- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Imaging VIPER-labeled Cellular Proteins by Correlative Light and Electron Microscopy
Published: Vol 9, Iss 21, Nov 5, 2019 DOI: 10.21769/BioProtoc.3414 Views: 6844
Original research article
The authors used this protocol in:

Dec 2018



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Advances in fluorescence microscopy (FM), electron microscopy (EM), and correlative light and EM (CLEM) offer unprecedented opportunities for studying diverse proteins and nanostructures involved in fundamental cell biology. It is now possible to visualize and quantify the spatial organization of cellular proteins and other macromolecules by FM, EM, and CLEM. However, tagging and tracking cellular proteins across size scales is restricted by the scarcity of methods for attaching appropriate reporter chemistries to target proteins. Namely, there are few genetic tags compatible with EM. To overcome these issues we developed Versatile Interacting Peptide (VIP) tags, genetically-encoded peptide tags that can be used to image proteins by fluorescence and EM. VIPER, a VIP tag, can be used to label cellular proteins with bright, photo-stable fluorophores for FM or electron-dense nanoparticles for EM. In this Bio-Protocol, we provide an instructional guide for implementing VIPER for imaging a cell-surface receptor by CLEM. This protocol is complemented by two other Bio-Protocols outlining the use of VIPER (Doh et al., 2019a and 2019b).
Keywords: Protein tagBackground
Multiple protein targets can be imaged at once by fluorescence microscopy (FM), electron microscopy (EM), or correlative light and EM (CLEM) (Giepmans et al., 2005; Lucas et al., 2012; Philimonenko et al., 2014; Johnson et al., 2015; Kim et al., 2015). FM enables multi-color microscopy in both living and fixed cells, and acquiring data can be relatively fast and easy. However, EM offers better resolution for imaging nanoscale features, including cell receptors, membrane boundaries, neuronal connections (Hildebrand et al., 2017), chromatin organization (Ou et al., 2017), or the endocytic machinery (Sochacki et al., 2017). We anticipate an increased reliance on multi-color, cross-platform imaging for investigating proteins associated with normal cell function and human diseases (Megason and Fraser, 2007; Lichtman et al., 2008; Milne and Subramaniam, 2009; Muller and Heilemann, 2013; Plaza et al., 2014; Kremer et al., 2015; Lucocq et al., 2015; Karreman et al., 2016; Laine et al., 2016; Romero-Brey and Bartenschlager, 2017). Currently, most high-resolution imaging studies obtain protein-specific contrast with immunolabeling, which has known shortcomings (Berglund et al., 2008; Bordeaux et al., 2010; Baker, 2015; Bradbury and Pluckthun, 2015).
The central obstacle that has limited progress in multi-scale microscopy is the shortage of genetic tags for labeling proteins. There are a number of protein tags available for imaging proteins by FM, including fluorescent proteins and self-labeling enzyme tags (Sunbul and Yin, 2009; Hinner and Johnsson, 2010). However, there are few genetic tags for EM or CLEM (Ellisman et al., 2012). Most EM tags rely on the oxidation of diaminobenzidine (DAB) to form a polymer that is stained with osmium tetroxide to generate contrast. Examples include APEX (Martell et al., 2012; Lam et al., 2015), miniSOG (Shu et al., 2011), the tetracysteine tag (Gaietta et al., 2002), and others (Kuipers et al., 2015; Liss et al., 2015). Among the DAB-reliant EM tags, miniSOG, FLIPPER (Kuipers et al., 2015), and the tetracysteine tag are compatible with CLEM. However, DAB staining is finicky, and it can be difficult to localize the stain sufficiently to resolve targets. Further progress in multi-scale microscopy will require new methods for labeling proteins with EM- and CLEM-compatible reporters, such as quantum dots (Qdots) (Giepmans et al., 2005) or FluoroNanogoldTM particles (Takizawa et al., 2015).
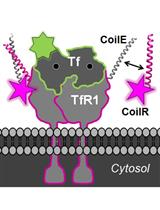
We recently described a new class of tags for multiscale microscopy called Versatile Interacting Peptide (VIP) tags (Tane et al., 2017; Doh et al., 2018). VIP tags use heterodimerizing coiled-coil peptides to label proteins. One coil is expressed as a fusion to the protein of interest. This coil has a partner, the probe peptide, that is conjugated to a reporter molecule to deliver protein-specific contrast. The probe peptide can be conjugated to a number of reporters, such as fluorophores, small molecules (e.g., biotin), or nanoparticles.
In this Bio-Protocol, we outline the use of VIPER to image a transmembrane receptor by CLEM. VIPER is a VIP tag comprised of CoilE tag and CoilR probe peptide. In Procedure A, we describe the plating and transfection of cells to express a CoilE-tagged receptor: transferrin receptor 1 (TfR1-CoilE). Procedure B describes how to label a cell receptor with CoilR-biotin for subsequent detection with streptavidin-Qdot655. In Procedure C we have an illustrated guide on how to mount ITO coverslips to a slide holder for correlative fluorescence imaging. Procedure D describes image acquisition with a commercially available CLEM microscope, the FEI CorrSightTM. In Procedures E and F we describe methods for preparing samples for EM. Procedure G details the acquisition of SEM micrographs on a Helios NanolabTM 660 EM. We additionally developed a quantitative image analysis pipeline for automated image segmentation on high magnification SEM images (see the Data Analysis section). This program runs in Matlab and reports the number of nanoparticles within a field of view in SEM micrographs.
This protocol is published in tandem with two supporting publications detailing the use of VIPER (Figure 1). Doh et al. (2019a) details how to generate CoilR probe peptides, including biotinylated CoilR. Doh et al. (2019b) describes imaging VIPER-labeled proteins in cells by FM.
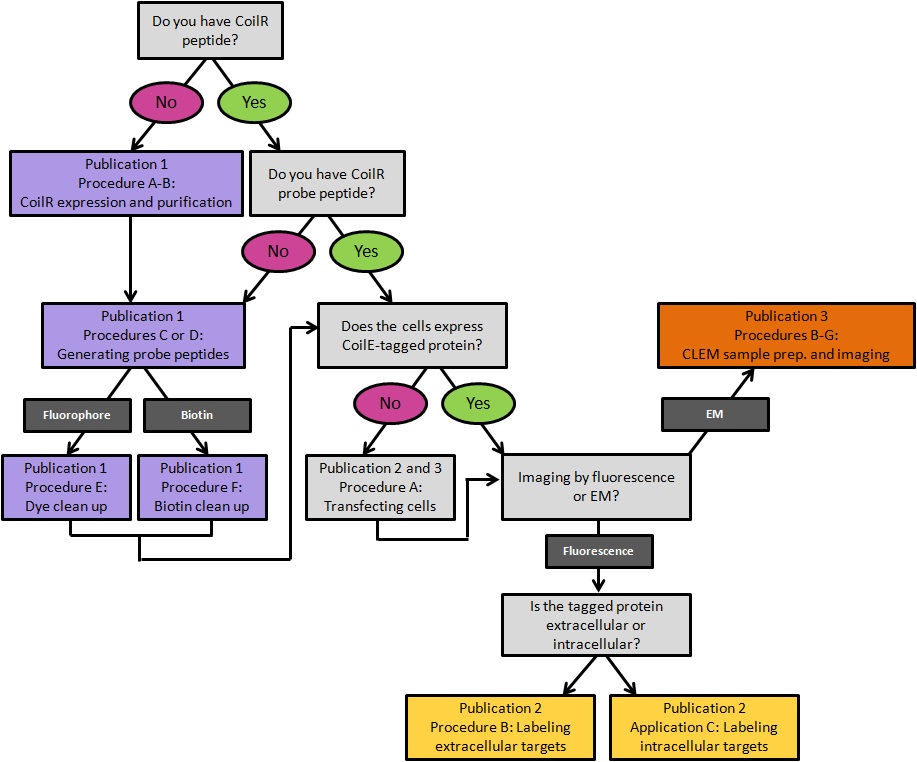
Figure 1. A decision tree for implementing VIPER for labeling cellular proteins. Procedures are color-coded by the publication in which they appear. Publication 1: Doh et al., 2019a; Publication 2: Doh et al., 2019b; Publication 3: this article.
Materials and Reagents
Note: “*” indicates a brand that is critical to the success of the experiment.
