- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Platelet Isolation and Activation Assays

Published: Vol 9, Iss 20, Oct 20, 2019 DOI: 10.21769/BioProtoc.3405 Views: 12817
Reviewed by: Meenal SinhaXiaoyi ZhengAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Apr 2019
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Platelets regulate hemostasis and are the key determinants of pathogenic thrombosis following atherosclerotic plaque rupture. Platelets circulate in an inactive state, but become activated in response to damage to the endothelium, which exposes thrombogenic material such as collagen to the blood flow. Activation results in a number of responses, including secretion of soluble bioactive molecules via the release of alpha and dense granules, activation of membrane adhesion receptors, release of microparticles, and externalization of phosphatidylserine. These processes facilitate firm adhesion to sites of injury and the recruitment and activation of other platelets and leukocytes, resulting in aggregation and thrombus formation. Platelet activation drives the hemostatic response, and also contributes to pathogenic thrombus formation. Thus, quantification of platelet-associated responses is key to many pathophysiologically relevant processes. Here we describe protocols for isolating, counting, and activating platelets, and for the rapid quantification of cell surface proteins using flow cytometry.
Keywords: PlateletsBackground
Platelets are small circulating cells with key roles in the normal hemostatic response to vascular injury, and also in pathogenic thrombus formation resulting from, for example, atherosclerotic plaque rupture. Both hemostasis and plaque rupture result in exposure of thrombogenic material to the circulation, including collagen, von Willebrand factor (vWF), and also a variety of soluble platelet agonists released from damaged cells. Thrombin, generated as part of the coagulation cascade, is a potent platelet agonist that activates platelets via protease-activated receptors. Once activated, platelets undergo a number of processes, including shape change, degranulation of alpha and dense granules, secretion of bioactive molecules (including platelet agonists, such as thromboxane A2, ADP and serotonin), phosphatidylserine exposure, and receptor activation. Released soluble platelet agonists regulate autocrine and paracrine activation via GPCR signaling, resulting in the activation and recruitment of other platelets, and formation of a thrombus (Li et al., 2010).
Activation also results in upregulation of a number of adhesive receptors on the platelet surface. Principally, activation results in the ‘inside-out’ signaling processes that activate the fibrinogen receptor, integrin αIIbβ3 (glycoprotein IIb-IIIa), which is responsible for platelet-platelet interactions as a result of binding to fibrinogen. Activated αIIbβ3 also binds to vWF, further associating platelets to sites of vascular damage (Bennett, 1996). Activation also results in the externalization of P-selectin, which regulates leukocyte binding via its ligand P-selectin glycoprotein ligand-1. This results in the formation of platelet-leukocyte aggregates, which are essential for the delivery of pro-inflammatory cytokines to the damaged endothelium (Yun et al., 2016). Activation also causes loss of phospholipid asymmetry and exposure of phosphatidylserine (PS) on the outer leaflet of the membrane. This supports the formation of the prothrombinase complex (Monroe et al., 2002) and therefore the conversion of prothrombin into active thrombin (Lane et al., 2005).
In addition to hemostatic roles, soluble factors released from activated platelets also influence other processes. For example, interleukin-1 from activated platelets has proinflammatory roles in arthritis (Boilard et al., 2010), and cerebrovascular disease (Thornton et al., 2010). Therefore, the importance of measuring factors found on the surface and released from activated platelets is important to many areas of biology.
Here we describe several commonly used protocols that can be used to isolate and activate platelets, and quantify the proteins they express. These protocols can be easily adapted to look at other proteins of interest on platelets.
