- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Implementing VIPER for Imaging Cellular Proteins by Fluorescence Microscopy
Published: Vol 9, Iss 21, Nov 5, 2019 DOI: 10.21769/BioProtoc.3413 Views: 5940
Original research article
The authors used this protocol in:

Dec 2018
Advertisement




Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






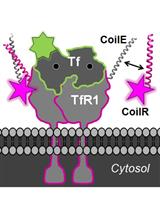

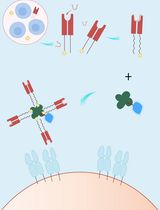
Abstract
Genetically-encoded tags are useful tools for multicolor and multi-scale cellular imaging. Versatile Interacting Peptide (VIP) tags, such as VIPER, are new genetically-encoded tags that can be used in various imaging applications. VIP tags consist of a coiled-coil heterodimer, with one peptide serving as the genetic tag and the other (“probe peptide”) delivering a reporter compatible with imaging. Heterodimer formation is rapid and specific, allowing proteins to be selectively labeled for live-cell and fixed-cell imaging. In this Bio-Protocol, we include a detailed guide for implementing the VIPER technology for imaging receptors on live cells and intracellular targets in fixed cells. This protocol is complemented by two other Bio-Protocols outlining the use of VIPER (Doh et al., 2019a and 2019b).
Keywords: Protein tagBackground
Fluorescence microscopy (FM) enables researchers to observe the architecture and assembly of proteins in cells dynamically and in multicolor (Liu et al., 2015; Valm et al., 2017; Guo et al., 2018; Liu et al., 2018). Fluorescence imaging relies on strategies for labeling target proteins with bright, fluorescent reporter molecules. Labeling can be achieved in a number of ways (Giepmans et al., 2006; Crivat and Taraska, 2012; Vandemoortele et al., 2019), including immunolabeling, fluorescent proteins (e.g., green fluorescent protein) (Shaner et al., 2005; Snapp, 2009), chemical stains (e.g., DAPI, MitoTracker, or phalloidin conjugates) (Cottet-Rousselle et al., 2011; Bucevičius et al., 2018), and self-labeling tags (Keppler et al., 2003; Gautier et al., 2008; Los et al., 2008). The most useful tags can be used to deliver diverse chemical reporters with optimal properties, such as spectrally-distinct colors, high quantum yield and extinction coefficient (“brightness”), and photostability (Dempsey et al., 2001; Li and Vaughan, 2018). Tags that can bind to a variety of bright fluorophore ligands include the SNAP tag (Keppler et al., 2003; Gautier et al., 2008), Halo tag (Los et al., 2008), TMP tag (Miler et al., 2005), and FAPs (Szent-Gyorgyi et al., 2008). However, these protein tags are large (18-33 kDa), which can change protein folding, trafficking, and function (Brock et al., 1999; Costantini and Snapp, 2013; Huang et al., 2014; Johnson et al., 2015). A few peptide tags have been described for cell imaging, as exemplified by the tetracysteine tag, but these can have non-specific interactions and limited color choices (Griffin et al., 1998; Gaietta et al., 2002; Cohen et al., 2011; Liu et al., 2014).
An alternative approach is to use a peptide tag that forms a heterodimeric coiled-coil with a reporter peptide. This is the approach that we (Zane et al., 2017; Doh et al., 2018) and others (Tsutsumi et al., 2009; Nomura et al., 2010; Tsutsumi et al., 2011; Reinhardt et al., 2014 and 2015; Lotze et al., 2018) have used to label cellular proteins (Lotze et al., 2016; Yano and Matsuzaki, 2019). One advantage of this approach is that the genetic tag is small–just 4 to 7 kDa. A second advantage is that reporter peptide labeling is typically restricted to the cell surface, which is useful for labeling and tracking transmembrane receptors (Yano et al., 2008) (i.e., in pulse-chase experiments (Doh et al., 2018; Lotze et al., 2018)). We named our coiled-coil tags Versatile Interacting Peptide (VIP) tags. Our first tag, VIP Y/Z, enabled the selective fluorescent labeling of target proteins in cell lysates and on live cells (Zane et al., 2017). Next we described VIPER, which is comprised of a CoilE tag and a CoilR probe peptide. We showed that the probe peptide can be customized to the imaging application by conjugation to one of a number of reactive fluorophores and small molecules (i.e., biotin). VIPER labeled sub-cellular structures in fixed cells and transmembrane receptors on live cells. Proteins could be imaged by FM or correlative light and EM (CLEM) (Doh et al., 2018).
For any genetic tag, it is important to insert the tag at a location in the amino acid sequence where it will not interfere with the binding interactions, localization, folding, or function of the protein of interest. It is beyond the scope of this paper to dictate the location of the genetic tag for all feasible protein targets. We recommend reading Erik Snapp’s paper “Design and Use of Fluorescent Fusion Proteins in Cell Biology” for a discussion on choosing a tag insertion site (Snapp, 2005). For VIP tags, we offer the following suggestions and recommendations for tag placement.