Materials
- Aluminum coverslip holder
Note: We used a custom machined aluminum plate with a hole for a 22 x 22 mm coverslip. This plate is 76 x 26 x 1.5 mm with a 12 mm diameter hole in the center. - *Indium tin-oxide 22 x 22 mm coverslips (2SPI, catalog number: 06486-AB)
- Tape (Scotch® MagicTM Tape)
- Desiccator cabinet (Thermo Scientific NalgeneTM, catalog number: 53170070)
- Desiccant (DrieriteTM, catalog number: D1085)
- Kimwipes (Kimtech, catalog number: 34120)
- *Conductive silver paint “Leitsilber” (Ted Pella, catalog number: 16035)
- *SEM pin stub specimen mount (Ted Pella, catalog number: 16144)
- *Carbon thread (Leica, catalog number: 16771511116)
- LDPE 500 ml squeeze wash bottle (Thermo Scientific, catalog number: 24010500)
- Transfer bulb pipette (VWR, catalog number: 16001-182)
- CHO TRVb cells (courtesy of Prof. Timothy McGraw, Cornell University, Ithaca, New York) (McGraw et al., 1987)
- Ham’s F-12 Medium (Life Technologies, GibcoTM, catalog number: 11765062)
- Dulbecco’s phosphate-buffered saline without calcium or magnesium; DPBS (GibcoTM, catalog number: 14190144)
- Trypsin-EDTA (0.25%) (Life Technologies, GibcoTM, catalog number: 25200056)
- Fetal bovine serum (FBS) (GE, HycloneTM, catalog number: SH30910.03)
- *LipofectamineTM 2000 (Thermo Scientific, catalog number: 11668019)
- Opti-MEM (Life Technologies, GibcoTM, catalog number: 31985070)
- *Streptavidin-QdotTM 655 conjugate (Invitrogen, catalog number: Q10121MP)
- Anhydrous ethanol (Decon Labs, catalog number: 2716)
- 20% (v/v) paraformaldehyde (PFA) stock (Electron Microscopy Sciences, catalog number: 15713S)
- Live Cell Block solution (see Recipes)
- Qdot Block solution (see Recipes)
- Qdot Labeling solution (see Recipes)
Equipment
- Hemocytometer (Hausser Scientific, catalog number: 1475)
- Humidified CO2 Incubator (New Brunswick Galaxy 170S, catalog number: C0170S-120-0000)
- Tissue culture hood (Thermo Scientific, model: 1300 Series A2)
- Tissue culture inverted light microscope (Carl Zeiss, Zeiss Primovert)
- Fine point tweezers (Ted Pella Dumostar Biology, catalog number: 525-PS)
- Diamond-tipped scribe (Ted Pella, catalog number: 54468)
- Stainless steel crinkle washers (Tousimis Washers, catalog number: 8767-01)
- Orbital Shaker (Stovall Belly DancerTM, catalog number: BDRAA1158)
- Spinning disk confocal fluorescence microscope (FEI CorrSightTM)
- 63x objective lens (Carl Zeiss, 1.4 NA Plan-Apochromat M27, catalog number: 420780-9900-000)
- 5x objective lens (Carl Zeiss, 0.16 NA EC Plan-Neofluar M27, catalog number: 420330-9901-000)
- Scanning electron microscope (FEI Helios NanolabTM 660 SEM)
- Critical point dryer (Leica EM CPD300)
- High Vacuum Flash Carbon Coating Machine (Leica EM ACE600)
Software
- FEI MAPS (FEI version 2.1.38.1199 and version 3.0)
- FEI Helios NanolabTM xT Microscope Control (FEI version 5.5.1 and version 10.1.7)
- Matlab (Mathworks Software Version R2017b)
Procedure
- Transfecting cell lines to express a VIPER-tagged protein
This protocol describes the transfection of a tissue culture cell line to express a VIPER-tagged transmembrane receptor (i.e., TfR1-CoilE). We used the CHO TRVb cell line because it does not express TfR1 or transferrin receptor 2 (McGraw et al., 1987). After transfection, all TfR1 receptor will encode the C-terminal CoilE tag on the extracellular domain.
We recommend optimizing plating density and transfection conditions for each cell line. Refer to Thermo Fisher’s Protocol Pub No. MAN0007824 Rev 1.0 (Reference 23) for more information on transfection with Lipofectamine 2000. In our protocol, we recommend passaging cells on Day 1, transfecting cells on Day 2, and VIPER labeling cells on Day 3 (see Procedure B).
Recommendations for Protocol A:- Use sterile technique and work within a tissue culture (laminar flow) hood when working with live cells.
- When transfecting cells with a vector encoding a CoilE-tagged protein (e.g., pcDNA3.1_TfR1-CoilE), we also recommend transfecting cells with an untagged protein to compare labeling specificity (e.g., pcDNA3.1_TfR1). Untransfected cells also serve as a control for labeling specificity.
- The sequence for TfR1-CoilE can be found in the Sequences (Supplemental file).
- Day 1: Passage cells and plate onto indium tin oxide (ITO) coverslips
- Before starting, visually inspect cells on a tissue culture microscope to confirm that cells are adherent, healthy, and 80-90% confluent.
- Place single 22 x 22 mm ITO coverslips into each well of a sterile 6-well polystyrene tissue culture plate.
- The ITO coverslips are provided in a small box with the conductive side oriented face up. Transfer the coverslip to the 6-well plate without flipping the coverslip over. Maintain this orientation during transfection, labeling, and processing.
- Pre-coating coverslips with poly-L-lysine or other cell-surface treatments may be necessary to adhere cells to glass. CHO TRVb cells adhere to glass without additional support.
- Seed 1 x 106 cells per well (for CHO TRVb). Grow cells in Ham’s F12 medium supplemented with 5% FBS. Do not include antibiotics in the media.
- For other cell lines, seed at a density that will result in 80-90% confluency after 24 h.
- Incubate cells overnight in a humidified tissue culture incubator (37 °C with 5% CO2).
- Day 2: Prepare the transfection mixture
- Dilute vector DNA into Opti-MEM:
- For a 6-well plate, use 2 µg DNA in 100 μl of Opti-MEM per well.
- The DNA quality is paramount for a high transfection efficiency. Quantify DNA quality by UV spectroscopy and verify that the 260/280 nm ratio falls between 1.8 and 2.0.
- Keep the volume of DNA to less than 10% of the total volume in the transfection mixture.
- Dilute Lipofectamine 2000 into Opti-MEM and incubate for 5 min: For a 6-well plate, use 4 µg Lipofectamine2000 in 100 μl of Opti-MEM per well.
- Combine equal volumes of the Lipofectamine2000 and DNA solutions and mix by pipetting. The final ratio of DNA to Lipofectamine2000 will be 1:2. Incubate the mixture for at least 30 min at room temperature.
- Dilute vector DNA into Opti-MEM:
- Transfect cells:
- Aspirate media from cells.
- Add Opti-MEM to wells. For 6-well dishes, use 800 μl per well.
- Add 200 μl of the transfection mixture to each well (1:5 dilution). The total volume of fluid in each well should be 1 ml.
- Gently mix the solution by pipetting up and down or by rocking the plate.
- Incubate cells in the transfection mixture for 4 h in a tissue culture incubator (37 °C, 5% CO2).
- This time frame is recommended for transfecting CHO TRVb cells.
- Monitor cells by viewing on a TC microscope. Cell size and shape should appear unchanged during transfection. If cells contract or detach, then adjust the transfection conditions.
- Aspirate to remove the transfection media and wash once with complete media (e.g., media with FBS).
- Add complete media and grow in a tissue culture incubator (37 °C, 5% CO2) for at least 24 h. Cells will be ready for use between 24 and 48 h after transfection.
- Labeling VIPER-tagged receptors with Qdots on ITO coverslips
Protocol B describes a method for labeling a transmembrane receptor (TfR1-CoilE) with a biotinylated probe peptide (CoilR-biotin). Biotinylated receptor is subsequently detected with a streptavidin-Qdot655 conjugate. The biotinylated receptor could be labeled with other streptavidin conjugates, such as streptavidin-gold. We counter-stained with fluorescent transferrin (Tf-AF488), the ligand of TfR1, to enable the rapid identification of transfected cells. Lastly, live cells were cooled to 4 °C to pause endocytosis during labeling of the cell-surface receptor.- Day 3: Visually inspect each well on a tissue culture microscope to confirm that transfected cells are adherent, healthy, and 80-90% confluent.
- Remove media and add 500 μl of Live Cell Block Solution (Recipe 1) to each well. Return to the tissue culture incubator for 30 min.
- Use a pipette or transfer bulb pipette to aspirate media from wells, taking care to always leave enough media to keep the cells and coverslips hydrated.
- While the cells are in Live Cell Block Solution, prepare the CoilR labeling solution.
- Dilute CoilR-biotin probe peptide and 50 μg/ml Tf-AF488 into pre-chilled Ham’s F12 media without serum. Prepare 500 μl of the labeling solution per well.
- We recommend testing a range of probe peptide concentrations to obtain optimal signal to noise (e.g., 100 nM to 500 nM). For CHO TRVb cells expressing TfR1-CoilE, we recommend 100 nM CoilR-biotin in Ham’s F12 media.
- Other labeling reagents can be added at this time.
- Remove Block Solution 1 from each well.
- Add 500 μl of the labeling solution to each well. Label cells for 30 min at 4 °C, protected from light.
- Labeling is done at 4 °C in pre-chilled media to restrict endocytosis of VIPER-labeled receptors. Accessibility of the receptors on the cell surface will be especially critical for the subsequent SEM detection of CoilR-biotin with streptavidin-Qdots.
- Wash each well three times with ice-cold DPBS.
- Fix the cells using ice-cold 4% v/v PFA in DPBS. Incubate cells in fixative for 15 min at 4 °C.
- Wash each well twice with DPBS to remove fixative.
- Block the ITO coverslips with 800 μl Qdot Block Solution (Recipe 2) for 1 h at room temperature.
- While cells are in Qdot Block Solution, prepare the Qdot labeling solution:
- The Qdots are supplied by Invitrogen as a 1 μM stock solution. Before using, centrifuge (17,000 x g, 5 min) the streptavidin-Qdot655 conjugate to remove aggregated Qdots from solution.
- A range of concentrations should be tested to ensure optimal labeling.
- We used 10 nM streptavidin-Qdot655 in Qdot Labeling Solution (Recipe 3) for CHO TRVb transfected with TfR1-CoilE.
- Prepare 500 μl of labeling solution per well (e.g., 3 ml of solution for 6 samples).
- Remove Qdot Block Solution from each well.
- Add 500 μl of the streptavidin-Qdot655 labeling solution to each well and label for 1 h at room temperature. Protect samples from light during this step.
- After Qdot labeling, wash three times with DPBS.
- Image and map ITO coverslips by confocal FM (e.g., with an FEI CorrSightTM) before processing samples for CLEM (Procedures C and D).