Materials and Reagents
- Glass tubes (Fisher, catalog number: 11852363)
- Pipette tips (Gilson, catalog numbers: F167104; F167103; F167101)
- Pipettes
- 1 ml syringe (BD, catalog number: 309628)
- 10 ml syringe (BD, catalog number: 305959)
- 50 ml syringe (BD, catalog number: 300865)
- 0.22 μm syringe filter (Merck, catalog number: SLGP033RS)
- 21 G syringe needle (Sarstedt, catalog number: 85.1162)
- 23 G syringe needle (BD, catalog number: 300800)
- 27 G syringe needle (BD, catalog number: 302200)
- 14 ml round-bottomed tubes (Falcon, catalog number: 352059)
- 1.5 ml microcentrifuge tubes (Trefflab, catalog number: 96.07811.9.03)
- 2 ml microcentrifuge tubes (Trefflab, catalog number: 96.09329.01)
- EDTA coated capillary tube (Sarstedt, catalog number: 16.444)
- Aggregometry cuvettes (aggregometer specific; e.g., Helena, catalog number: 1473)
- Mouse (Jackson Laboratory, catalog number: 000664)
- Bovine Serum Albumin (Sigma-Aldrich, catalog number: A3059)
- Collagen (Helena, catalog number: 5368)
- Thrombin (Merck, catalog number: 69671-3)
- ADP (Sigma-Aldrich, catalog number: A2754)
- Platelet Activating Factor (Sigma-Aldrich, catalog number: 511075)
- Platelet surface markers:
- Anti-human CD41-PE (for detection of Integrin alpha-IIb, Biolegend, clone: HIP8, catalog number: 303706)
- Anti-human CD42b-PE (for detection of GPIb, Biolegend, clone: HIP1, catalog number: 303905)
- Activated platelet markers:
- PAC-1-FITC (for detection of activated integrin αIIbβ3, Thermofisher, clone: PAC-1, catalog number: MA5-28564)
- Anti‐CD62P‐FITC (for detection of alpha granule release, Biolegend, clone: AK4, catalog number: 304903)
- Anti-CD63-AF488 (for detection of dense granule release, Thermofisher, clone: MEM-259, catalog number: MA5-18149)
- Anti-human Annexin V-FITC (Biolegend, catalog number: 640914)
- Anti-human IL-1α-FITC (R&D, clone: 3405, catalog number: FAB200F)
- Rat monoclonal anti-mouse CD42b (Emfret, catalog number: R300)
- Sodium citrate (Sigma-Aldrich, catalog number: C8532)
- Atroxin (Sigma-Aldrich, catalog number: 11335)
- Hank’s Balanced Salt Solution (HBSS) (Sigma-Aldrich, catalog number: H9394)
- EDTA (Sigma-Aldrich, catalog number: E5134)
- Calcium Chloride (Sigma-Aldrich, catalog number: C7902)
- Cross-linked Collagen related peptide (CRP-XL) (Professor Richard Farndale, Dept. Biochemistry, Cambridge, https://collagentoolkit.bio.cam.ac.uk/thp/generic)
- Phosphate Buffered Saline (Sigma-Aldrich, catalog number: D8537)
- Sodium Azide (Sigma-Aldrich, catalog number: S2002)
- β-mercaptoethanol (Sigma-Aldrich, catalog number: M6250)
- Tris base (Sigma-Aldrich, catalog number: T6066)
- SDS (Sigma-Aldrich, catalog number: 75746)
- Bromophenol blue (Sigma-Aldrich, catalog number: 114391)
- Glycerol (Sigma-Aldrich, catalog number: G5516)
- Liquid nitrogen
- Deionized water
- Hydrochloric acid
- Serum-free DMEM (Sigma-Aldrich, catalog number: 05671)
- Calcium-free Tyrode’s buffer (Boston BioProducts, catalog number: BSS-350)
- Sodium citrate for anticoagulation of blood (see Recipes)
- Hank’s Balanced Salt Solution (HBSS) with EDTA (see Recipes)
- FACs buffer (see Recipes)
- Laemmli buffer (see Recipes)
Equipment
- -20 °C freezer
- Centrifuge with swing out rotor suitable for 15 ml tubes (Eppendorf, model: 5810R)
- Sonicator (Diagenode, Bioruptor, model: UCD-200)
- Accuri flow cytometer (BD, model: C6)
- Liquid nitrogen dewar
- Microbalance
- 37 °C incubator
- Warming box for rodents (Able Scientific, model: ASHE011AR)
- Mouse restrainer (Braintree Scientific, model: SHORTI STD)
- Weighing scales
- AggRAM Aggregometer (Helena Biosciences, model 110/220)
- Protein gel electrophoresis equipment
Software
- C6 Analysis Software (BD Accuri, catalog number: 653122), or other appropriate analysis software.
