatial patterns of mechanical behavior and differentiation of liver progenitor cells. This method can be applied to other stem cell systems especially where spatial patterning and collective mechanical behavior is of interest. It can also be applied to investigations of how the mechanical behavior of cancer relates to its microenvironment and further impacts proliferation or response to drugs. Although here we used contact microarray printing, this protocol can easily be applied to other micro patterned systems such as those with non-contact printing (Romanov et al., 2014) and Polydimethylsiloxane (PDMS) stamped substrates (Kane et al., 1999). In this application, we utilize the XY position within the array to know the condition of that island. This protocol could be extended to other systems where spatial position of cells on their substrate is linked to an experimental variable or substrates such as with gradients of stiffness (Hadden et al., 2017) or biomolecules (Dertinger et al., 2002).
Materials and Reagents
- Aluminum foil
- Forceps
- Pipettes
- 1 ml syringe
- 6-well polystyrene microplates (Fisher Scientific, catalog number: 08-772-1B)
Note: Other 6-well microplates may be suitable, however the 35 mm glass bottom Petri dishes fit well into these plates and do not fit into all 6-well plates. - Far-red fluorescent beads, 0.2% v/v (Life Technologies, catalog number: F-8816)
- 12 mm glass coverslip (Electron Microscopy Sciences, catalog number: 72231-01)
- 384-well polypropylene plate (USA Scientific, catalog number: 1823-8400)
- 35 mm glass bottom dish with 13 mm well and #1.5 German cover glass (Cell E&G, catalog number: GBD00002-200)
- Ligands
Fc-recombinant DLL1, mouse (R&D Systems, catalog number: 5026-DL-050)
Fc-recombinant DLL4, mouse (AdipoGen, catalog number: AG-40A-0145-C050)
Fc-recombinant JAG1, rat (R&D Systems, catalog number: 599-JG-100)
Human IgG (R&D Systems, catalog number: 1–001-A)
Protein A/G (Life Technologies, catalog number: 21186)
Note: Cell-cell signaling ligands can be arrayed on the surface in combination with ECM proteins. Retention of smaller molecules in the gel may differ, therefore it may be necessary to conjugate ligands to an additional molecule. We used Fc-recombinant notch ligands conjugated to protein A/G, and IgG as a control. If using this strategy, ligands should be combined with protein A/G at a molar ratio of 6:1 or greater. Ligands should be reconstituted per manufacturer recommendations and stored at -80 °C. - Extracellular Matrix proteins
Collagen I, rat tail (EMD Millipore, catalog number: 08-115MI), stored at 4 °C
Collagen III, human (EMD Millipore, catalog number: CC054)
Collagen IV, human (EMD Millipore, catalog number: CC076)
Fibronectin, human (Sigma-Aldrich, catalog number: F2006)
Laminin, mouse (EMD Millipore, catalog number: CC095)
Note: Numerous ECM proteins are suitable for this process. Choice of ECM protein will depend on the application. Here we have listed those used in our in various studies, which were prepared at 1 mg/ml, and stored at -20 °C unless otherwise noted. - 3-TPM (Sigma-Aldrich, catalog number: 440159-100ML)
Note: Exposure of 3-TPM to air could negatively impact silanization of glass substrates and should be stored per manufacturer instructions. The 3-TPM described here is packaged in a Sure/SealTM container, which requires a syringe and needle for use. - NaOH (Sigma-Aldrich, catalog number: 415413-1L)
- Ethanol 200 Proof (Decon Labs, catalog number: UN1170)
- Acrylamide (Sigma-Aldrich, catalog number: A3553-100G)
- Bis-acrylamide (Sigma-Aldrich, catalog number: M7279-25G)
- Irgacure 2959 (BASF Corporation, catalog number: 55047962)
- Methanol (Sigma-Aldrich, catalog number: 179957-1L)
- Sodium acetate (Sigma-Aldrich, catalog number: S2889-250G)
- Ethylenediaminetetraacetic (EDTA) (Sigma-Aldrich, catalog number: ED-100G)
- Glycerol (Sigma-Aldrich, catalog number: M6145-25ML)
- Glacial acetic acid (Sigma-Aldrich, catalog number: 695092-500ML)
- Phosphate-buffered saline (PBS) (Fisher Scientific, catalog number: SH3001302)
- 3-[(3-Cholamidopropyl) dimethylammonio]-1-propanesulfonate (CHAPS) (Sigma-Aldrich, catalog number: C3023-1G)
- Dextran-rhodamine (Life Technologies, catalog number: D-1841)
- 100x penicillin-streptomycin solution (Fisher Scientific, catalog number: SV30010)
- Bovine serum albumin (Sigma-Aldrich, catalog number: A2153-100G)
- Sodium dodecyl sulfate (Fisher Scientific, catalog number: BP166-100)
- Triton X-100 (Sigma-Aldrich, catalog number: X100-1L)
- 2x ECMP Printing Buffer (see Recipes)
- 2x Growth Factor (GF) Printing Buffer (see Recipes)
- Polyacrylamide Prepolymer Solution (see Recipes)
Equipment
- Vacuum Desiccator (Fisher Scientific, catalog number: 08-642-5)
- OmniGrid Micro arrayer (Digilab)
Note: Other microarrayers with similar capabilities can be used instead. Follow manufacturer’s instructions to operate arrayer. - Stealth or solid pins (ArrayIt, catalog number: SMP3 or SSP015)
Note: Other pin types may be suitable for use ArrayIt, SMP3 prints spots approximately 150 μm in diameter. SSP015 pins print spots of approximately 600 μm in diameter. Select pins that produce an appropriate island size for your application. - UV crosslinker, UVP, CL-1000
- Vortex
- Fluorescence microscope, Zeiss Axiovert 200M with a computer running Zeiss Zen image acquisition software
Note: Here we use a Zeiss Axiovert 200M, however other fluorescent microscopes are suitable. The microscope should be equipped with a robotic stage and the ability to mark and find locations. We also advise environmental controls (i.e., 37 °C and 5% CO2 since TFM involves live cell imaging. The provided MATLAB code is designed to directly load and read metadata from the Zeiss proprietary format (.czi). However, because it utilizes tools from Bio-Formats (Linkert et al., 2010), files from most common microscopes can be loaded with little or no modifications.