- Mounting ITO coverslips for CLEM imaging
This procedure describes the methods for handling ITO coverslips prepared in Procedure B. This procedure uses a custom-machined aluminum slide to hold the ITO coverslips during imaging. This slide was created by Ingo Gestmann (FEI). The dimensions of the slide are 76 x 26 x 1.5 mm. The hole is 12 mm in diameter, which is compatible with the 22 x 22 mm ITO coverslips. There are likely commercially-available coverslip holders, but we have not tested any. The custom-machined aluminum slide (Figure 2) has a piece of coverglass glued with epoxy to one side (Side B). The other side remains open, with a circular chamber (Side A).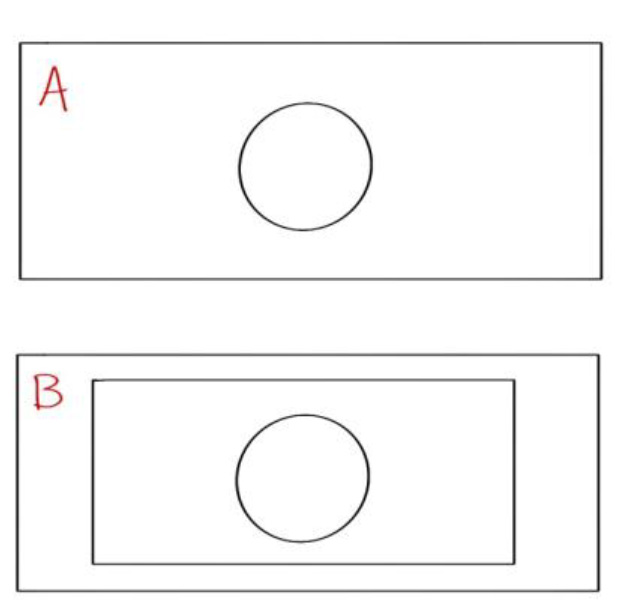
Figure 2. Diagram of the aluminum coverslip holder. Side A shows a circular hole. Side B has a rectangular piece of coverglass glued to cover the circular hole. This set-up allows the user to create an enclosed chamber of buffer by taping an ITO coverslip to side A.- Using a diamond-tipped scribe, scratch a small “F” into the center of the ITO coverslip to create a fiducial marker. Make strokes roughly 1 cm in length. A larger fiducial generates better correlation later on during SEM, but it should still be fully visible through the hole in the aluminum slide (see Figure 3). The coverslip can be stabilized during this process by holding it down with fine point tweezers.
- For multiple samples, use the scribe to make additional marks to enable quick differentiation of your samples. An example scheme would be:
- Sample 1: “F” only; no notch,
- Sample 2: notch below “F”,
- Sample 3: notch above “F”,
- Sample 4: notch to the left of “F”,
- Sample 5: notch to the right of “F”,
- Sample 6: notch below and above “F”.

Figure 3. An “F” carved into the center of a 22 x 22 mm ITO coverslip. Note the size of the “F” relative to the size of the coverslip. - Place the aluminum slide on the laboratory bench with side A facing up.
- Pipette DPBS into the circular chamber drop-wise until the chamber is full. Add one more drop to create a raised meniscus (Figure 4).
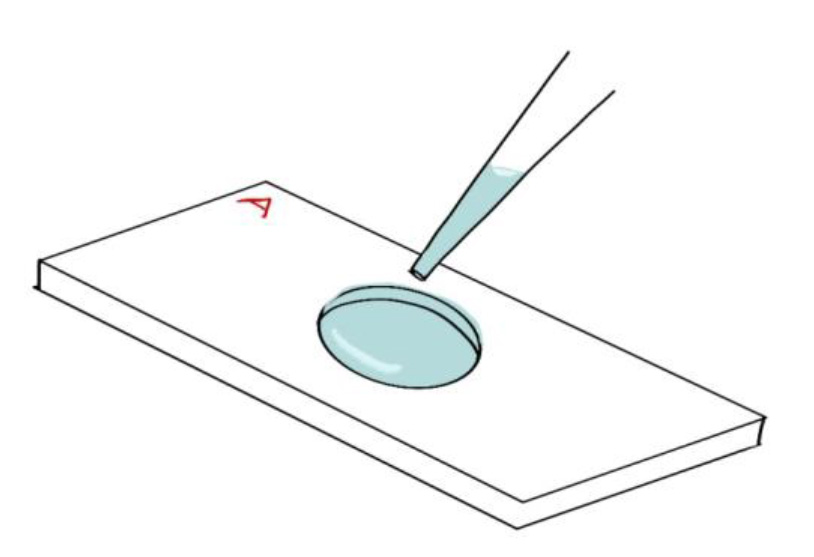
Figure 4. The aluminum coverslip holder with the circular chamber filled with buffer - Remove the labeled ITO coverslip from the 6-well plate using fine point tweezers.
- Allow the excess DPBS to drip off but do not let the coverslip dry out. The ITO coverslip should be wet for the next step.
- Position one edge of the wet ITO coverslip against the aluminum coverslip holder with the cells facing down, towards the surface of Side A (Figure 5).
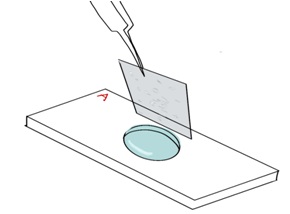
Figure 5. Positioning the ITO coverslip on the aluminum coverslip holder. The side of the ITO coverslip with the adhered cells is positioned facing towards the chamber. - Use tweezers to carefully lower the ITO coverslip onto Side A. The ITO coverslip should be floating on a thin layer of fluid (Figure 6). Do this quickly to avoid trapping air in the chamber.
- There should be no bubbles in the chamber between the ITO coverslip and the glass on Side B of the slide. Air bubbles will dry out the cells. Small bubbles are permissible if they do not contact the ITO coverslip when the slide is inverted.
- If a large air bubble is trapped under the ITO coverslip, use tweezers to lift the ITO coverslip at a shallow angle before dropping the ITO coverslip onto the DPBS again.
- The coverslip can also be nudged gently off of the slide with tweezers to free the bubble from the side of the coverslip.
- The position of the coverslip can be adjusted at this point as long as it is free-floating.
- Make sure you can see the entire fiducial marker (“F”) through the hole of the chamber.
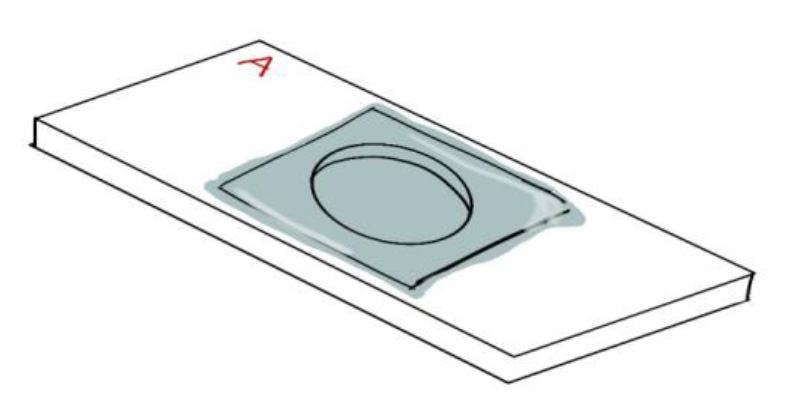
Figure 6. The ITO coverslip floating on a thin layer of DPBS over the circular chamber on Side A of the aluminum coverslip holder - Blot excess fluid from the slide. Fold a Kimwipe in half and touch the folded edge to the side of the coverslip to blot excess fluid (Figure 7). Repeat for all four sides until dry. If there is fluid on top of the coverslip you can lay a piece of tissue on top of the coverslip to blot the remainder. Only do this step after blotting the sides.
- This will “seal” the coverslip to the slide. Moving or re-positioning the ITO coverslip at this point could damage the cells.
- If there is a bubble in the circular chamber or the ITO coverslip is poorly positioned after this step, DPBS can be added gently to the sides of the coverslip. Wait for the DPBS to flow underneath the coverslip and then lift it off the slide.
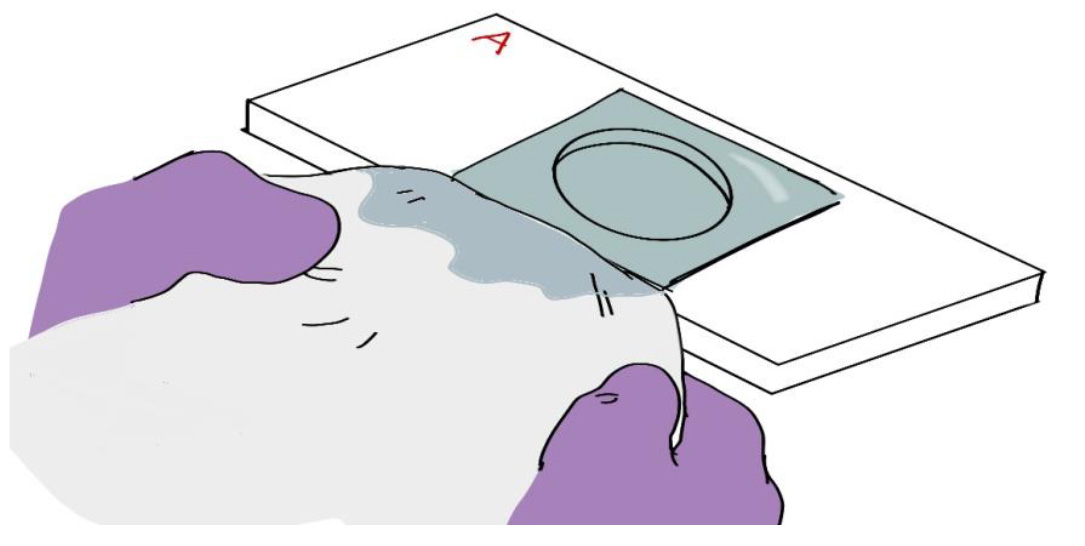
Figure 7. Removing excess DPBS from the aluminum coverslip holder by blotting with a Kimwipe - Tear a strip of clear tape (e.g., 3M ScotchTM tape) in half and secure the coverslip to the slide by taping at the sides. Repeat for all sides (Figure 8).