This protocol includes methods for implementing VIPER for imaging cellular proteins by FM. These imaging experiments are performed on transfected mammalian tissue culture cells. In Procedure A we provide a transfection protocol for introducing plasmid DNA encoded VIP-tagged constructs into cells. In Procedure B, we describe labeling a transmembrane receptor on the surface of living cells. In Procedure C, we describe methods for imaging intracellular targets in fixed cells. This publication is complemented by Doh et al. (2019a), which outlines how to generate CoilR probe peptide, and Doh et al. (2019b), which discusses using VIPER to image proteins by CLEM. A decision tree for implementing these methods is provided in Figure 1.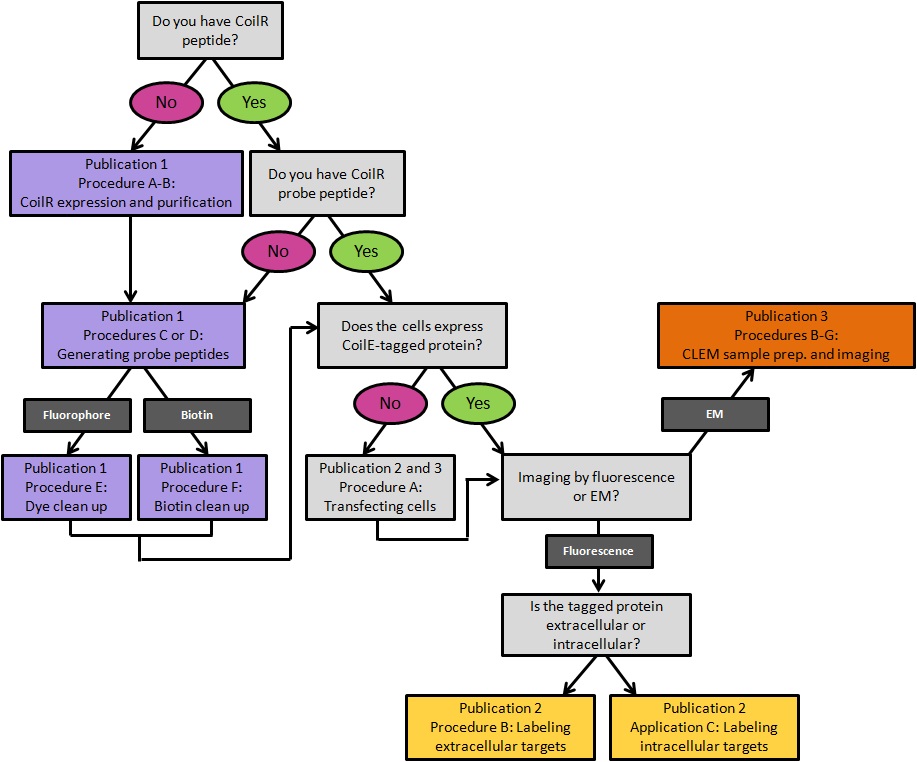
Figure 1. A decision tree for implementing VIPER for labeling cellular proteins. Procedures are color-coded by the publication in which they appear. Publication 1: Doh et al., 2019a; Publication 2: this article; Publication 3: Doh et al., 2019b.
Materials and Reagents
Note: “*” indicates a brand that is critical to the success of the experiment.
Materials
- Polystyrene 10 cm tissue culture dish (Thermo Scientific, NuncTM, catalog number: 12565020)
- Polystyrene 6-well tissue culture plate (Thermo Scientific, BioLiteTM, catalog number: 12-556-004)
- 8-well chambered coverglass with #1.5 glass (i.e., Cellvis, catalog number: C8-1.5H-N or Thermo Scientific, NuncTM Lab-TekTM, catalog number: 155411)
- LDPE 500 ml squeeze wash bottle (Thermo Scientific, catalog number: 24010500)
- Transfer bulb pipette (VWR, catalog number: 16001-182)
- U-2 OS cells (ATCC, catalog number: HTB-96)
- CHO TRVb cells (Courtesy of Prof. Timothy McGraw, Cornell University, Ithaca, New York) (McGraw et al., 1987)
- Plasmid DNA of VIP-tagged protein (Generated by the user)
- mEmerald-actin-C18 (Addgene, catalog number: 53978)
- Human transferrin Alexa FluorTM 488 conjugate; Tf-AF488 (Invitrogen, catalog number: T13342)
- *LipofectamineTM 2000 (Thermo Scientific, catalog number: 11668019)
- Ham’s F-12 Medium (Life Technologies, GibcoTM, catalog number: 11765062)
- McCoy’s 5A Medium (Life Technologies, GibcoTM, catalog number: 16600082)
- Dulbecco’s phosphate-buffered saline without calcium or magnesium; DPBS (GibcoTM, catalog number: 14190144)
- Trypsin-EDTA (0.25%) (Life Technologies, GibcoTM, catalog number: 25200056)
- Fetal bovine serum; FBS (GE, HycloneTM, catalog number: SH30910.03)
- Opti-MEM (Life Technologies, GibcoTM, catalog number: 31985070)
- Hoechst 33342 nucleic acid stain (Invitrogen, catalog number: H1399)
- Bovine serum albumin (BSA) fraction V (Research Products International, catalog number: A30075100.0)
- Immersion oil (Carl Zeiss ImmersolTM 518 F, catalog number: 4449620000000)
- Sodium azide (Sigma-Aldrich, catalog number: 71289-5G)
- Triton X-100 (Sigma-Aldrich, catalog number: 93443-100ML)
- Saponin (Sigma-Aldrich, catalog number: 558255100GM)
- Ham’s F12 media
- Glycerol (Thermo Fisher, Fisher BioReagentsTM, catalog number: BP229 1)
- Tris (Thermo Fisher, Fisher BioReagentsTM, catalog number: BP152-500)
- Sucrose (Thermo Fisher, Fisher Chemical, catalog number: D16-500)
- Live Cell Block Solution (see Recipes)
- Mounting Medium (see Recipes)
- Fixed Cell Block Solution (see Recipes)
Equipment
- Hemocytometer (Hausser Scientific, catalog number: 1475)
- Humidified CO2 Incubator (New Brunswick Galaxy 170S, catalog number: C0170S-120-0000)
- Tissue culture hood (Thermo Scientific, 1300 Series A2)
- Tissue culture inverted light microscope (Carl Zeiss, Zeiss Primovert)