Software
- MATLAB version 2018b, Mathworks
- MATLAB scripts from https://github.com/UnderhillLab/High-Throughtput-TFM
Note: The provided scripts are intended to be able to be used by those with basic MATLAB experience, however adjusting the scripts for the additional needs of a given user may require more advanced MATLAB expertise.
Procedure
- Substrate fabrication
Polyacrylamide gels are prepared as adapted from protocols in literature (Tse and Engler, 2010; Wen et al., 2014).- Silanization of glass bottom dishes. This functionalizes the glass surface so that the polyacrylamide hydrogel will covalently bind to it.
- Add 250 μl of a 0.2 N NaOH solution to the glass area of each glass bottom dish. Ensure NaOH solution coats entire glass area but not the plastic surface as shown in Figure 1A.
- Incubate on a gently rocking shaker for 1 h.
- Rinse dishes 3 times with deionized water (dH2O).
- Use compressed, filtered air to dry the dishes. Bake the dishes at 80 °C on aluminum foil on a hot plate until completely dry. Overheating the dishes may cause adhesive on the glass to fail.
- Add 0.2 ml of 3-(Trimethoxysilyl) Propyl Methacrylate (3-TPM) to 9.8 ml of 100% ethanol.
- Apply 100 ul of 3-TPM solution to the glass area of each dish. Ensure 3-TPM solution coats entire glass area but not the plastic surface as shown in Figure 1A.
- Incubate on a gently rocking shaker for 30 min.
- Add 3 ml of ethanol to each dish. Incubate on a gently rocking shaker for 5 min.
- Remove ethanol. Use compressed air to dry the dishes.
- Bake the dishes at 80 °C on aluminum foil on a hot plate until completely dry.
Note: At this stage, silanized glass bottom dishes can be stored for up to one month in a dark, dry location.
Figure 1. Glass bottom dish silanization and gel fabrication. A. Proper coverage for NaOH and 3-TPM solutions. B. Droplet of prepolymer solution. C. Coverslip floated on prepolymer droplet. D. Inverted dish to allow beads to settle. E. Dish filled with deionized water following UV exposure. F. Dish with coverslip removed and gel dehydrated.
- Polyacrylamide gel fabrication. The polyacrylamide gel provides the deformable hydrogel substrate for cells to bind to as part of TFM preparation.
- Prepare a polyacrylamide pre-polymer solution based on desired stiffness (Recipe 3). You will need 20 μl of solution for each dish.
- Add 0.1 g of Irgacure 2959 (light-sensitive) to 0.5 ml 100% methanol to produce a 20% w/v Irgacure solution. Vortex until solution is clear.
- Combine the pre-polymer solution and Irgacure solution at a 9:1 ratio. Prepare enough.
- Sonicate the bead suspension for 15 min.
- Add beads to the working solution at a 1:500 ratio (i.e., 1 μl bead solution to 500 μl working solution). Vortex to ensure complete mixing.
- Degas the full working solution in a vacuum desiccator for 15 min.
- Pipette 20 μl of the working solution into the center a glass bottom dish. Avoid bubbles in the droplet as shown in Figure 1B.
- Carefully float a 12 mm coverslip onto the droplet. The solution should spread the edges of the droplet as shown in Figure 1C.
- Repeat with the remaining dishes.
- Invert the dishes, as shown in Figure 1D, and let stand for 15 min to allow beads to migrate towards the surface.
- Expose dishes to 365 nM UVA for 15 min in the UV crosslinker.
- Fill dishes with deionized water as shown in Figure 1E. Let sit for 3-24 h.
- Carefully remove coverslips using forceps. Avoid twisting or shearing the coverslip which may cause ripping in the gel surface.
Note: Use of clean coverslips and careful removal after 24 h of soaking in dH2O typically results in successful removal of the coverslip without damaging the gel. For very soft gels, it may be necessary to further passivate the coverslip surface. Strategies include wiping the surface with Rain‑X® using a delicate task wiper, or vapor deposition of Dichlorodimethylsilane. - Dehydrate gels at 50 °C on a hot plate until all water has evaporated from the gel. A dehydrated gel is shown in Figure 1A. Gel dishes can be stored for up to one month in a dark, dry location.
- Silanization of glass bottom dishes. This functionalizes the glass surface so that the polyacrylamide hydrogel will covalently bind to it.
- Microarray printing
This process prints an array of circular spots of biomolecules onto the polyacrylamide substrates. These array spots are where cells will adhere forming circular islands.- If needed, prepare a suitable 2x printing buffer (Recipe 1)
Note: ECM protein printing (ECMP) buffer is appropriate for most ECM molecules. Growth factor (GF) printing buffer is suitable for other classes of molecules such as growth factors or ligands, where a low pH could cause issues. - Prepare a microarraying source plate.
- In a 384-well V-bottom microplate, prepare printing solutions which should include equal volumes of 2x printing buffer with biomolecule solution at double the target concentration. For many ECM proteins, 250 µg/m is a suitable concentration. Optimal concentrations will vary depending on the molecule, its retention, and its function. The total volume in each well should be between 5 and 15 µl.