Note: Avoid applying an excess of tape because it will need to be removed after fluorescence imaging.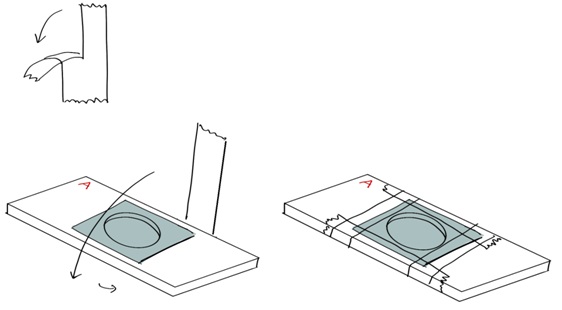
Figure 8. Attaching the coverslip to the aluminum coverslip holder using clear tape - Repeat this procedure for each ITO coverslip.
- Image the prepared samples on an inverted confocal fluorescence microscope. We imaged an FEI CorrSightTM in the following Procedure D.
Note: We recommend imaging soon after preparing the samples because some evaporation may occur.
- Using a diamond-tipped scribe, scratch a small “F” into the center of the ITO coverslip to create a fiducial marker. Make strokes roughly 1 cm in length. A larger fiducial generates better correlation later on during SEM, but it should still be fully visible through the hole in the aluminum slide (see Figure 3). The coverslip can be stabilized during this process by holding it down with fine point tweezers.
- Fluorescence imaging and mapping cells on an FEI CorrSightTM System
This procedure describes using the FEI CorrSightTM imaging system to collect micrographs of labeled cells. Samples prepared in Procedures A-C are first imaged by confocal FM to identify fluorescent cells. Confocal FM is used to image VIPER fluorescence (Qdot655) and Tf-AF488 on the cell surface. During FM imaging, the FEI MAPS software enables the user to select and “map” cells for subsequent high-resolution imaging by scanning EM (SEM). Figure 9 provides an overview of the FEI MAPS software.
Note: Once the slide is placed on the stage and imaging commences, the slide must remain in place until it is fully mapped. Even small adjustment to the slide placement will shift the slide and make the map inaccurate, which makes it difficult to re-locate cells for SEM imaging.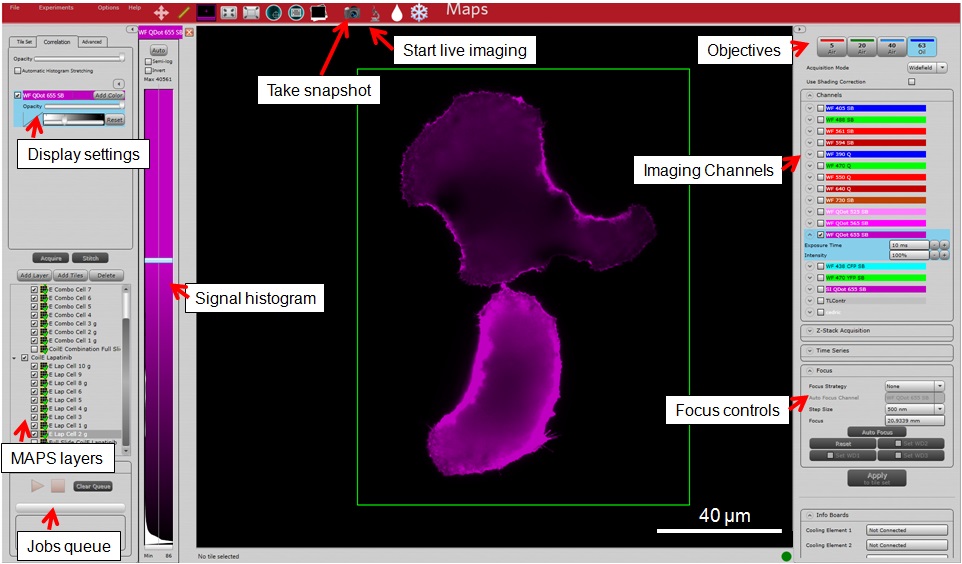
Figure 9. Annotated screen capture of the FEI MAPS 2.1.38 software- Place immersion oil on the 63x objective lens. Then switch to the 5x objective lens. This can be controlled in the panel labeled “Objectives”. Refer to Figure 9 for software controls from this point on. Additional information can be found in the FEI CorrSightTM user manual (Reference 9).
Note: This is a critical step. Removing the slide after 5x imaging to load oil on the 63x objective will offset the mapping. This will make re-locating cells difficult by SEM. - Place the aluminum coverslip holder on the microscope stage with the ITO coverslip (Side A) facing downwards towards the objective.
- Turn off all the fluorescence channels using the panel labeled “Imaging Channels” by unchecking all the boxes in that panel. Image the sample using transmitted light.
- In the panel labeled “Imaging Channels,” turn on the “TLControl” (transmitted light) channel by checking its box.
- In the panel labeled “Focus Controls,” set the focus to 20 mm.
- Start live imaging by clicking the button labeled “Live Imaging”.
- Adjust the focus with scroll wheel of the mouse until the cells and the fiducial marker (“F”) are in focus. You can adjust the step size of the focusing in the panel labeled “Focus controls.”
Note: The focal plane will vary across the coverslip. Therefore, it is important is to keep the fiducial “F” in focus as you move the stage.
- Drive the stage to the edge of the circular hole in the aluminum coverslip holder.
- To do this, left click on the live image where you want the stage to move.
- Once you have located an edge, take a snapshot (Figure 10) by clicking the button labeled “Take snapshot”. The “Take snapshot” function loads the data into an aggregate layer called “PreviewImages” in the layers panel.
Note: This is not a viable place to store image data. However, these preview snapshots allow you to reference the edges of the circular chamber. These boundaries are needed in order to draw the tile-set that will capture the whole ITO coverslip. - Continue taking snapshots until you can determine of the boundaries of the circular chamber. This can generally be done with 4 snapshots placed at the top, bottom, left, and right of the circle.

Figure 10. A snapshot of the ITO coverslip (light gray) showing the boundary of the circular chamber in the aluminum coverslip holder (black)
- Left click and drag over the slide hole (using the snapshots of the hole boundaries to guide you) and right-click “Add tiles here”. This will create a tile array to image the entire slide, capturing the fiducial F.
- Execute the acquisition in the “Jobs queue” panel to generate a stitched tile image of the coverslip by transmitted light (Figure 11).

Figure 11. A stitched tile image showing the ITO coverslip (light gray) mounted on the aluminum coverslip holder (black). Note that the entire “F” fiducial, denoted by a blue arrowhead, was captured and in focus. Small air bubbles can also be seen, with a few denoted by a red arrowhead. - Prepare to image cells by FM.
- Check box on the “WF 488 SB” channel in the “Imaging Channels” panel. This setting excites Tf-AF488 at 488 nm and collects emission through a 525/50 nm filter.
- Check box on the “WF Qdot 655 SB” channel. This setting excites Qdot655 at 405 nm and collects emission through a 690/50 nm filter.
- You can modify the intensity of each channel in the “Imaging Channels” panel by clicking on the channel name. Leave the intensity at 100% and set the exposure time appropriately by imaging live in these channels.
- “Good” acquisition settings generate signal that occupies more than 50% of the dynamic range of the 16-bit detector, without saturated signal in any pixels. This can be observed in the panel labeled “Signal histogram.”
- The settings may require some adjustment after switching to the 63x objective lens.
- Cells can now be imaged and mapped at 63x magnification.
- During image acquisition use the 5x objective to locate cells by low magnification. Then switch to the 63x objective oil lens to collect high-resolution images. When a cell is “mapped”, the fluorescence image is acquired and the image is automatically saved with the coordinate location.
- We recommend selecting cells for imaging based on their Tf-AF488 signal to avoid biasing cell selection towards the brightest Qdot655 signal (i.e., the brightest VIPER-labeled cells).
- We recommend taking 5x fluorescence images of the slide. These images are helpful for locating the mapped cells by SEM. This is especially true for samples with high cell density, where finding individual cells based on size and shape alone is difficult.
- Repeated switching between 5x and 63x can create air bubbles in the immersion oil. These do not affect imaging at 63x, but the bubbles will appear in the 5x images.
- To map cells, locate a desired cell at 63x magnification using the live-imaging function. Left click and drag a single tile over the cell location and right-click “Add tiles here”. This will create a tile to image the cell and register its location.
- Execute the acquisition in the “Jobs queue panel”.
Note: The FEI MAPS acquisition software will remember the absolute position of cells imaged at 63x relative to the full size of the coverslip mapped at 5x magnification. - Create a new layer for each cell. You can group samples by creating Layer groups. For each layer, make the names descriptive (e.g., Cell 1 VIPER Qdot slide A). The names will become the folder names where data is stored.
Note: All of the individual images collected by the FEI MAPS software are automatically named “Tile_000_0001”. Therefore, descriptive layer names are important. - After imaging and mapping, remove the slide from the FEI CorrSightTM microscope.
- Clean the slide thoroughly with lens paper to remove immersion oil. Then clean with lens paper using 70% ethanol.
- Remove the tape gently and return the ITO coverslip to DPBS in a 6-well plate for further processing (Figure 12).
- The ITO coverslip should be oriented with cells facing upwards in the 6-well plate.
- The tape can be scored with a razor blade to help remove it from the aluminum slide.
- If the sample is “stuck” on the aluminum slide after tape removal, apply a few drops of DPBS around the edges of the slide and wait for the fluid to move under the coverslip, lifting it from the slide. Forcing the coverslip off with tweezers can break the coverslip or damage the cells by sliding them against the aluminum slide.