- Spinning disk confocal fluorescence microscope (Zeiss Yokogawa CSU-X1 on Zeiss AxioObserver)
- Line scanning (Airyscanning) fluorescence microscope (Zeiss LSM880 on inverted microscope stand)
Software
- Fiji (ImageJ Version 2.0.0-rc-46) (Schindelin et al., 2012)
Procedure
- Transfecting cell lines to express a VIPER-tagged protein
VIP tags have been used in a number of cell lines successfully. Receptor imaging using VIPER (Procedure B) was done in Chinese hamster ovary (CHO) TRVb cells. CHO TRVb cells do not express TfR1 or transferrin receptor 2 (McGraw et al., 1987) which makes it a useful cell line for imaging TfR1-CoilE. Labeling intracellular targets in fixed cells (Procedure C) was done in U-2 OS, a human osteosarcoma cell line. We have used VIP tags in other cell lines not discussed in this publication (e.g., HEK293 and AU565).
We recommend optimizing plating density and transfection conditions for each cell line. Refer to ThermoFisher’s Protocol Pub No. MAN0007824 Rev 1.0 (Reference 24) for more information on transfection with Lipofectamine 2000. In our procedure, we recommend passaging cells on Day 1, transfecting cells on Day 2, and VIPER labeling cells on Day 3 for imaging (see Procedures B-C).
Recommendations for Procedure A:
•Use sterile technique and work within a tissue culture (laminar flow) hood when working with live tissue culture cell lines.
•When transfecting cells with a vector encoding a CoilE-tagged protein (e.g., pcDNA3.1_TfR1-CoilE), we also recommend transfecting cells with an untagged protein to compare labeling specificity (e.g., pcDNA3.1_TfR1). Untransfected cells are also useful controls for labeling specificity.
•The amino acid sequence for TfR1-CoilE and mEmerald-CoilE-actin-C18 can be found in the Sequences (Supplemental file).
•The DNA quality is paramount for high transfection efficiency. Quantify DNA quality by UV spectroscopy and verify that the 260/280 nm ratio falls between 1.8 and 2.0.
•There are many different methods for introducing genes into cells and we encourage researchers to use methods that have been optimized for their cell lines.- Day 1: Passage cells and seed into an 8-well chambered coverslip.
- Before starting, visually inspect cells on a tissue culture microscope (e.g., by bright field illumination, low magnification) to confirm that cells are adherent, healthy, and 80-90% confluent.
- Seed 5 x 104 cells per well (for U-2 OS or CHO TRVb) into NuncTM Lab-TekTM 8-well chambered coverslips.
- For other cell lines, seed at a density that will result in 80-90% confluency after 24 h.
- Do not include antibiotics in the media.
- For U-2 OS, grow cells in McCoy’s 5A medium supplemented with 10% FBS.
- For CHO TRVb cells, grow cells in Ham’s F-12 medium supplemented with 5% FBS.
- Incubate cells overnight in a humidified tissue culture incubator (37 °C with 5% CO2).
- Day 2: Prepare the transfection mixture:
- Dilute vector DNA into Opti-MEM.
- For an 8-well chambered coverslip, use 500 ng DNA per well.
- Keep the volume of DNA to less than 10% of the total volume in the transfection mixture.
- Dilute Lipofectamine 2000 into Opti-MEM and incubate for 5 min:
- For 8-well chambered coverslips, use 1,000 ng Lipofectamine 2000 per well.
- Combine equal volumes of the Lipofectamine 2000 and DNA solutions and mix by pipetting. The final ratio of DNA to Lipofectamine 2000 will be 1:2. Incubate the mixture for at least 30 min at room temperature.
- Dilute vector DNA into Opti-MEM.
- Transfect cells:
- Aspirate media from cells.
- Add Opti-MEM to wells. For 8-well chambered coverslips, use 200 μl per well.