- In your source plate, you should also include a solution with a fluorescent marker which will be used to convey the orientation of the array to determine the locations of each condition. We recommend rhodamine-conjugated dextran at a final concentration of 2.5 mg/ml. The source plate configuration will differ based on the arrayer, pin configuration, and desired array layout.
- Mix each well thoroughly by pipetting. Take care to avoid generating bubbles. Centrifuge the source microplate for 1 min at 1,000 x g. Source plates can be used immediately or stored at 4 °C for 1 day before until microarray fabrication. If storing, cover source plate with an adhesive seal.
- Clean and prepare pins according to the manufacturer's instructions. Load cleaned pins into the microarrayer printhead.
- Prepare the microarrayer and arraying program using the manufacturer’s software. The set-up and programming will differ based on the arrayer and desired array layout. The program should be devised such that the array orientation is unambiguous, and the locations of each arrayed conditions are known and could be determined by the location relative to the fluorescent marker in any orientation. An example of this is provided in Figure 2.
Notes:- The example array layout shown was designed for printing 6 replicates of 4 conditions, which were simultaneously printed by four SSP015 pins which produce 600 μm diameter islands. The number of islands and/or conditions can be scaled up when using pins that produce smaller islands.
- Be sure to record how the layout corresponds to the arrayed conditions. Include wash steps using both water and dimethyl sulfoxide (DMSO) between each condition in order to prevent carry-over and cross-contamination.

Figure 2. Example layout of an array of 4 ECM conditions with dextran rhodamine markers. When the islands and dextran rhodamine spots are visible, the orientation of the array can be discerned whether rotated or if viewed from above or below.
- Turn on the humidifier and ensure it is adjusted to 65% RH (non-condensing). Wait until the rheometer matches the set point.
- Place the source plate in the arrayer in an appropriate adaptor.
- Dehydrate hydrogel substrates at 50 °C for 15 min. Protect from light due to the presence of the fluorescent beads.
- Place dishes into an appropriate adaptor. If the arrayer can fit a standard multiwell plate, the 6-well plate is suitable to hold the dishes.
Note: See the note under Materials and Reagents #5. - Begin array fabrication. Check frequently that the humidity has not dropped below 65% RH (non-condensing).
- When the program is complete, store fabricated arrays covered with aluminum foil at room temperature and 65% RH (non-condensing) overnight. While the array spots are visible, it is helpful to visibly mark the top or bottom of the array so the orientation is known when placing on the microscope. For some hydrogel and pin combinations, it may be necessary to store arrays at ambient temperature and humidity for an additional two days to ensure arrays have dried completely. Arrays can be stored for up to 7 days before use.
- If needed, prepare a suitable 2x printing buffer (Recipe 1)
- Seeding cells on microarrays
Here cells are transferred from their normal culture condition on to the microarrayed hydrogel substrates for TFM.- To sterilize the gels, add 3 ml of PBS with 1% v/v penicillin/streptomycin. Expose to UV C for 30 min. Exchange penicillin/streptomycin solution for cell culture media.
- Collect and count cells following the cell appropriate procedure. Resuspend cells in culture media at an appropriate concentration for seeding. This will differ based on cell type but will likely range between 170 x 103 and 7 x 105 cells/ml. Add 3 ml of cell suspension to each dish. Incubate dishes at 37 °C and 5% CO2 for 2-24 h, or until confluent cell islands have formed. Seeding density and time may need to be optimized for your cells and application. Agitation of the dishes every 15-60 min may also aid in forming consistent, confluent islands.
- Once islands have formed, rinse arrays twice with 3 ml prewarmed media. At this stage, add any experimental treatments such as growth factors or inhibitors. Change media every 1-2 days until time to perform TFM, or as your cell culture protocol suggests, maintaining any treatment concentrations at each exchange. Figure 3 shows an example of an array with cellular islands.

Figure 3. 35 mm glass bottom dish with cellular island array after 24 h of culture
- Acquiring images for traction force microscopy
- Prepare a solution of 1% v/v bovine serum albumin (BSA) and 1% v/v sodium dodecyl sulfate (SDS) in PBS. This will be used to dissociate cells during image acquisition.
- The microscopy process will vary based on the microscope used, but the following steps should broadly apply. Some additional details are provided for use with Zeiss Zen software. The microscope should have a robotic stage and a chamber which provides incubation and humidity control which have been appropriately set and allowed to come to equilibrium. Place one dish on the stage of the inverted fluorescent microscope. It is helpful to use the mark provided after array fabrication to place the dish with the array’s XY axis approximately aligned along that of the microscope stage. Secure the dish in place so that it does not shift when the stage moves.
- Using the appropriate fluorescent channel, locate and mark (in the software) the XY positions of the fluorescent arrayed markers. In Zeiss Zen Blue 2, this is accomplished in the acquisition section by checking the tiles box and adding a single position for each island, as shown in Figure 4.