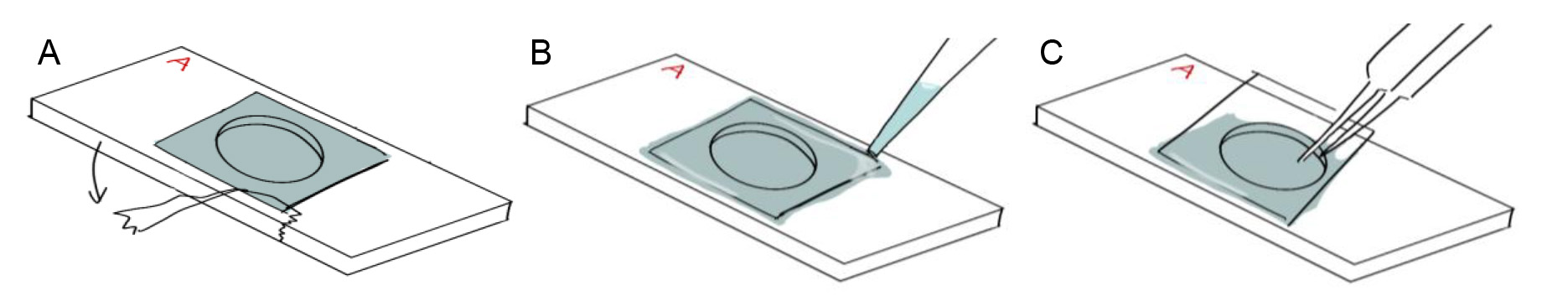
Figure 12. Removing the ITO coverslip on the aluminum coverslip holder. A. Remove the tape by pulling towards the side, instead of by pulling up, to minimize force applied to coverslip. B. Add fluid to the edges of the ITO coverslip and wait for it to penetrate underneath the coverslip. C. Use fine point tweezers to gently lift the coverslip off from the edges.
- Place immersion oil on the 63x objective lens. Then switch to the 5x objective lens. This can be controlled in the panel labeled “Objectives”. Refer to Figure 9 for software controls from this point on. Additional information can be found in the FEI CorrSightTM user manual (Reference 9).
- Processing ITO coverslips for SEM imaging
This procedure describes the methods used to dehydrate samples for SEM imaging on the Helios NanolabTM 660. The methods continue from the last step of Procedure D, after samples have been imaged by FM and ITO coverslips are placed in a 6-well plate.- Prepare solutions of ethanol that will be used to dehydrate samples.
- Dilutions should be prepared with anhydrous ethanol and sterile DI water.
- Prepare 10-15 ml stocks of: 25%, 50%, 75%, and 90% (v/v) ethanol. Undiluted (“100% ethanol”) will also be used for dehydrating samples.
Note: For SEM imaging, it is critical to fully remove water from the cells when dehydrating because residual water can cause cell breakage and deformation (Figure 13). Using a fresh bottle of anhydrous ethanol every time minimizes this risk.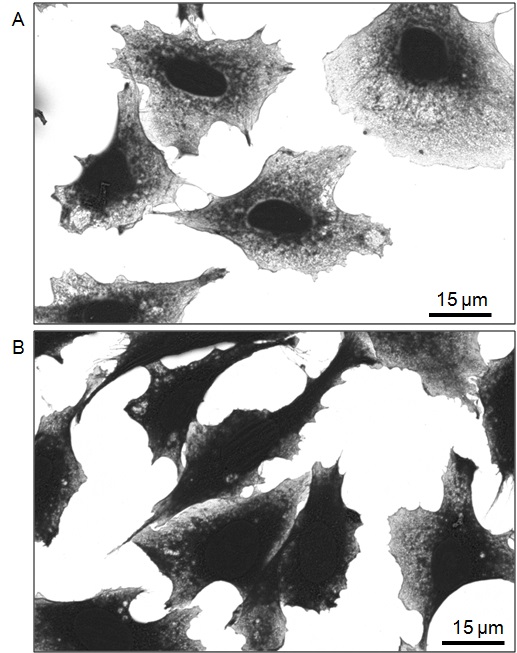
Figure 13. Micrographs of dehydrated CHO TRVb cells processed for SEM imaging. SEM micrographs were acquired in backscattered mode on an FEI Helios NanolabTM 660 SEM at 3,500x magnification (horizontal field width: 119 μm). The ITO coverslip appears bright white in this acquisition mode. A. Micrographs of damaged cells that were dehydrated with an ethanol gradient prepared using an old bottle of ethanol (opened and reused over several months). Cells are light gray and have a lace-like appearance with pronounced nuclei. The white ITO substrate can be seen from behind cells that are damaged, resulting in cells that are light gray around the nucleus. B. Micrographs of well-preserved cells that were dehydrated with an ethanol gradient prepared using a new bottle of anhydrous ethanol. In contrast to cells in A, these cells are intact, appear dark gray, and are more raised.
- Withdraw most of the DPBS from the ITO coverslips using a micropipette or transfer pipette. Leave a small layer of fluid over the cells. If the samples dry out in air, the cells may break or appear damaged by SEM. To minimize this risk, never let the ITO coverslips go dry. This is especially important when using high percentages of ethanol, which evaporates quickly.
- Wash the ITO coverslips with DI water (1.5 ml/well) to remove salts. Wash 5 min at room temperature with gentle agitation.
Note: Gentle agitation can be achieved by hand or on a rocking stage. - Withdraw most of the water from the wells, again working to ensure that the coverslips remain wet.
- Add 25% (v/v) ethanol (1.5 ml/well) to the slides to start the dehydration gradient. Incubate for 5 min at room temperature with gentle agitation.
- Withdraw most of most of the 25% (v/v) ethanol from the wells, leaving enough solution so that the slides stay covered.
- Repeat Steps E5 and E6 with: 50%, 75%, 90%, and 100% ethanol.
Note: This ethanol step-gradient dehydrates the cells slowly to minimize cell shrinking and structural damage during dehydration. - Incubate coverslips a second time in 100% ethanol to ensure that all water is removed from the samples.
- Coverslips are now ready to be loaded into the critical point dryer.
- Use a critical point dryer (Leica EM CPD300) to dehydrate samples.
- Open the sample chamber and load the ITO coverslips.
- Using fine point tweezers, transfer the first layer of coverslips with the cells facing up.
- ITO coverslips can be stacked on top of each other through the use of stainless steel crinkle washers. Place a washer gently on top of the ITO coverslip, and then add the next coverslip (Figure 14). The washers are large enough that the cells of interest should be in the hole of the washer and not in contact with the washer itself. A total of 4 layers of coverslips can fit inside the sample chamber.
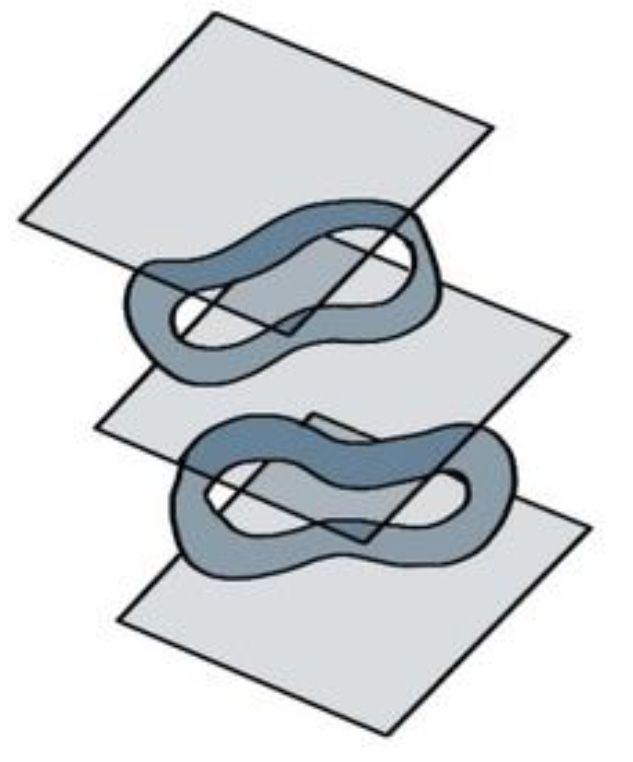
Figure 14. Using stainless steel crinkle washers to separate ITO coverslips during dehydration. The washers prevent the coverslips from adhering to each other during the final dehydration steps.
- Use the critical point dryer to dehydrate the samples. On the Leica system, we programmed the following dehydration conditions:
Stirring: 50%
Automatic exchanging: on,
Speed of CO2 injection: slow,
Fillers: 1 stage (fillers are solid plastic pieces that are placed in the chamber to take up volume and minimize the drying volume),
Delay of CO2 injection: 120 s,
Speed of CO2 exchange: 1 (out of 10),
Number of CO2 exchange cycles: 25,
Heating for CO2 gassing out: slow,
Speed of CO2 gassing out: slow.
- Open the sample chamber and load the ITO coverslips.
- Dried samples are now ready for mounting to pin stub specimen mounts (Procedure F).
- Prepare solutions of ethanol that will be used to dehydrate samples.
- Mounting coverslips and carbon-coating for SEM imaging
This procedure describes methods used to mount dehydrated ITO coverslips on SEM pin stub specimen mounts (Ted Pella), referred to herein as “SEM mounting pins” or “pins”. It also describes the method used to carbon coat the samples. This procedure starts with dehydrated samples prepared in Procedure E.- Brush on a small amount of conductive silver paint around the edges of an SEM mounting pin.
- Apply the paint near to where the contours of the ITO coverslip will be when it is placed on the pin.
- The silver paint must fully off-gas and dry before it is placed under vacuum in the coating machine. Gluing coverslips by the edges rather than the center makes the drying process go faster in Step F3.
- The SEM mounting pin can be labeled with the sample identity by writing on the underside.
- While the silver paint is wet, place the ITO coverslip with the cells facing up on the SEM mounting pin using fine point tweezers (Figure 15).