- Dilute the transfection mixture into each well (1:5 dilution) by adding 50 μl of the transfection mixture. The total volume of fluid in each well should be 250 μl.
- Gently mix the solution by pipetting up and down or by rocking the chambered slide.
- Incubate cells in the transfection mixture for 4 h in a tissue culture incubator (37 °C, 5% CO2).
- This time frame is recommended for U-2 OS and CHO TRVb cells.
- Monitor cells by viewing on a TC microscope. Cell size and shape should appear unchanged during transfection. If cells contract or detach, then adjust the transfection conditions.
- Aspirate to remove the transfection media and wash once with complete media (e.g., media with FBS).
- Add complete media and grow in a tissue culture incubator (37 °C, 5% CO2) for at least 24 h. Cells will be ready for use between 24 and 48 h after transfection.
- Day 1: Passage cells and seed into an 8-well chambered coverslip.
- Application: Labeling a transmembrane receptor with VIPER on living cells
Investigations into cell surface receptors are enabled by VIP tags, which can be used to tag and track receptors in living cells. When imaging live cells, VIPER labeling is restricted to the cell surface because the probe peptide (e.g., CoilR-Cy5) is live cell-impermeant. This feature is an advantage for observing receptor trafficking or population changes in localization over time.
Procedure B describes a method for imaging iron uptake in cells. Briefly, iron uptake in the cell is mediated by a transmembrane receptor named transferrin receptor 1 (TfR1). This receptor binds and transports iron-bound transferrin (Tf) into cells through clathrin-mediated endocytosis. We used VIPER to observe this process. Specifically, we describe a method for labeling TfR1-CoilE with red-fluorescent CoilR-Cy5 for imaging by confocal FM. As a counter-stain for the receptor, we also treat cells with fluorescent Tf (i.e., Tf-AF488), which binds to TfR1. The cells are labeled live at 4 °C in order to halt the process of endocytosis. The methods described here could be used to image other cell receptors.
Receptor labeling is performed on live cells and TfR1 can be imaged either live or post-fixation. In Procedure B, we specify that the cells are fixed prior to imaging. This is done for convenience, and is recommended for users optimizing labeling conditions. Alternatively, as described in our publication, cells can be imaged live (i.e., by time-lapse) to observe receptor trafficking (Doh et al., 2018).
Procedure B commences on Day 3 with transfected cells in 8-well chambered slides, generated as described in Procedure A. We recommend including unlabeled or single-labeled controls for imaging. We also recommend assessing VIP labeling specificity by including an untagged protein control. An example slide layout is provided in Figure 2.
Figure 2. Layout of an 8-well chambered coverslip for imaging CHO TRVb cells expressing TfR1. Top row: Cells expressing Coil-E tagged receptor (TfR1-CoilE). Bottom Row: Cells expressing untagged receptor (TfR1). The first line in each well indicates the vector used in the transfection. The subsequent lines indicate the labeling reagents, which are color-coded based on their fluorescence.
Recommendations for Procedure B:
• Preserving cell morphology and health is important for optimal labeling and imaging.
• Never let the cells go dry when processing cells for imaging. The preferred method for removing fluid from 8-well coverslips is to briskly invert the slide in a flicking motion. The surface tension of water will ensure an adequate layer of fluid remains in the well after it has been emptied.
•Reduce cell loss by gently dispensing solutions against the walls of the well. DPBS can be stored in a squeeze wash bottle for convenience when washing.
•Optimize labeling with the probe peptide. VIPER labeling efficiency will depend on the quality of the probe peptide, incubation temperature and time, and the target protein properties (abundance and localization). We have successfully labeled and imaged target proteins with 1 nM to 1 µM probe peptide. Previous experiments featuring competition with unlabeled peptide showed that 100 nM of peptide was enough to label most of an abundant protein target (Doh et al., 2018).- Day 3: Visually inspect transfected cells on a tissue culture microscope (e.g., by bright field illumination, low magnification) to confirm that cells are adherent, healthy, and 80-90% confluent.
- Remove media and add 100 μl of Live Cell Block Solution (Recipe 1) to each well. Return to the tissue culture incubator for 30 min.
Note: We include this step to reduce non-specific binding to live-cell surfaces. - While the cells are in Live Cell Block Solution, prepare the labeling solution in pre-chilled Ham’s F-12 media without serum.
- Dilute CoilR probe peptide and 50 μg/ml Tf-AF488 into Ham’s F-12 media. Prepare at least 100 μl solution per well. We recommend first trying 100 nM CoilR-Cy5.
- Analogous to a standard immunolabeling optimization, we recommend testing a range of probe peptide concentrations to obtain optimal signal to noise (e.g., 1 nM to 500 nM CoilR-Cy5).
- Other labeling reagents can be added to the labeling solution at this time, such as ligands, antibodies, or organelle stains.
- Remove Live Cell Block Solution from each well.
- Add 100 μl cold labeling solution to each well. Label cells for 30 min at 4 °C, protected from light.
- Wash each well three times with ice-cold DPBS.
- Optional step: Add complete Ham’s F-12 media to each well and incubate the sample at 37 °C for 5 to 30 minutes to enable labeled receptors to traffic (e.g., endocytose).