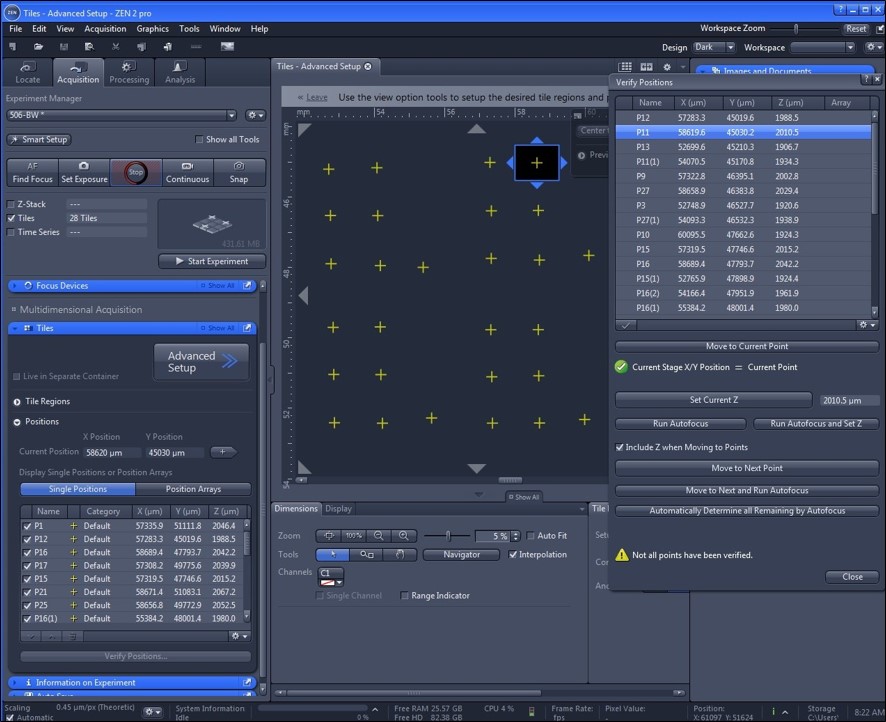
Figure 4. Zeiss Zen Blue 2 environment. To mark locations, in acquisition, mark Tiles and enter advanced setup. Set individual island positions as shown by the yellow markers. The verify positions tool can be used to move through positions to set the Z position without altering the XY position. - Using phase contrast or bright field, locate and mark the XY positions of the cell islands. Either imaging modality may be suitable as long as the image can provide the boundary of the cell island in subsequent analysis. Individually find and save the Z plane focus of each island. An example image phase contrast is shown in Figure 5A. The verify positions tool can be used to move through positions to set the Z position without altering the XY position. The saved XY positions of the islands can be saved and reused for subsequent dishes.
- Begin the automated imaging of the phase contrast images of the cell islands. Save this file with a suitable name that notes the experiment details as well as that it is the phase contrast image.
- Switch to the appropriate fluorescent channel for the beads. Individually find and save the Z plane focus of the top surface of the gel under each island. Take care to avoid changing the XY positions. Save this file with a suitable name that notes the experiment details as well as that it is the pre-dissociation image. An example image is shown in Figure 5B.
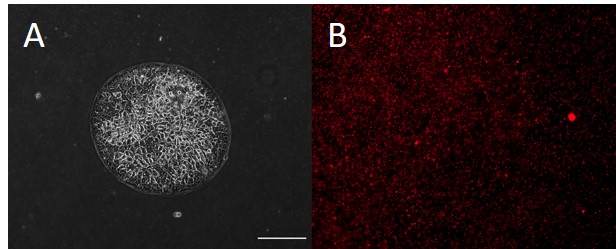
Figure 5. Example images from TFM data collection. A. Phase contract image of a cellular island. Image should be clear enough to define island boundaries. B. Fluorescence image of the beads at the surface of the substrate under the island. Scale bar: 200 μm. - Begin the automated imaging in the fluorescent channel of the pre-dissociation bead images. Save this file with the same experiment details as well as that it is the pre-dissociation image.
- Carefully add 150 µl of the SDS solution to the dish, taking care to not bump or move the dish. Monitor dissociation of the cell islands using the phase contrast channel. Wait until the islands have completely dissociated from the substrate at which time there the island locations, when viewed in phase contrast channel, should appear mostly blank.
Note: Some cells may require addition of more SDS solution, or higher concentration. Additionally, Triton-X may be used instead. - Return to the island positions and using the fluorescent channel check that the beads at the top of the surface are still in focus. If the beads have moved out-of-plane due to relaxion of the cell induced deformation, correct and save the Z-coordinate of the focus plane but do not alter the XY positions. Repeat automated imaging of the islands to capture the post-dissociation images of the beads. The displacement of the beads will likely be difficult to discern, meaning the post-dissociation fluorescent bead image should appear very similar to the pre-dissociation image. Save this file with a suitable name that notes the experiment details as well as that it is the post-dissociation image.
- Repeat Steps D2-D9 for each dish.
Data analysis
- Process images to estimate displacement fields and calculate traction fields
Note: Images collected per the previous section may be exported and used with available TFM code packages. Here we provide the TFM code we have implemented, and its use is explained in the following sections. The TFM and analysis are designed to be used with arrays of cells with the same geometry to enable assessing replicates of the same condition. These scripts allow the user to generate TFM data and create basic visualizations. Further manipulation and analysis of the data for a user’s specific application may require generation of additional scripts.- Ensure the provided MATLAB code has been saved to an appropriate location. Navigate to the folder where this directory has been saved.
- Make sure all image files have been saved or transferred to a folder available from the computer to be used for analysis.
- From the command window, run the function run_island_tfm with no inputs.
- You will be prompted to select the file with the phase contrast image. Navigate to and select the file with the phase contrast image.
- A new file selection window will open in the same directory as the selected file. Now select the file with the pre-dissociation fluorescent image. Finally, when prompted, select the post-dissociation image.
- A GUI will appear for entering the experimental information for the image files. This GUI is shown in Figure 6. The graph panel shows the XY position of each image collected. The numbers correspond to how the images are indexed in the image file. Enter all relevant experiment information listed above the “Data file name” field. The pixel size will be read from the metadata of the image but can be changed. The “number of arrayed conditions” refers to the number of different conditions in the imaged array. For the example layout in this protocol, there are 4 arrayed conditions. This number is used to group the islands into their conditions.