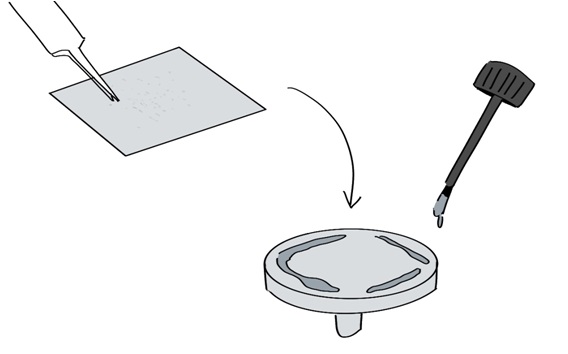
Figure 15. Gluing the ITO coverslip to the SEM mounting pin using conductive silver paint. The paint is applied sparsely to the edges of the SEM mounting pin. - Store the pin-mounted ITO coverslips under desiccation and dry the silver paint overnight.
Note: We store pin-mounted coverslips in a desiccator cabinet with desiccant (i.e., DrieriteTM). - After the silver paint has dried, transfer samples to a high vacuum flash coating machine (Leica EM ACE600):
- Vent the chamber of the coating apparatus. When the chamber reaches atmospheric pressure (100 mbar), open the door.
- Place samples inside the chamber by seating the pins in the carousel holder. Do not overlap samples. Close the door.
- Load the carbon thread into the coating unit above the sample chamber.
- Place the coating unit back in the coating machine.
- Coat the samples under vacuum:
- Run program “Pulse sgh coater”. Use these settings:
Coat thickness: 10 nm,
Sample height: 3 nm,
Tilt: 0 degrees.
- Run program “Pulse sgh coater”. Use these settings:
- Once the carbon coating is finished and the sample chamber is vented, the samples can be removed and are ready for imaging by EM (Procedure G).
Note: SEM mounting pins can be reused by gently prying off the ITO coverslip and washing the pins with 100% ethanol. Let pins fully dry before adding silver paint.
- Brush on a small amount of conductive silver paint around the edges of an SEM mounting pin.
- Operating the FEI Helios NanolabTM 660 SEM instrument for SEM imaging
This procedure uses samples that were prepared through the end of Procedure F. These samples should contain cells on ITO coverslips that were mapped, dehydrated, glued to SEM mounting pins, and carbon coated.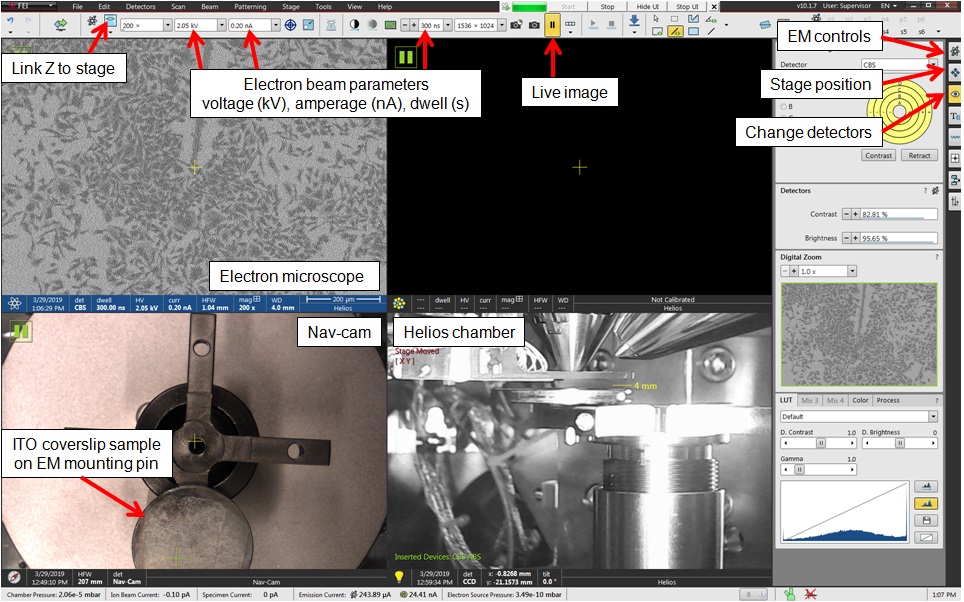
Figure 16. Annotated screen capture of the FEI Helios NanolabTM xT Microscope Control software (version 10.1.7)- Refer to Figure 16 for an annotated screen capture of the software controls. Additional information can be found in the FEI Helios NanolabTM 660 user manual (Reference 10).
- Vent the sample chamber of the FEI Helios NanolabTM 660 SEM. A button labeled “Vent” is found in the panel labeled “EM controls.”
- Once it is fully vented the chamber can be opened and the samples can be loaded onto the stage (up to 2 due to the size of the pins used). You will know when the chamber is fully vented when the SEM makes a hissing sound. You will also not be able to physically open the chamber door until it is fully vented.
- Fasten the samples to the sample holder using the screw driver.
- Swing out the camera head (the “Nav-cam”) and take a photo of the stage with the samples loaded. This is done by pressing the single button on the camera head.
- Retract the Nav-cam head and close the chamber.
- Pump down the chamber to reach optimal vacuum. This is done by pressing the button labeled “Pump” found in “EM controls”.
- Open the FEI MAPS 3.0 software and load the MAPS files from the previous FEI CorrSightTM mapping session.
- Prepare for operation of the SEM. The SEM is set to acquire on the Everhart-Thornley Detector (ETD) by default.
- Once the chamber is under vacuum, turn on the electron beam in “EM controls”. Set the beam voltage to 3.0 kV and 0.2 nA. These are conditions that balance sample preservation with image quality.
- Using the Nav-cam photograph of the stage (bottom left, Figure 16), drive the microscope to the sample by double left-clicking on the sample.
- Zoom in on a feature at > 2,000x magnification and focus the microscope using the controller dashboard attached to the SEM. Click “Link Z to stage” in the software.
- Set the stage height to 4 mm in the panel labeled “Stage position” in preparation for Step G7.
- Switch to the circular backscatter (CBS) detector in the panel labeled “Change detectors”. The live camera view of the chamber should show a metal detector arm (the CBS detector) swing under the electron gun.
Note: Inappropriate stage height can damage your sample and the detector arm (see Step G6d). - Use the Alignment Wizard to globally align the SEM to the coverslip light image collected in Procedure D (Figure 11 and Figure 17).
- Left click on “Global Alignment” in the FEI MAPS software.
- Drive the SEM to different landmarks of the “F” fiducial marker.
- Take snapshots by right-clicking the center marker in the FEI MAPS software and select “Snapshot here”. The snapshots will load into FEI MAPS.
- Use the global alignment panel to match the features of the fiducial “F” by SEM to the “F” on the transmitted light image of the ITO coverslip.
- Run the alignment command. If done correctly, the SEM images collected should now align perfectly with the “F” on the transmitted light image collected in Procedure D (Figure 18).
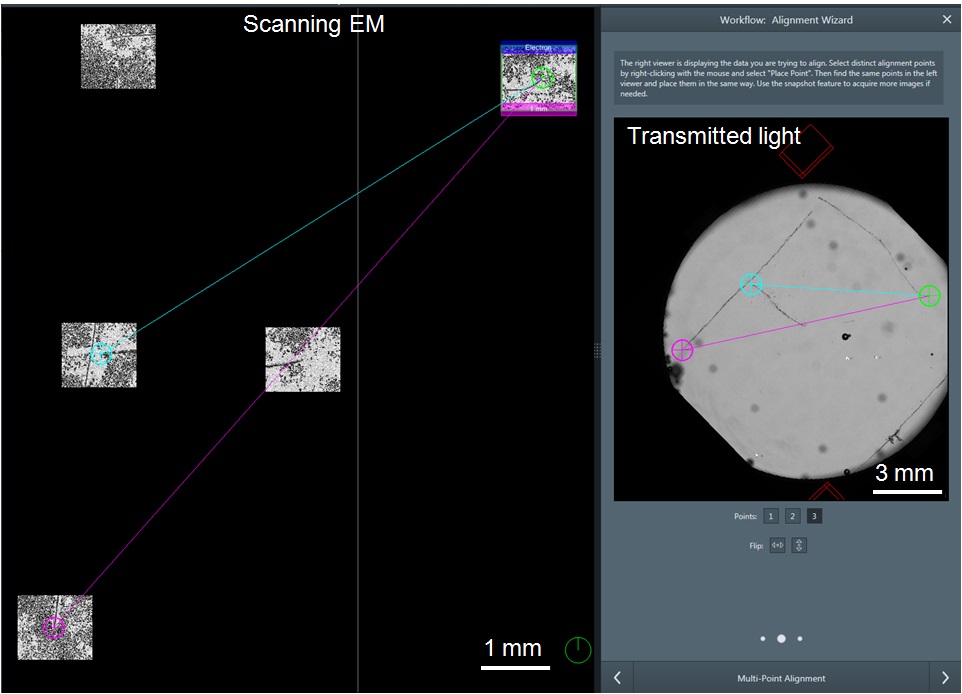
Figure 17. Aligning the FEI Helios NanolabTM 660 SEM stage to the map generated by the FEI CorrSightTM using the Global Alignment tool. Backscatter SEM snapshots of the fiducial marker “F” are aligned to the transmitted light image of the same “F” using a 3-point alignment. The user manually matches three landmarks on the “F” between the SEM and transmitted light image. The location marked by a green circle in the SEM is the same location as the green circle in the transmitted light view. The same process is repeated for the blue and pink circles.
Figure 18. The FEI Helios NanolabTM 660 SEM snapshots of the fiducial marker “F” aligned with the transmitted light image from the FEI CorrSightTM. After SEM image alignment with light-based images generated by the FEI CorrSightTM, the FEI Helios NanolabTM 600 SEM is now aligned with the FEI MAPS software. The user can now select anywhere on the micrographs collected by FM to drive the SEM stage to approximately the same location. Then the same cell can be imaged at high magnification by SEM.