- Optional step: For pulse-chase labeling instructions, refer to our prior publication (Doh et al., 2018).
- Fix the cells using ice-cold 4% v/v paraformaldehyde (PFA) in DPBS for 15 min at 4 °C.
- Prepare the fixative by diluting 20% v/v PFA 5-fold into DPBS. Make fresh fixative for every experiment.
- An opened solution of 20% v/v PFA can be stored in a sealed vial for up to 2 weeks.
- Wash each well twice with DPBS.
- Optional Step: Immunolabeling can be done at this point in the procedure.
- Stain the nuclei with Hoechst 33342 (10 μg/ml) in DPBS for 5 min at 37 °C.
- Wash wells once with DPBS.
- Add Mounting Medium (Recipe 2) to wells.
- Image cells by FM. See Doh’s Supporting Information for more information on the imaging system and acquisition parameters (Doh et al., 2018). The imaging parameters that we used are summarized below:
- Image cells using 63x/1.4 NA oil immersion objective lens on a Zeiss Yokogawa CSU-X1 spinning disk confocal microscope.
- Capture single confocal slices with identical acquisition settings optimized for each channel.
- Hoechst 33342 was imaged using 405 nm excitation and a 450/50 nm emission filter.
- AF488 was imaged using 488 nm excitation and a 525/50 nm emission filter.
- Cy5 was imaged using 633 nm excitation and a 670/30 emission filter.
- Application: Labeling an intracellular target with VIPER in cells post-fixation
In Procedure C, we describe methods for imaging intracellular targets in fixed cells using VIPER. VIPER enables selective labeling of cellular proteins associated with organelles in fixed cells, including the cytoskeleton, nucleus, and mitochondria (Doh et al., 2018). We used expression vectors that encoded target proteins fused to mEmerald, a green fluorescent protein. Fusion to a fluorescent protein was used to confirm that VIP-tagged proteins localized correctly. For fusion proteins, the CoilE tag was inserted between mEmerald and the target protein (e.g., β-actin or histone H2B). This approach can also be used to label other intracellular targets (Doh et al., 2018). Labeling intracellular targets requires that cells be fixed before treatment with a CoilR probe peptide because the peptide does not cross live cell membranes. We provide a different recipe for blocking fixed cells to reduce non-specific labeling. Similar to Procedure B, we recommend handling live cells carefully to retain viability and reduce cell loss.
In Procedure C, we describe a method for highlighting actin in U-2 OS cells expressing a fusion protein: mEmerald-CoilE-actin-C18. Fixed cells are treated with CoilR-Cy5 probe peptide. This procedure starts with transfected cells in 8-well chambered coverslips on Day 3, which can be prepared as described in Procedure A. We recommend including unlabeled or single-labeled controls for each fluorophore (i.e., mEmerald, Hoechst, and Cy5) and including an untagged protein control (mEmerald-actin-C18). An example slide layout is provided in Figure 3.
Figure 3. Layout of an 8-well chambered coverslip for imaging U-2 OS cells expressing either mEmerald-CoilE-actin-C18 (top row) or mEmerald-actin-C18 (bottom row). The first line in each well indicates the vector used in the transfection. The subsequent lines indicate labeling reagents, which are color-coded based on their fluorescence. In this setup, three probe peptide concentrations (i.e., 10, 100, and 200 nM CoilR-Cy5) are included to enable the identification of the optimal labeling conditions.- Day 3: Visually inspect each well on a tissue culture microscope to confirm that cells are adherent, healthy, and 80-90% confluent.
- Remove media and wash each well once with DPBS.
- Fix cells with 4% v/v PFA in DPBS for 10 min at room temperature.
- Wash each well twice with DPBS to remove fixative.
- Permeabilize cells with 0.1% (v/v) Triton X-100 in DPBS for 10 min at room temperature. Alternatively, cells can be permeabilized with 0.1% (w/v) saponin in DPBS.
- Rinse wells twice with DPBS to remove detergent.
- Remove DPBS and add 100 μl of Fixed Cell Block Solution (Recipe 3) to each well. Incubate for 30 min at room temperature.
- During the block step, prepare the labeling solution in Fixed Cell Block Solution:
- Use 1 to 500 nM CoilR probe peptide (i.e., CoilR-Cy5) in Fixed Cell Block Solution.
- We recommend testing a range of probe peptide concentration to obtain an optimal signal to noise ratio.
- Prepare 100 μl solution per well. Other labeling reagents can be added to the labeling solution at this time, such as ligands, primary antibodies, or organelle stains.
- Remove Fixed Cell Block Solution and add the prepared CoilR labeling solution to each well. Protect the slide from light and incubate for 30 min at room temperature.
- Wash the wells 3 times with DPBS.
- Stain the nuclei with Hoechst 33342 (10 μg/ml) in DPBS for 5-10 min at room temperature.
- Wash wells once with DPBS.
- Add Mounting Medium to wells.