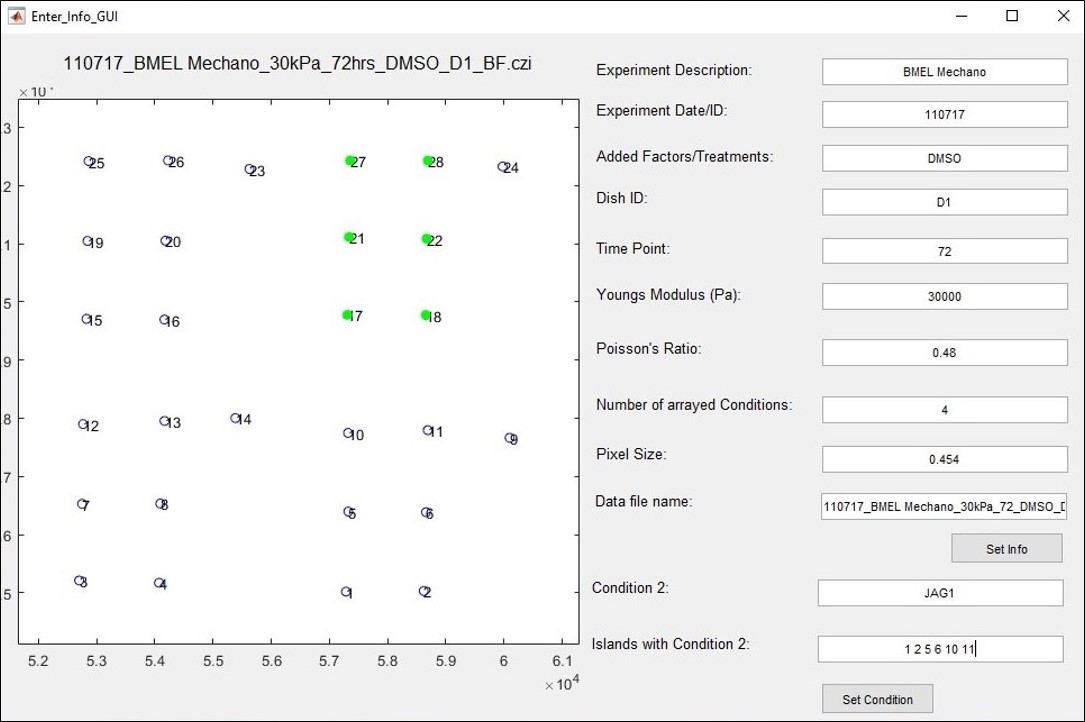
Figure 6. GUI for entering experiment and dish information. Chart panel shows XY position of each island, labeled by the index of the island in the file. Chart title is the image file name. Pixel size is pulled from the image metadata but can be set manually. All other fields are set by the user. - Once these fields are completed, click “Set Info.” This will populate the “Data file name” field using the entered information which will be the file name of the output file. This field can be changed manually. Two additional fields will appear. Enter a name for the first condition in the “Condition 1” field. Enter the numbers of all the islands assigned this condition in the “Islands with Condition 1” field as a list of numbers. Click “Set Condition.” If there are additional conditions, repeat these steps for each of the remaining conditions. Once all conditions have been set, the “Done” button will appear. Confirm that all experiment information is correct, then click “Done.” All islands assigned a condition in this step will be analyzed. To analyze only some islands, set the number of conditions to the number conditions represented by the islands to be analyzed, and only list those islands in the “Islands with Condition” field.
- The script will now cycle through the conditions and islands. It will first run a script to correct frame shifts between the before and after dissociation.
Note: The correct shift function is to remove full frame shifts between the images caused stage drift between collecting the images. This is done by performing digital image correlation on regions of the image far enough from the cells where there is no expected deformation. The script automatically selects four regions near the four corners of the image. To manually select these regions, change the ‘n’ to a ‘y’ in the frame_shift function call.