- After completing the global alignment, any cell from a prior fluorescent image can be selected in FEI MAPS and the SEM will drive to the location of the cell for high magnification SEM imaging.
- Due to the difficulty of aligning high magnification images, the cell will likely be off from the position selected by 10-100 microns.
- Use the shape of the cells or the 5x fluorescent images to locate the exact cell you want to image by SEM.
- Image at 3,500x magnification (horizontal field width: 119 μm) to capture the entire cell in a single field-of-view.
- Similar to FEI CorrSightTM operation, create a new layer for each cell and each set of images. Samples can be grouped by creating Layer groups. Make layer names descriptive because they will be the folder names where your data is kept. All of the individual images collected by the FEI MAPS software are automatically named “Tile_000_0001” so descriptive layer names are important.
- Image at 65,000x (horizontal field width: 6.37 μm) or 100,000x (horizontal field width: 4.14 μm) magnification to capture the cell surface and to resolve individual Qdots.
- The electron beam is destructive and will ablate cells (i.e., damage cells and introduce holes). While adjusting the imaging conditions (i.e., focus, stigmatism), examine an area adjacent to the area containing the cell that will be imaged. This will help preserve cell morphology for SEM imaging at 65,000x or higher magnification. See Figure 19 for micrographs acquired at 3,500x, 63,000x, and 100,000x magnifications.
- Optionally, quantify the Qdots in the SEM micrographs as described in “Data analysis.”
- Acquire multiple images per cell, and multiple cells per condition. We recommend at least 2 images per cell and at least 3 cells.
- Transient transfection produces cell-cell variability in protein expression. Therefore, it is important to include replicates and image multiple cells to compensate for this variability.
- For quantification of Qdots in SEM micrographs, we imaged 6 cells per condition with 2 images per cell, resulting in 12 images per condition.

Figure 19. SEM micrographs of cells imaged at 3,500x (horizontal field width: 118 µm), 63,000x (horizontal field width: 6.38 µm) and 100,000x magnification (horizontal field width: 4.14 µm). 5x digital magnification of each capture is shown in the right column, highlighting the area outlined by a green box in the corresponding micrograph. Qdots are visible in the high-magnification micrographs as light gray circular particles.
Data analysis
Quantification of particles in SEM images
VIPER labeling with Qdots is stoichiometric, meaning that there will be one Qdot per VIPER tag. This feature enables labeled proteins to be quantified by algorithmic segmentation and counting of the Qdots. We recommend acquiring SEM images at high magnification (65,000x or 100,000x) for optimal particle detection and segmentation.
We developed a custom pipeline in Matlab (ver. R2017b) for automated detection and quantitation of particles in backscattered SEM images. We first detected bright objects of interest on a dark background by using morphological top-hat filtering. The object segmentation was performed using a succession of mathematical morphology operations. Briefly, automatic intensity thresholding was performed to detect Qdots. Clustered objects were separated using a seeded watershed transformation from the ultimate eroded results. Next, we counted the segmented single particles (n = 1), dimers (n = 2), and multimers (n > 2). A representative micrograph before and after segmentation is provided in Figure 20.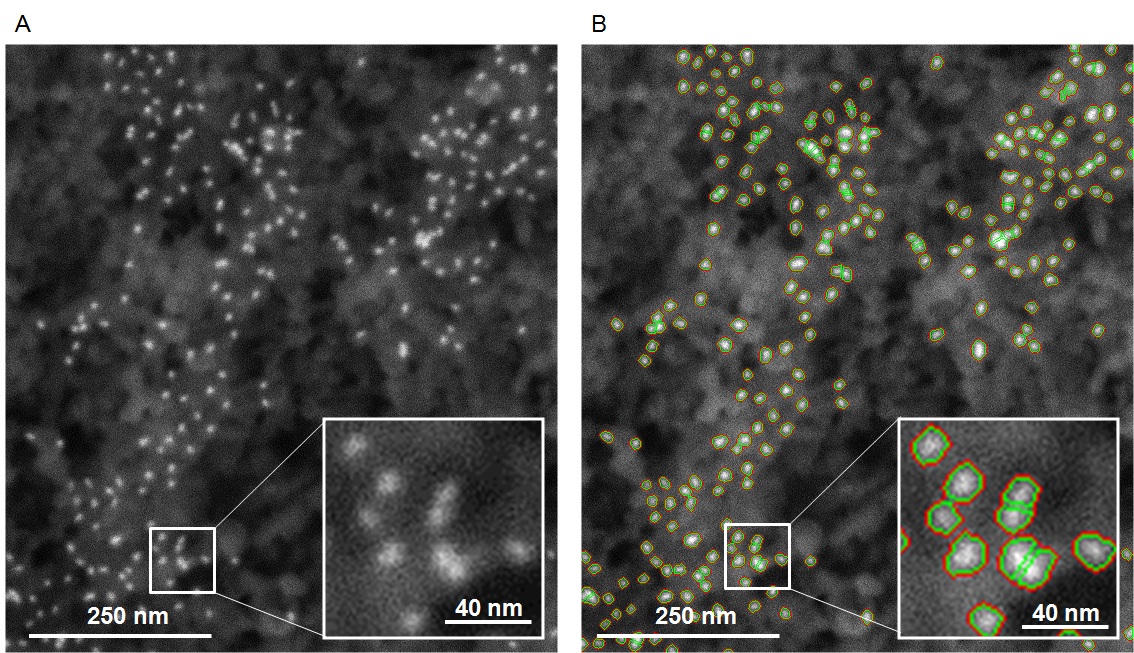
Figure 20. Algorithmic segmentation and automated counting of Qdots on the cell surface. A. The original, unprocessed SEM micrograph acquired at 100,000x magnification is shown. Qdots appear as bright white dots on the cell surface, which appears dark gray. B. The micrograph in A was processed in Matlab to generate a counting mask. The red mask (red outline) indicates the objects detected by morphological top-hat filtering. The green mask (green outline) indicates the objects detected and separated using watershed transformation. Insets show a 3x magnification of the region indicated.
After the automated image processing, we visually inspected the segmentation to refine parameters and exclude objects falsely annotated as Qdots. False-annotations were rare, but typically result from irregular background, intensity variations, background artifacts, or errors in segmentation overlooked by the automated procedure described above.
The parameter sets were optimized to quantify Qdot655 in SEM micrographs acquired at 65,000x-100,000x magnification on an FEI Helios NanolabTM 660 SEM instrument using a CBS detector. These parameters may need to be adjusted to identify particles depending on the image quality, resolution, or type of particle (e.g., 10 nM gold or Qdot555). The “VIPER_object_detection.m" code requires two input parameters related to: (i) intensity (i.e., “int_threshold” in VIPER_object_detection.m) and (ii) object size (“obj_size”, “obj_size_filter” in VIPER_object_detection.m). The intensity-related parameter should be changed if there are variations in intensity across the images. The object size parameter (“obj_size”) will need to be adjusted if the detection particles are a different physical size (e.g., large gold particles) or if the resolution varies (i.e., pixel size). Also, the code includes filtering options for removing small objects. The “obj_size_filter” can be adjusted to remove undersized annotations. These are the parameters that we used for our analysis of Qdot655 particles:
int_threshold = 60; % intensity parameter;
obj_size = 10; % object size parameter;
obj_size_filter = 5; % filter for small size object.
Here is a brief tutorial that describes how to run our custom analysis pipeline to detect and count particles (i.e., Qdots) and to count the segmented particles:
- Copy the file 'VIPER_object_detection.m' into the desired computer directory.
- Within the same computer directory, make a folder titled './img/'.
- Copy SEM images ('tiff' file format) under './img/'.
- Run 'VIPER_object_detection.m' in the command window in Matlab.
- The program will start segmentation and count the objects.
- The output file will provide the following information:
qdot_count.csv: number of objects (single Qdots, dimers, multimers, N_total ≥ N_1 + N_2 x 2 + N_more x 3),
[filename]-PTmask.mat: object mask,
[filename]-Overlay.png: overlaid image with boundaries (red: objects, green: separated objects).
Recipes
- Live Cell Block solution
10% (v/v) FBS, 6% (w/v) BSA in Ham’s F12 media - Qdot Block solution
10% FBS and 6% BSA in DPBS - Qdot Labeling solution
6% (w/v) BSA in DPBS
Acknowledgments
This work was funded by the OHSU School of Medicine and the National Institutes of Health (R01 GM122854). JKD was partially funded by the Portland Chapter of Achievement Rewards for College Scientists (ARCS). The protocols described herein were originally described in Doh et al. (2018). CHO TRVb cells were graciously provided by Prof. Timothy McGraw. EM and CLEM experiments were performed at the Multiscale Microscopy Core at OHSU and supported by the Advanced Multiscale Microscopy Shared Resource at the OHSU Knight Cancer Institute (NIH P30 CA069533). The aluminum coverslip holder was fabricated by Ingo Gestmann (FEI).
Competing interests
The authors declare no financial or non-financial competing interests. An international patent application is pending on the VIP technology (PCT/US17/60609).
References
- Baker, M. (2015). Reproducibility crisis: Blame it on the antibodies. Nature 521(7552): 274-276.
- Berglund, L., Bjorling, E., Oksvold, P., Fagerberg, L., Asplund, A., Szigyarto, C. A., Persson, A., Ottosson, J., Wernerus, H., Nilsson, P., Lundberg, E., Sivertsson, A., Navani, S., Wester, K., Kampf, C., Hober, S., Ponten, F. and Uhlen, M. (2008). A genecentric Human Protein Atlas for expression profiles based on antibodies. Mol Cell Proteomics 7(10): 2019-2027.