Procedure
Note: Perform all protocol steps for live platelets at room temperature–do not put platelets on ice. A Schematic overview of the whole process is shown in Figure 1.
Figure 1. Schematic overview of the processes outlined in these protocols
- Collection of human blood
Note: Sodium citrate anticoagulation is recommended to produce serum, but other anticoagulants can be used for these protocols. See Notes section for further details.- Draw 9 ml of venous blood into a 10 ml syringe with a 21 G syringe needle from a consenting adult donor. This should be performed by a competent phlebotomist.
- Remove needle from syringe and immediately and gently transfer blood into a round-bottomed 14 ml centrifuge tube containing 1 ml 3.8% sodium citrate (see Recipes).
- Mix blood with sodium citrate by gently inverting the tube three times.
- Collection of mouse blood
Note: Sodium citrate anticoagulation is recommended to produce serum, but other anticoagulants can be used for these protocols. See Notes section for further details.- Collect 75 µl blood from the mouse dorsal pedal vein, or another venous non-terminal collection site. For blood collection from the pedal vein, place mouse into a restrainer and puncture the vein with a 23 G syringe needle. Collect the blood by holding an EDTA coated capillary tube to the puncture site. Pre-warming the mice greatly aids identification of the pedal vein.
- Alternatively, collect 900 µl of blood by cardiac puncture under terminal anesthesia with a 23 G needle and 1 ml syringe. Remove needle from syringe and transfer blood immediately to a microcentrifuge tube containing 100 µl of 3.8% sodium citrate.
- Enumeration of platelets in whole blood by flow cytometry
Note: Avoid bubbles and transfer of any excess volume during pipetting.- Mix whole blood by gently inverting the tube several times.
- Using a 20 µl pipette tip, collect 10 µl of blood. Wipe outside of tip with tissue to remove excess volume.
- Add 10 µl of blood to 190 µl of FACs buffer (see Recipes). Wash out pipette tip into buffer by gently pipetting up and down.
- Mix well by gently pipetting, and discard 100 µl. Add 0.5 µl of anti-CD41-PE (or other fluorophore-conjugated platelet marker antibody) to the remaining 100 µl and mix gently.
- Incubate for 20 min at RT in the dark.
- Mix well by gently pipetting.
- Remove 40 µl with a clean pipette tip. Wipe the outside of tip with a tissue to remove excess blood. Transfer to 1,960 µl of FACs buffer.
- Mix gently by inverting the tube six times.
- Analyze immediately by flow cytometry, collecting 50 µl with an FSC-H threshold of 25,000 (or the maximum threshold that removes noise without losing platelet events) on medium speed. Backflush between each sample and wipe the sample injection port with tissue between samples to remove any excess volume and blood.
Note: Use counting beads for flow cytometers that cannot perform volume measurements. - Display data as FSC-A vs. FL2-A (or the appropriate fluorescence channel for the antibody used).
- Record the count of the positively stained platelet population by adding together the counts from the platelet gate and the platelet/RBC coincidence gate (Figure 2).