A GUI will now appear to aid in drawing a boundary around the cell island. This GUI is shown in Figure 7. The software will attempt to draw a boundary around the cell island, which is plotted in red. The slides can be used to adjust the parameters of the trace which will cause the trace to rerun in real time. You can also change the values of these parameters by entering numbers into the text boxes and then hit the “Rerun” button. Alternatively, you can draw the boundary manually by clicking the “draw manual” button. Left click around the island until the boundary is closed. Double click inside the boundary to create the shape. To avoid sharp corners, after the polygon is drawn, a blurring and rounding is applied using the current value in the “Blur” field. To avoid this rounding, set this field to 1. The manual draw function can be used to trace multiple areas. To reset the manual boundaries, click “Clear boundaries,” or click “Rerun” to repeat the automated tracing. When satisfied with the boundary, click “Done.”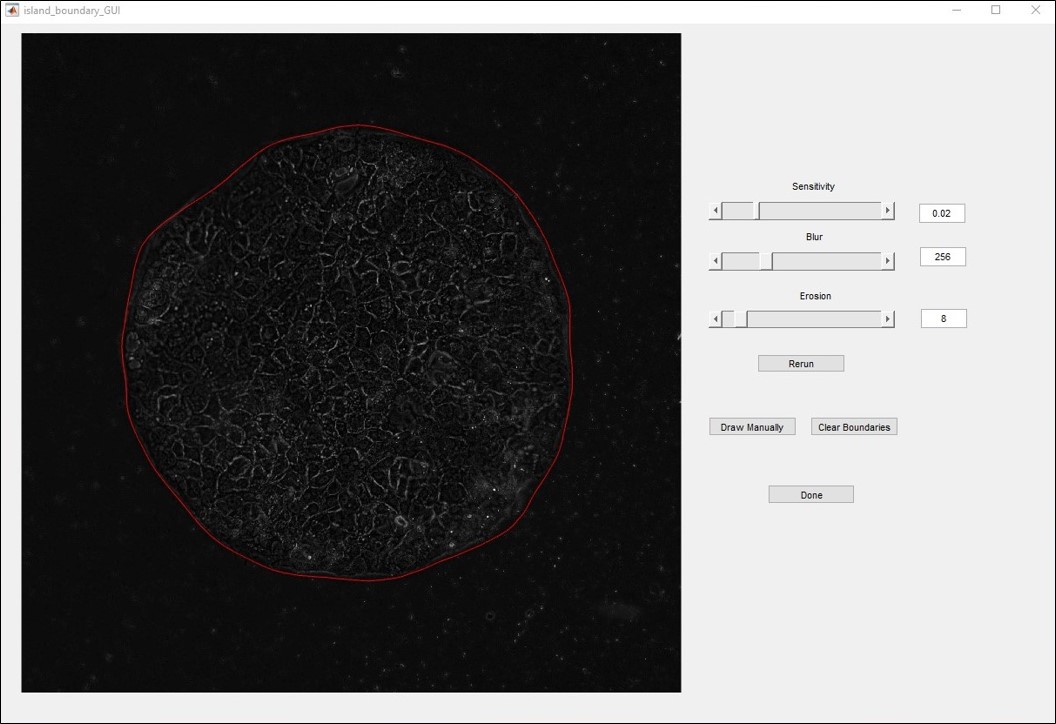
Figure 7. GUI for selecting cell island boundary. Parameters can be set with the sliders which will update the boundary in real time, or by typing in values and clicking Rerun. You can also manually draw the boundary. - This will bring up the next island. Repeat this process for all islands in the file selected for analysis.
- When the boundary trace has been completed for each island, the data will be saved. The program will then move to calculate the displacement and traction fields for each island. This process can take up to several hours depending on the number of images, image size, and processors on the computer. Data is saved after each island to limit data loss in the case of issues during analysis. Data is saved in the folder “data out.”
- Most of the computation time is the displacement field estimation. To rerun the traction field calculation with different settings on a file which already has the boundaries identified and displacement field calculated, run the command run_island_tfm(‘rerun’). You will be prompted to choose a data file which will be reanalyzed.
- View data and generate summaries
- In the code provided, the output is saved in the folder “data out” with the file name established during analysis. To explore data from a single file, data can be loaded into the workspace by double clicking in the Current Folder explored or using the load function.
- This loads the cell array “all_cell_data” into the workspace. Each cell corresponds to a single island from the analysis. The output is intended to provide easy access to all relevant data for the user to explore and analyze in MATLAB or export to other programs for analysis as appropriate for their application. Table 1 provides the organization of the data stored within the output file. Data structure elements can be accessed using dot notation of the form structName.fieldName.
Note: See https://www.mathworks.com/help/matlab/matlab_prog/access-data-in-a-structure-array.html for more information on accessing data in structures. - A set of functions are provided to view and analyze data. Example code to use these functions is provided in data_analysis_examples. To view the results from a single island, use view_one_island. See code for further documentation. An example of the output is shown in Figure 8.
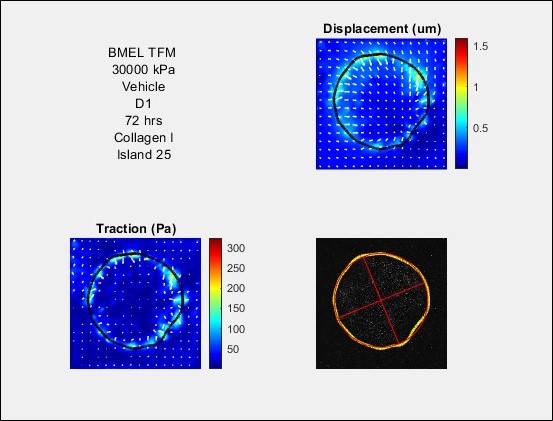
Figure 8. Example output from view_one_island. Island information is displayed. Vector fields are plotted over the magnitudes of the displacement filed (in μm) and traction field (in Pa). The island boundary is plotted in black. The phase contrast image is also displayed, with the boundary plotted in yellow. The best fit ellipse is shown in red, with the major and minor axes. - Using the boundary traced of the island, the script finds a best fit ellipse. This ellipse can be used to align compare many replicates of islands with the same geometry.
Note: The provided scripts assume the islands can be represented by an ellipse. This can be adapted to other geometries by altering how the center and angle of rotation are identified.
Use view_one_island_rotandcen to view the data centered and rotated according to the best fit ellipse. - The function collect_island_data is provided to collect data from multiple islands across multiple TFM runs. The output of this function is a table with data and information on each of the islands loaded from the files (see Table 1), and a structure holding the displacement and traction data, indexed according to the “summ_ind” field of the summary table This table can be exported to excel or similar. When the function is run without an input, you will be prompted to select data files to load and consolidate.