- Bordeaux, J., Welsh, A., Agarwal, S., Killiam, E., Baquero, M., Hanna, J., Anagnostou, V. and Rimm, D. (2010). Antibody validation. Biotechniques 48(3): 197-209.
- Bradbury, A. and Pluckthun, A.(2015). Reproducibility: Standardize antibodies used in research. Nature 518(7537): 27-29.
- Doh, J. K., Enns, C. A. and Beatty, K. E. (2019a). Implementing VIPER for imaging cellular proteins by fluorescence microscopy. Bio-protocol 9(21): e3413.
- Doh, J. K., Tobin, S. J. and Beatty, K. E. (2019b). Generation of CoilR probe peptides for VIPER-labeling of cellular proteins. Bio-protocol 9(21): e3412.
- Doh, J. K., White, J. D., Zane, H. K., Chang, Y. H., López, C. S., Enns, C. A. and Beatty, K. E. (2018). VIPER is a genetically encoded peptide tag for fluorescence and electron microscopy. Proc Natl Acad Sci U S A 115(51): 12961-12966.
- Ellisman, M. H., Deerinck, T. J., Shu, X. and Sosinsky, G. E.(2012). Coeerlative light and electron microscopy. In Methods in cell biology. Academic Press 111:139-155.
- FEI CorrSightTM user manual. FEI. (Accessed 12-Oct, 2019, at http://www.fei.co.jp/_documents/CorrSightDatasheet.pdf.)
- FEI Helios NanolabTM 660 user manual. FEI. (Accessed 12-Oct, 2019, at https://engineering.unl.edu/downloads/files/SOP-Helios660FIB-2019.pdf)
- Gaietta, G., Deerinck, T. J., Adams, S. R., Bouwer, J., Tour, O., Laird, D. W., Sosinsky, G. E., Tsien, R. Y. and Ellisman, M. H. (2002). Multicolor and electron microscopic imaging of connexin trafficking. Science 296(5567): 503-507.
- Giepmans, B. N. , Deerinck, T. J., Smarr, B. L., Jones, Y. Z. and Ellisman, M. H.(2005). Correlated light and electron microscopic imaging of multiple endogenous proteins using Quantum dots. Nat Meth 2(10): 743-749 .
- Hildebrand, D. G. C., Cicconet, M., Torres, R. M., Choi, W., Quan, T. M., Moon, J., Wetzel, A. W., Scott Champion, A., Graham, B. J., Randlett, O., Plummer, G. S., Portugues, R., Bianco, I. H., Saalfeld, S., Baden, A. D., Lillaney, K., Burns, R., Vogelstein, J. T., Schier, A. F., Lee, W. A., Jeong, W. K., Lichtman, J. W. and Engert, F. (2017). Whole-brain serial-section electron microscopy in larval zebrafish. Nature 545(7654): 345-349.
- Hinner, M. J. and Johnsson, K. (2010). How to obtain labeled proteins and what to do with them. Curr Opin Biotechnol 21(6): 766-776.
- Johnson, E., Seiradake, E., Jones, E. Y., Davis, I., Grunewald, K., Kaufmann, R. (2015). Correlative in-resin super-resolution and electron microscopy using standard fluorescent proteins. Sci Rep 5: 9583.
- Karreman, M. A., Mercier, L., Schieber, N. L., Solecki, G., Allio, G., Winkler, F., Ruthensteiner, B., Goetz, J. G. and Schwab, Y. (2016). Fast and precise targeting of single tumor cells in vivo by multimodal correlative microscopy. J Cell Sci 129(2): 444-456.
- Kim, D., Deerinck, T. J., Sigal, Y. M., Babcock, H. P., Ellisman, M. H. and Zhuang, X. (2015). Correlative stochastic optical reconstruction microscopy and electron microscopy. PLoS One 10(4): e0124581.
- Kremer, A., Lippens, S., Bartunkova, S., Asselbergh, B., Blanpain, C., Fendrych, M., Goossens, A., Holt, M., Janssens, S., Krols, M., Larsimont, J. C., Mc Guire, C., Nowack, M. K., Saelens, X., Schertel, A., Schepens, B., Slezak, M., Timmerman, V., Theunis, C., R, V. A. N. B., Visser, Y. and Guerin, C. J. (2015). Developing 3D SEM in a broad biological context. J Microsc 259(2): 80-96.
- Kuipers, J., van Ham, T. J., Kalicharan, R. D., Veenstra-Algra, A., Sjollema, K. A., Dijk, F., Schnell, U., Giepmans, B. N.(2015). FLIPPER, a combinatorial probe for correlated live imaging and electron microscopy, allows identification and quantitative analysis of various cells and organelles. Cell Tissue Res 360(1): 61-70.
- Laine, R. F., Kaminski Schierle, G. S., van de Linde, S. and Kaminski, C. F. (2016). From single-molecule spectroscopy to super-resolution imaging of the neuron: a review. Methods Appl Fluoresc 4(2): 022004.
- Lam, S. S., Martell, J. D., Kamer, K. J., Deerinck, T. J., Ellisman, M. H., Mootha, V. K. and Ting, A. Y. (2015). Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat Methods 12(1): 51-54.
- Lichtman, J. W., Livet, J. and Sanes, J. R. (2008). A technicolour approach to the connectome. Nat Rev Neurosci 9(6): 417-422.
- Lipofectamine® 2000 Reagent Protocol 2013. (2013). (Accessed 12-Oct, 2019, at tools.thermofisher.com/content/sfs/manuals/Lipofectamine_2000_Reag_protocol.pdf
.) - Liss, V., Barlag, B., Nietschke, M. and Hensel, M. (2015). Self-labelling enzymes as universal tags for fluorescence microscopy, super-resolution microscopy and electron microscopy. Sci Rep 5: 17740.
- Lucas, M. S., Gunthert, M., Gasser, P., Lucas, F. and Wepf, R. (2012). Bridging microscopes: 3D correlative light and scanning electron microscopy of complex biological structures. Methods Cell Biol 111: 325-356.
- Lucocq, J. M., Mayhew, T. M., Schwab, Y., Steyer, A. M. and Hacker, C. (2015). Systems biology in 3D space--enter the morphome. Trends Cell Biol 25(2): 59-64.
- Martell, J. D., Deerinck, T. J., Sancak, Y., Poulos, T. L., Mootha, V. K., Sosinsky, G. E., Ellisman, M .H.and Ting A. Y. (2012). Engineered ascorbate peroxidase as a genetically encoded reporter for electron microscopy. Nat Biotech 30(11): 1143-1148.
- McGraw, T. E., Greenfield, L. and Maxfield, F. R. (1987). Functional expression of the human transferrin receptor cDNA in Chinese hamster ovary cells deficient in endogenous transferrin receptor. J Cell Biol 105(1): 207-214.
- Megason, S. G. and Fraser, S. E. (2007). Imaging in systems biology. Cell 130(5): 784-795.
- Milne, J. L. and Subramaniam, S. (2009). Cryo-electron tomography of bacteria: progress, challenges and future prospects. Nat Rev Microbiol 7(9): 666-675.
- Muller, B. and Heilemann, M. (2013). Shedding new light on viruses: super-resolution microscopy for studying human immunodeficiency virus. Trends Microbiol 21(10): 522-533.
- Ou, H. D., Phan, S., Deerinck, T. J., Thor, A., Ellisman, M. H. and O'Shea, C. C. (2017). ChromEMT: Visualizing 3D chromatin structure and compaction in interphase and mitotic cells. Science 357(6349).
- Philimonenko, V. V., Philimonenko, A. A., Sloufova, I., Hruby, M., Novotny, F., Halbhuber, Z., Krivjanska, M., Nebesarova, J., Slouf, M. and Hozak, P. (2014). Simultaneous detection of multiple targets for ultrastructural immunocytochemistry. Histochem Cell Biol 141(3): 229-239.
- Plaza, S. M., Scheffer, L. K. and Chklovskii, D. B. (2014). Toward large-scale connectome reconstructions. Curr Opin Neurobiol 25: 201-210.
- Romero-Brey, I. and Bartenschlager, R. (2017). Viral infection at high magnification: 3D electron microscopy methods to analyze the architecture of infected cells. Viruses 7(12): 6316-6345
- Shu, X., Lev-Ram, V., Deerinck, T. J., Qi, Y., Ramko, E. B., Davidson, M. W., Jin, Y., Ellisman, M. H. and Tsien, R. Y. (2011). A genetically encoded tag for correlated light and electron microscopy of intact cells, tissues, and organisms. PLoS Biol 9(4): e1001041.
- Sochacki, K. A., Dickey, A. M., Strub, M. P. and Taraska, J. W. (2017). Endocytic proteins are partitioned at the edge of the clathrin lattice in mammalian cells. Nat Cell Biol 19(4): 352-361.
- Sunbul, M. and Yin, J. (2009). Site specific protein labeling by enzymatic posttranslational modification. Org Biomol Chem 7(17): 3361-3371.
- Takizawa, T., Powell, R. D., Hainfeld, J. F. and Robinson, J. M. (2015). FluoroNanogold: an important probe for correlative microscopy. J Chem Biol 8(4): 129-142.
- Tane, H. K., Doh, J. K., Enns, C. A. and Beatty, K. E. (2017). Versatile interacting peptide (VIP) tags for labeling proteins with bright chemical reporters. Chembiochem 18(5): 470-474.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Doh, J. K., Chang, Y. H., Enns, C. A., Lόpez, C. S. and Beatty, K. E. (2019). Imaging VIPER-labeled Cellular Proteins by Correlative Light and Electron Microscopy. Bio-protocol 9(21): e3414. DOI: 10.21769/BioProtoc.3414.
Category
Cancer Biology > Proliferative signaling > Cell biology assays > Chemoresistance
Cell Biology > Cell imaging > Fluorescence
Biochemistry > Protein > Labeling
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.