- Image cells by FM. Figure 4 provides representative micrographs of cells expressing mEmerald-CoilE-actin-C18 or mEmerald-actin-C18. See Doh et al.’s Supporting Information for more information on the imaging system and acquisition parameters (Doh et al., 2018), which are summarized below:
- Image cells using a 63x/1.4 NA oil immersion objective lens on a Zeiss Yokogawa CSU-X1 spinning disk confocal microscope (or similar system).
- Capture single confocal slices with identical acquisition settings optimized for each channel.
- Hoechst 33342 was imaged using 405 nm excitation and a 450/50 nm emission filter.
- mEmerald was imaged using 488 nm excitation and a 525/50 nm emission filter.
- Cy5 was imaged using 633 nm excitation and a 670/30 emission filter.

Figure 4. Highlighting actin in U-2 OS cells using VIPER. U-2 OS cells were transfected with vectors encoding mEmerald-CoilE-Actin-C18 (mEm-CoilE-Actin; top row) or mEmerald-Actin-C18 (mEm-Actin; bottom row), labeled post-fixation by treatment with CoilR-Cy5 (100 nM), and then imaged by confocal FM. CoilR-Cy5 labeling was specific to CoilE-tagged protein, with Cy5 (magenta) and mEmerald (green) fluorescence appearing co-localized (white) in the merged image. Cy5 labeling was not observed in cells expressing mEmerald-actin-C18. The merge includes the nucleus false-colored blue (Hoechst 33342).
Recipes
Notes:
- Buffers are made in autoclaved DI water unless otherwise stated.
- The pH of Tris buffers changes with temperature, and the pH should be measured at room temperature.
- The pH of DPBS is 7.0.
- Live Cell Block Solution
10% (v/v) FBS and 6% (w/v) BSA in Ham’s F-12 media
Note: Block solution can be prepared in other media, such as McCoy’s 5A media for U-2 OS cells. - Mounting Medium
90% (v/v) glycerol
10 mM Tris pH 7.0
0.1% (w/v) sodium azide - Fixed Cell Block Solution
10% (v/v) FBS
5% (w/v) sucrose
2% (w/v) BSA in DPBS
Acknowledgments
Research for this project was supported by the OHSU School of Medicine and the National Institutes of Health (R01 GM122854). JKD was partially funded by the Portland Chapter of Achievement Rewards for College Scientists (ARCS). The protocols described herein were originally described in Doh, Zane, et al. (Doh et al., 2018). Fluorescent organelle constructs (Actin, H2B, and Mito) were acquired from the Michael Davidson Fluorescent Protein library from Addgene. CHO TRVb cells were provided by Prof. Timothy McGraw. We are grateful to our colleagues at OHSU for their contributions to this work, particularly Dr. Hannah Zane and Dr. Jonathan White.
Competing interests
The authors declare no financial or non-financial competing interests. An international patent application is pending on the VIP technology (PCT/US17/60609).
References
- Argos, P. (1990). An investigation of oligopeptides linking domains in protein tertiary structures and possible candidates for general gene fusion. J Mol Biol 211(4): 943-958.
- Brock, R., Hamelers, I. H. and Jovin, T. M. (1999). Comparison of fixation protocols for adherent cultured cells applied to a GFP fusion protein of the epidermal growth factor receptor. Cytometry 35(4): 353-362.
- Bucevičius, J., Lukinavičius, G. and Gerasimaitė, R. (2018). The use of hoechst dyes for DNA staining and beyond. Chemosensors 6(2): 18.
- Chen, X., Zaro, J. L. and Shen, W. C. (2013). Fusion protein linkers: property, design and functionality. Adv Drug Deliv Rev 65(10): 1357-1369.
- Cohen, J. D., Thompson, S. and Ting, A. Y. (2011). Structure-guided engineering of a Pacific Blue fluorophore ligase for specific protein imaging in living cells. Biochemistry 50(38): 8221-8225.
- Costantini, L. M. and Snapp, E. L. (2013). Fluorescent proteins in cellular organelles: serious pitfalls and some solutions. DNA Cell Biol32(11):622-627.
- Cottet-Rousselle, C., Ronot, X., Leverve, X. and Mayol, J. F. (2011). Cytometric assessment of mitochondria using fluorescent probes. Cytometry Part A 79(6),:405-425.
- Crasto, C. J. and Feng, J. A. (2000). LINKER: a program to generate linker sequences for fusion proteins. Protein Eng 13(5): 309-312.
- Crivat, G. and Taraska, J. W. (2012). Imaging proteins inside cells with fluorescent tags. Trends Biotechnol 30(1): 8-16.
- Dempsey, G. T., Vaughan, J. C., Chen, K. H., Bates, M. and Zhuang, X. (2011). Evaluation of fluorophores for optimal performance in localization-based super-resolution imaging. Nat Methods 8(12): 1027-1036.
- Doh, J. K., Tobin, S. J. and Beatty, K. E. (2019a). Generation of CoilR probe peptides for VIPER-labeling of cellular proteins. Bio-protocol 9(21): e3412.
- Doh, J. K., Chang, Y. H., Enns, C. A., Lopes, C. S. and Beatty, K. E. (2019b). Imaging VIPER-labeled cellular proteins by correlative light and electron microscopy. Bio-protocol 9(21): e3414.