Table 1. Structure of data output file and description of saved dataData Field Description cell_info. young Youngs Modulus of substrate pois Poisson Ratio of substrate pixelsize Size of original image pixels in micron experiment Description of experiment date_ID Identifier of experiment or run soluble Soluble factors or treatments added to dish dish_num Identifier of dish, e.g., if replicates of a unique condition time Time from seeding and TFM data collection arrayed_condition ECM or other condition of the island within the array Xposition X position of island within array Yposition Y position of island within array image_number Index of image within the original file file file containing original phase/brightfield image dm Final spacing, in pixels, of displacement/traction field output Images. Images{1} Pre-dissociation fluorescent image Images{2} Post-dissociation fluorescent image Images{3} Phase/Brightfield image cell_boundaries. mask mask of regions determined within island boundary boundary_points Cell containing the XY coordinates of each of the boundaries traced cny Sensitivity factor used to trace boundary sigma Blur factor used to trace boundary di Erosion factor used to trace boundary ellipse_fit. phi Angle of roation of the fit ellipse with respect to the major axis being horizontal X_center center at the X axis of the non-rotated ellipse Y_center center at the Y axis of the non-rotated ellipse X0 center at the X axis of the rotated ellipse Y0 center at the Y axis of the rotated ellipse major_axis major axis of ellipse (in pixels) minor_axis minor axis of ellipse (in pixels) rotated_boundary_points Cell containing the XY coordinates of each of the boundaries traced, rotated and centered according to the fit ellipse cell_displacements. raw_displacements. raw_displacements{1} X component of raw estimated displacement field in pixels raw_displacements{2} Y component of raw estimated displacement field in pixels raw_displacements{3} Magnitude of raw estimated displacements in pixels Displacements. displacements{1} X component of displacement field in micron displacements{2} Y component of displacement field in micron displacements{3} Magnitudes of displacements field in micron rotated_displacements displacements which have been rotated and centered using the fit ellipse cell_tractions. Tractions. tractions{1} X component of tractions in Pa tractions{2} Y component of tractions in Pa tractions{3} Magnitudes of tractions in Pa RMS Root mean square of traction field RMS_int Root mean square of traction field within cell island boundary Strain_Energy Strain Energy rotated_tractions Tractions which have been rotated and centered using the fit ellipse - The output of the collect_island_data function can further be used to create summary data. The function summarize_islands plots and outputs averaged displacement and traction fields, see Figure 9 for an example. This function uses the aligned the data from each island based on the best fit ellipse. The islands to include in the averaging can be selected by choosing a subset of set of islands of the summary_table by indexing on one or more variables. See data_analysis_examples for an example of using this function.
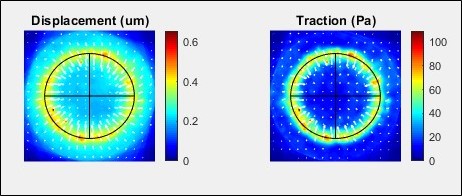
Figure 9. Example of average displacement and traction vector fields produced by summarize_islands - The circular islands described in this protocol and used in our work (Kaylan et al., 2018) have radial symmetry. Therefore, it was informative to analyze the traction a function of a single radial dimension as shown in Figure 10. The function summarize_islands_1D is provided to perform this analysis. Here, the XY position of each data point is converted to a radial coordinate, and the radial position is normalized by the measured radius of the island. The traction data is binned by radial coordinate, and a mean is taken. The data used for this analysis can also be selected using the summary table as discussed previously. The function also outputs the data table with the peak traction of each island amended to the relevant lines. See data_analysis_examples for an example of using this function.
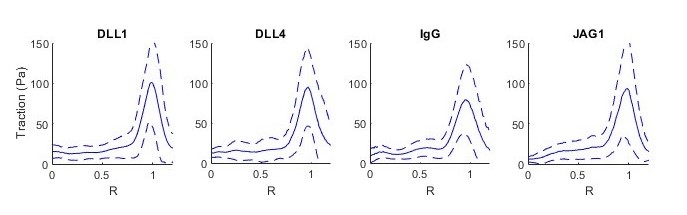
Figure 10. Tractions from 6 replicates of 4 conditions in one TFM dish plotted as a function of normalized radial coordinate. Dashed line shows standard deviation. In all conditions, traction peaks near the island periphery, where R = 1.
Recipes
- 2x ECMP Printing Buffer
- Add 164 mg sodium acetate and 37.2 mg EDTA to 5.9 ml dH2O
- Once dissolved, add 50 µl warmed Triton X-100 and 4 ml glycerol. Vortex until combined
- Add approximately 80 µl of glacial acetic acid, titrating to adjust the pH to 4.8
- Store at 4 °C until arraying
- 2x Growth Factor (GF) Printing Buffer
- Add 105.5 mg sodium acetate and 37.2 mg EDTA to 6 ml PBS
- Once dissolved, add 0.1 g CHAPS and 3.8 ml glycerol
- Store at 4 °C until arraying
- Polyacrylamide Prepolymer Solution
- Polyacrylamide solutions are prepared according to the desired stiffness. An example set of stiffness recipes is adapted from (Wen et al., 2014)
4 kPa Young’s modulus: 0.400 g acrylamide and 0.040 g bis-acrylamide
13 kPa Young’s modulus: 0.600 g acrylamide and 0.045 g bis-acrylamide
30 kPa Young’s modulus: 0.800 g acrylamide and 0.055 g bis-acrylamide - Add appropriate amount of acrylamide and bis-acrylamide to 10 ml dH2O
- Vortex until the solution is clear. Filter solution with 0.2 µm syringe
- Store solution at 4 °C for up to one month
- Polyacrylamide solutions are prepared according to the desired stiffness. An example set of stiffness recipes is adapted from (Wen et al., 2014)
Acknowledgments
We acknowledge the Carl R. Woese Institute for Genomic Biology, University of Illinois at Urbana–Champaign Core Facility for assistance with microscopy. Research reported in this publication was supported by the National Institute of Biomedical Imaging and Bioengineering of the National Institutes of Health under Award Number T32EB019944. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Competing interests
The authors declare that they have no competing financial interests.