- The final dilution is 1:1005, so platelet count in blood can be determined using the calculation:
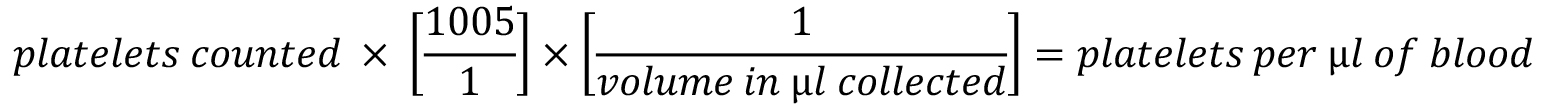
Note: If liquid anticoagulant is used, the dilution factor from this will have to be taken into account. For example, for sodium citrate 1 ml:9 ml of blood, the value from the above equation needs to be multiplied by [10/9]. Typical human and mouse platelet count is 150-450 x 103/µl, and 1,000 x 103/µl, respectively.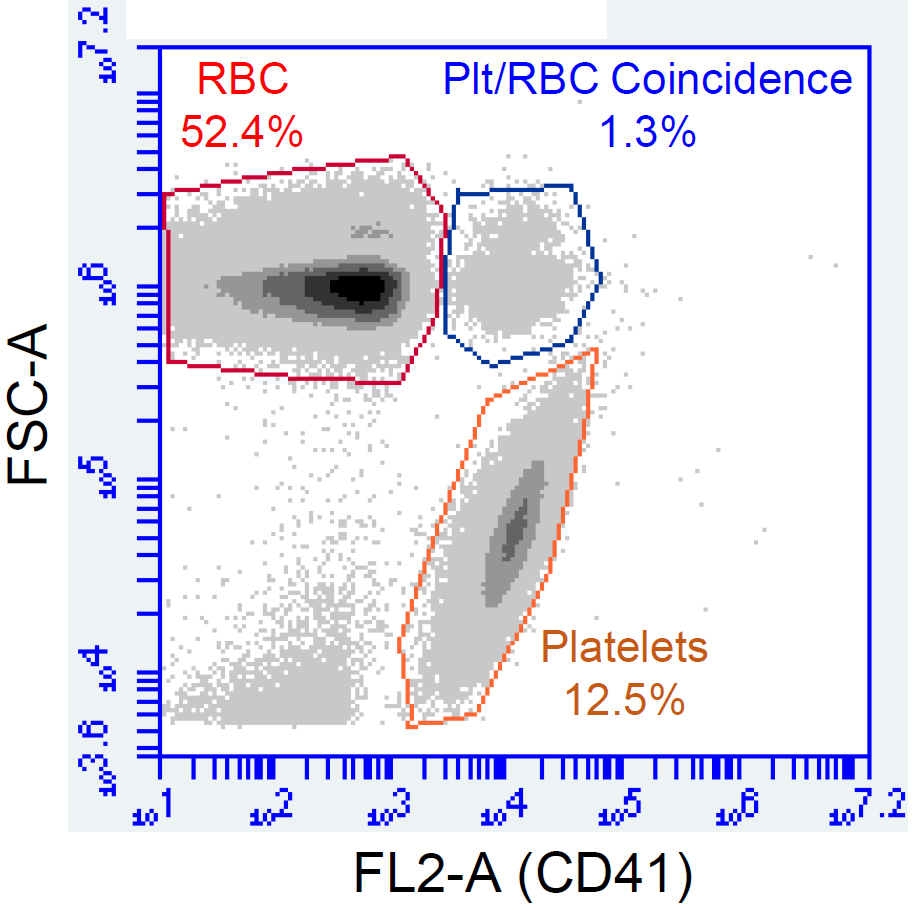
Figure 2. Example of results showing analysis of platelet population by flow cytometry. Whole blood diluted as above was stained with anti-CD41-PE prior to analysis by flow cytometry. The platelet and platelet/RBC coincidence populations are identified as having higher fluorescence in FL2.
- Production of platelet rich plasma from whole blood
- Centrifuge whole blood for 20 min at 350 x g, RT.
- The blood will have separated into two layers: the lower, dark layer of packed red blood cells, and the upper yellow layer of platelet rich plasma (PRP). The PRP volume will be approximately 50% of the whole blood volume.
- Carefully remove the upper PRP layer and transfer to a new round bottomed centrifuge tube.
- PRP may be used directly in aggregometry experiments. Further processing is required to provide serum, or to generate washed platelet suspensions.