- Doh, J. K., White, J. D., Zane, H. K., Chang, Y. H., López, C. S., Enns, C. A. and Beatty, K. E. (2018). VIPER is a genetically encoded peptide tag for fluorescence and electron microscopy. Proc Natl Acad Sci U S A 115(51): 12961-12966.
- Gaietta, G., Deerinck, T. J., Adams, S. R., Bouwer, J., Tour, O., Laird, D. W., Sosinsky, G. E., Tsien, R. Y. and Ellisman, M. H. (2002). Multicolor and electron microscopic imaging of connexin trafficking. Science 296(5567): 503-507.
- Gautier, A., Juillerat, A., Heinis, C., Correa, I. R., Jr., Kindermann, M., Beaufils, F. and Johnsson, K. (2008). An engineered protein tag for multiprotein labeling in living cells. Chem Biol 15(2): 128-136.
- George, R. A. and Heringa, J. (2002). An analysis of protein domain linkers: their classification and role in protein folding. Protein eng 15(11): 871-879.
- Giepmans, B. N., Adams, S. R., Ellisman, M. H. and Tsien, R. Y. (2006). The fluorescent toolbox for assessing protein location and function. Science 312(5771): 217-224.
- Griffin, B. A., Adams, S. R. and Tsien, R. Y. (1998). Specific covalent labeling of recombinant protein molecules inside live cells. Science 281(5374): 269-272.
- Guo, Y., Li, D., Zhang, S., Yang, Y., Liu, J. J., Wang, X., Liu, C., Milkie, D. E., Moore, R. P., Tulu, U. S., Kiehart, D. P., Hu, J., Lippincott-Schwartz, J., Betzig, E. and Li, D. (2018). Visualizing intracellular organelle and cytoskeletal interactions at nanoscale resolution on millisecond timescales. Cell 175(5): 1430-1442 e1417.
- Huang, L., Pike, D., Sleat, D. E., Nanda, V. and Lobel, P. (2014). Potential pitfalls and solutions for use of fluorescent fusion proteins to study the lysosome. PLoS ONE 9(2):e88893.
- Johnson, G. R., Buck, T. E., Sullivan, D. P., Rohde, G. K. and Murphy, R. F. (2015). Joint modeling of cell and nuclear shape variation. Mol Biol Cell 26(22): 4046-4056.
- Keppler, A., Gendreizig, S., Gronemeyer, T., Pick, H., Vogel, H. and Johnsson, K. (2003). A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat Biotechnol 21(1): 86-89.
- Li, H. and Vaughan, J. C. (2018). Switchable fluorophores for single-molecule localization microscopy. Chem Rev 118(18): 9412-9454.
- Lipofectamine® 2000 Reagent Protocol 2013. (2013). (Accessed 12-Oct, 2019, at
. tools.thermofisher.com/content/sfs/manuals/Lipofectamine_2000_Reag_protocol.pdf.) - Liu, D. S., Nivon, L. G., Richter, F., Goldman, P. J., Deerinck, T. J., Yao, J. Z., Richardson, D., Phipps, W. S., Ye, A. Z., Ellisman, M. H., Drennan, C. L., Baker, D. and Ting, A. Y. (2014). Computational design of a red fluorophore ligase for site-specific protein labeling in living cells. Proc Natl Acad Sci U S A 111(43): E4551-4559.
- Liu, Z., Lavis, D. L. and Betzig, E. (2015). Imaging live-cell dynamics and structure at the single-molecule level. Mol Cell 58(4): 644-659.
- Liu, T. L., Upadhyayula, S., Milkie, D. E., Singh, V., Wang, K., Swinburne, I. A., Mosaliganti, K. R., Collins, Z. M., Hiscock, T. W., Shea, J., Kohrman, A. Q., Medwig, T. N., Dambournet, D., Forster, R., Cunniff, B., Ruan, Y., Yashiro, H., Scholpp, S., Meyerowitz, E. M., Hockemeyer, D., Drubin, D. G., Martin, B. L., Matus, D. Q., Koyama, M., Megason, S. G., Kirchhausen, T. and Betzig, E. (2018). Observing the cell in its native state: Imaging subcellular dynamics in multicellular organisms. Science 360(6386).
- Los, G. V., Encell, L. P., McDougall, M. G., Hartzell, D. D., Karassina, N., Zimprich, C., Wood, M. G., Learish, R., Ohana, R. F., Urh, M., Simpson, D., Mendez, J., Zimmerman, K., Otto, P., Vidugiris, G., Zhu, J., Darzins, A., Klaubert, D. H., Bulleit, R. F. and Wood, K. V. (2008). HaloTag: a novel protein labeling technology for cell imaging and protein analysis. ACS Chem Biol 3(6): 373-382.
- Lotze, J., Reinhardt, U., Seitz, O. and Beck-Sickinger, A. G. (2016). Peptide-tags for site-specific protein labelling in vitro and in vivo. Mol Biosyst 12(6): 1731-1745.