References
- Butler, J. P., Tolic-Norrelykke, I. M., Fabry, B. and Fredberg, J. J. (2002). Traction fields, moments, and strain energy that cells exert on their surroundings. Am J Physiol Cell Physiol 282(3): C595-C605.
- Dertinger, S. K., Jiang, X., Li, Z., Murthy, V. N. and Whitesides, G. M. (2002). Gradients of substrate-bound laminin orient axonal specification of neurons. Proc Natl Acad Sci U S A 99(20): 12542-12547.
- Discher, D. E., Mooney, D. J. and Zandstra, P. W. (2009). Growth factors, matrices, and forces combine and control stem cells. Science 324(5935): 1673-1677.
- Flaim, C. J., Chien, S. and Bhatia, S. N. (2005). An extracellular matrix microarray for probing cellular differentiation. Nat Methods 2(2): 119-125.
- Hadden, W. J., Young, J. L., Holle, A. W., McFetridge, M. L., Kim, D. Y., Wijesinghe, P., Taylor-Weiner, H., Wen, J. H., Lee, A. R., Bieback, K., Vo, B. N., Sampson, D. D., Kennedy, B. F., Spatz, J. P., Engler, A. J. and Choi, Y. S. (2017). Stem cell migration and mechanotransduction on linear stiffness gradient hydrogels. Proc Natl Acad Sci U S A 114(22): 5647-5652.
- Han, S. J., Oak, Y., Groisman, A. and Danuser, G. (2015). Traction microscopy to identify force modulation in subresolution adhesions. Nat Methods 12(7): 653-656.
- Kane, R. S., Takayama, S., Ostuni, E., Ingber, D. E. and Whitesides, G. M. (1999). Patterning proteins and cells using soft lithography. Biomaterials 20(23-24): 2363-2376.
- Kaylan, K. B., Berg, I. C., Biehl, M. J., Brougham-Cook, A., Jain, I., Jamil, S. M., Sargeant, L. H., Cornell, N. J., Raetzman, L. T. and Underhill, G. H. (2018). Spatial patterning of liver progenitor cell differentiation mediated by cellular contractility and Notch signaling. Elife 7: e38536.
- Kaylan, K. B., Kourouklis, A. P. and Underhill, G. H. (2017). A high-throughput cell microarray platform for correlative analysis of cell differentiation and traction forces. J Vis Exp (121).
- Kourouklis, A. P., Kaylan, K. B. and Underhill, G. H. (2016). Substrate stiffness and matrix composition coordinately control the differentiation of liver progenitor cells. Biomaterials 99: 82-94.
- Kulkarni, A. H., Ghosh, P., Seetharaman, A., Kondaiah, P. and Gundiah, N. (2018). Traction cytometry: regularization in the Fourier approach and comparisons with finite element method. Soft Matter 14(23): 4687-4695.
- Landauer, A. K., Patel, M., Henann, D. L. and Franck, C. (2018). A q-Factor-Based digital image correlation algorithm (qDIC) for resolving finite deformations with degenerate speckle patterns. Exp Mech 58(5): 815-830.
- Linkert, M., Rueden, C. T., Allan, C., Burel, J. M., Moore, W., Patterson, A., Loranger, B., Moore, J., Neves, C., Macdonald, D., Tarkowska, A., Sticco, C., Hill, E., Rossner, M., Eliceiri, K. W. and Swedlow, J. R. (2010). Metadata matters: access to image data in the real world. J Cell Biol 189(5): 777-782.
- Mertz, A. F., Che, Y., Banerjee, S., Goldstein, J. M., Rosowski, K. A., Revilla, S. F., Niessen, C. M., Marchetti, M. C., Dufresne, E. R. and Horsley, V. (2013). Cadherin-based intercellular adhesions organize epithelial cell-matrix traction forces. Proc Natl Acad Sci U S A 110(3): 842-847.
- Polacheck, W. J. and Chen, C. S. (2016). Measuring cell-generated forces: a guide to the available tools. Nature Methods 13(5): 415-423.
- Romanov, V., Davidoff, S. N., Miles, A. R., Grainger, D. W., Gale, B. K. and Brooks, B. D. (2014). A critical comparison of protein microarray fabrication technologies. Analyst 139(6): 1303-1326.
- Sabass, B., Gardel, M. L., Waterman, C. M. and Schwarz, U. S. (2008). High resolution traction force microscopy based on experimental and computational advances. Biophys J 94(1): 207-220.
- Schwarz, U. S. and Soine, J. R. (2015). Traction force microscopy on soft elastic substrates: A guide to recent computational advances. Biochim Biophys Acta 1853(11 Pt B): 3095-3104.
- Tse, J. R. and Engler, A. J. (2010). Preparation of hydrogel substrates with tunable mechanical properties. Curr Protoc Cell Biol Chapter 10: Unit 10 16.
- Wen, J. H., Vincent, L. G., Fuhrmann, A., Choi, Y. S., Hribar, K. C., Taylor-Weiner, H., Chen, S. and Engler, A. J. (2014). Interplay of matrix stiffness and protein tethering in stem cell differentiation. Nat Mater 13(10): 979-987.
Article Information
Copyright
![]() Berg and Underhill. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Berg and Underhill. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Berg, I. C. and Underhill, G. H. (2019). High Throughput Traction Force Microscopy for Multicellular Islands on Combinatorial Microarrays. Bio-protocol 9(21): e3418. DOI: 10.21769/BioProtoc.3418.
Category
Cell Biology > Cell structure > Cell adhesion
Cell Biology > Cell imaging > Live-cell imaging
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.