Note: Platelets will be more concentrated in PRP than in whole blood. If the platelet count in PRP is required to make lysates or for platelet activation, the “enumeration of platelets in whole blood by flow cytometry” protocol may be used, using 10 µl of PRP at Step C3 instead of whole blood.
- Production of platelet poor plasma from platelet rich plasma
- To generate platelet poor plasma (PPP), centrifuge PRP for 10 min at 2,000 x g, RT.
- The PRP will have separated into an upper pale yellow layer (PPP) and a platelet pellet. Carefully remove the upper PPP layer, avoid disturbing the platelet pellet.
- PPP may be used as a control for aggregometry assays (see Section F), and to adjust PRP concentration. It may also be used to make PPP-derived serum, which lacks factors released from activated platelets.
- Aggregometry for assessment of platelet function
- Aliquot 250 µl of PPP into a glass aggregometry cuvette containing a stir bar.
- Insert the cuvette into the aggregometer and take a reading. This will calibrate the aggregometer to 100% aggregation. Remove the cuvette.
- Aliquot 250 µl of PRP or washed platelet suspension into a fresh aggregometry cuvette containing a stir bar. The concentration of platelets in washed platelet suspensions should be adjusted to a standardized level, with a recommended concentration of 2 x 108/ml, in calcium-free Tyrode’s buffer. For experiments using PRP, the platelet concentration can be adjusted using platelet poor plasma (PPP).
- Insert the cuvette into the aggregometer and begin recording changes in light transmission.
- After 30 s, add an agonist (see Table 1 below). Keep recording changes in light transmission for 15 min. Agonist concentrations should be adjusted to ensure that the volume of agonist does not exceed 1/100 of the platelet volume. Varying the agonist concentration can reveal information about the sensitivity of platelets to different agonists. An example aggregometry trace is shown in Figure 3.
Note: Recommended volumes may vary depending on aggregometer model, use volumes according to the manufacturer’s recommendation.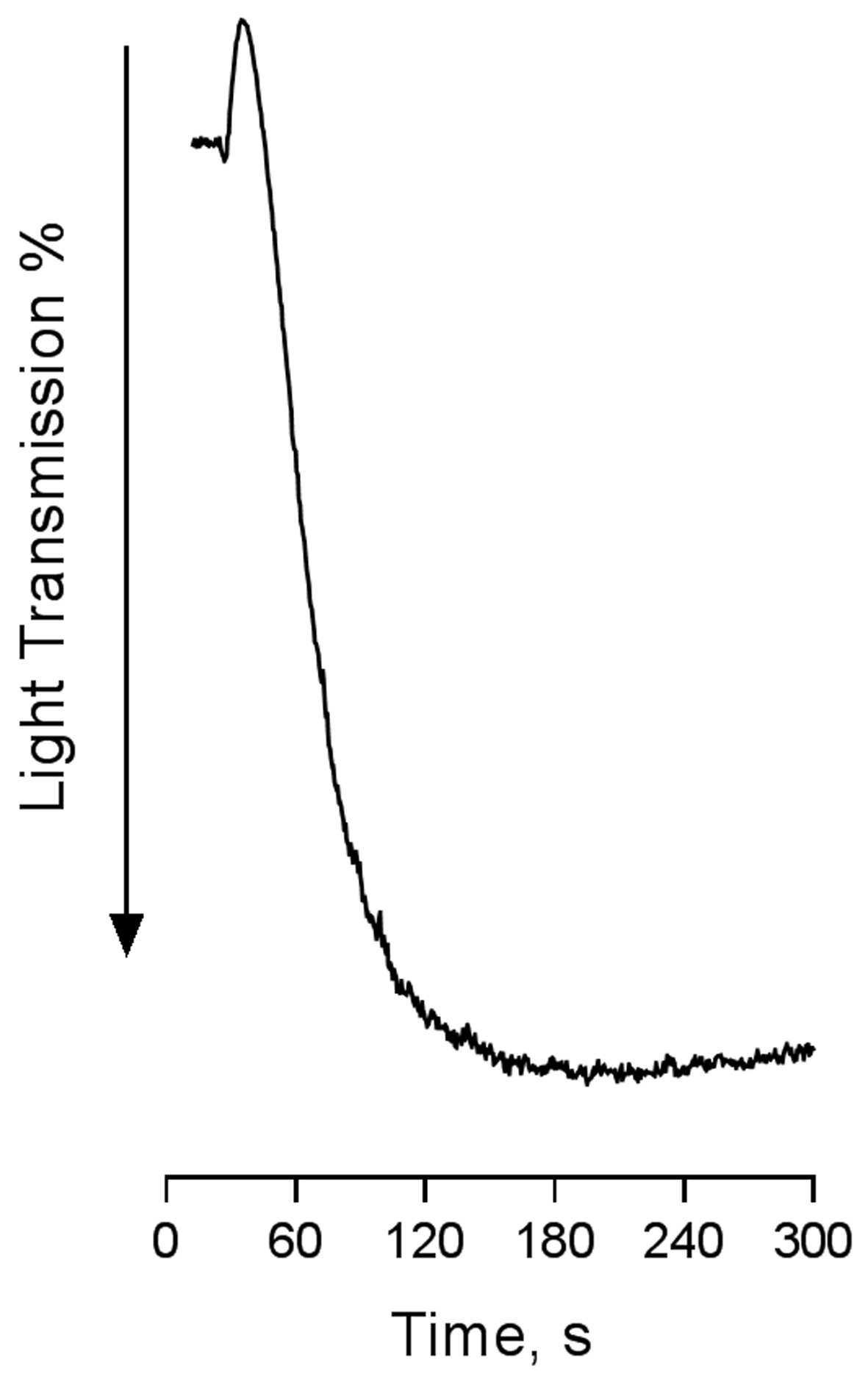