- Lotze, J., Wolf, P., Reinhardt, U., Seitz, O., Morl, K. and Beck-Sickinger, A. G. (2018). Time-Resolved Tracking of Separately Internalized Neuropeptide Y2 Receptors by Two-Color Pulse-Chase. ACS Chem Biol 13(3): 618-627.
- McGraw, T. E., Greenfield, L. and Maxfield, F. R. (1987). Functional expression of the human transferrin receptor cDNA in Chinese hamster ovary cells deficient in endogenous transferrin receptor. J Cell Biol 105(1): 207-214.
- Miller, L. W., Cai, Y., Sheetz, M. P. and Cornish, V. W. (2005). In vivo protein labeling with trimethoprim conjugates: a flexible chemical tag. Nat Methods 2(4): 255-257.
- Nomura, W., Mino, T., Narumi, T., Ohashi, N., Masuda, A., Hashimoto, C., Tsutsumi, H. and Tamamura, H. (2010). Development of crosslink-type tag-probe pairs for fluorescent imaging of proteins. Biopolymers 94(6): 843-852.
- Reinhardt, U., Lotze, J., Morl, K., Beck-Sickinger, A. G. and Seitz, O. (2015). Rapid covalent fluorescence labeling of membrane proteins on live cells via coiled-coil templated acyl transfer. Bioconjug Chem 26(10): 2106-2117.
- Reinhardt, U., Lotze, J., Zernia, S., Morl, K., Beck-Sickinger, A. G. and Seitz, O. (2014). Peptide-templated acyl transfer: a chemical method for the labeling of membrane proteins on live cells. Angew Chem Int Ed Engl 53(38): 10237-10241.
- Schindelin, J., Arganda-Carreras, I., Frise, E., Kaynig, V., Longair, M., Pietzsch, T., Preibisch, S., Rueden, C., Saalfeld, S., Schmid, B., Tinevez, J. Y., White, D. J., Hartenstein, V., Eliceiri, K., Tomancak, P. and Cardona, A. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9(7): 676-682.
- Shaner, N. C., Steinbach, P. A. and Tsien, R. Y. (2005). A guide to choosing fluorescent proteins. Nat Methods 2(12): 905-909.
- Snapp, E. (2005). Design and use of fluorescent fusion proteins in cell biology. Curr Protoc Cell Biol Chapter 21: 21.4.1-21.4.13.
- Snapp, E. L. (2009). Fluorescent proteins: a cell biologist's user guide. Trends Cell Biol 19(11): 649-655.
- Szent-Gyorgyi, C., Schmidt, B. F., Creeger, Y., Fisher, G. W., Zakel, K. L., Adler, S., Fitzpatrick, J. A., Woolford, C. A., Yan, Q., Vasilev, K. V., Berget, P. B., Bruchez, M. P., Jarvik, J. W. and Waggoner, A. (2008). Fluorogen-activating single-chain antibodies for imaging cell surface proteins. Nat Biotechnol 26(2): 235-240.
- Tsutsumi, H., Abe, S., Mino, T., Nomura, W. and Tamamura, H. (2011). Intense blue fluorescence in a leucine zipper assembly. Chembiochem 12(5): 691-694.
- Tsutsumi, H., Nomura, W., Abe, S., Mino, T., Masuda, A., Ohashi, N.,Tanaka, T., Ohba, K., Yamamoto, N., Akiyoshi, K. and Tamamura, H. (2009). Fluorogenically active leucine zipper peptides as tag–probe pairs for protein imaging in living cells. Angew Chem Int Ed Engl 48(48):9164-9166.
- Valm, A. M., Cohen, S., Legant, W. R., Melunis, J., Hershberg, U., Wait, E., Cohen, A. R., Davidson, M. W., Betzig, E. and Lippincott-Schwartz, J. (2017). Applying systems-level spectral imaging and analysis to reveal the organelle interactome. Nature 546(7656): 162-167.
- Vandemoortele, G., Eyckerman, S. and Gevaert, K. (2019). Pick a tag and explore the functions of your pet protein. Trends Biotechnol 37(10): 1078-1090.
- Yano, Y. and Matsuzaki, K. (2019). Live-cell imaging of membrane proteins by a coiled-coil labeling method-Principles and applications. Biochim Biophys Acta Biomembr 1861(5): 1011-1017.
- Yano, Y., Yano, A., Oishi, S., Sugimoto, Y., Tsujimoto, G., Fujii, N. and Matsuzaki, K. (2008). Coiled-coil tag--probe system for quick labeling of membrane receptors in living cell. ACS Chem Biol 3(6): 341-345.
- Zane, H. K., Doh, J. K., Enns, C. A. and Beatty, K. E. (2017). Versatile interacting peptide (VIP) tags for labeling proteins with bright chemical reporters. Chembiochem 18(5): 470-474.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Doh, J. K., Enns, C. A. and Beatty, K. E. (2019). Implementing VIPER for Imaging Cellular Proteins by Fluorescence Microscopy. Bio-protocol 9(21): e3413. DOI: 10.21769/BioProtoc.3413.
Category
Cancer Biology > Proliferative signaling > Cell biology assays > Chemoresistance
Cell Biology > Cell imaging > Fluorescence
Biochemistry > Protein > Labeling
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.