Figure 3. Representative aggregometry trace. The above aggregometry trace shows increased light transmission through a washed platelet suspension as they aggregate in response to 1 µg/ml CRP-XL.
- Production of serum from blood anticoagulated with sodium citrate
- For production of serum from PRP, add 22 µl of 1 M CaCl2 to 1 ml of PRP and incubate in a glass tube at 37 °C until a clot forms and retracts away from the sides of the tube. This takes approximately 30-45 min. Transfer the liquid phase (serum) into a clean microcentrifuge tube and store at -20 °C.
- For production of serum from PPP, add 22 µl of 1 M CaCl2 to 1 ml of PPP and incubate at 37 °C until coagulated. Coagulated PPP will form a loose clot that does not retract. Centrifuge the coagulated PPP at 10,000 x g for 1 min and transfer the upper liquid layer (serum) into a clean microcentrifuge tube. Store at -20 °C.
- Platelet washing
- Wash platelets by diluting PRP (see Procedure D) with 5 volumes of calcium-free HBSS, supplemented with 4 mM EDTA, pH 6.4 (see Recipes).
- Centrifuge for 20 min at 350 x g, RT.
- Washed platelets can be resuspended directly into media suitable for experiment.
Note: A low level of leukocytes will be present in washed platelets. Repeated rounds of the washing protocol will reduce leukocyte contamination.
- Platelet activation assays: preparation of platelets for flow cytometry or lysate production
- In order to produce results that are comparable with platelet aggregometry, conduct these experiments in an aggregometry cuvette under stirred conditions.
- Following centrifugation of platelets (see Section H), gently resuspend the platelet pellet into calcium-free, EDTA-free HBSS, pH 6.4 (see Recipes). Dilute platelets to 4 x 108/ml (giving 8 x 106 platelets in a working volume of 20 μl), adjust volumes according to the number of conditions and replicates.
- Activate platelets by the addition of platelet agonists (see Table 1 for examples). Ensure that you include a vehicle control.
- Stain platelets for flow cytometry, or make lysates for Westerns, etc.
Table 1. Examples of platelet agonists and concentrations for platelet activation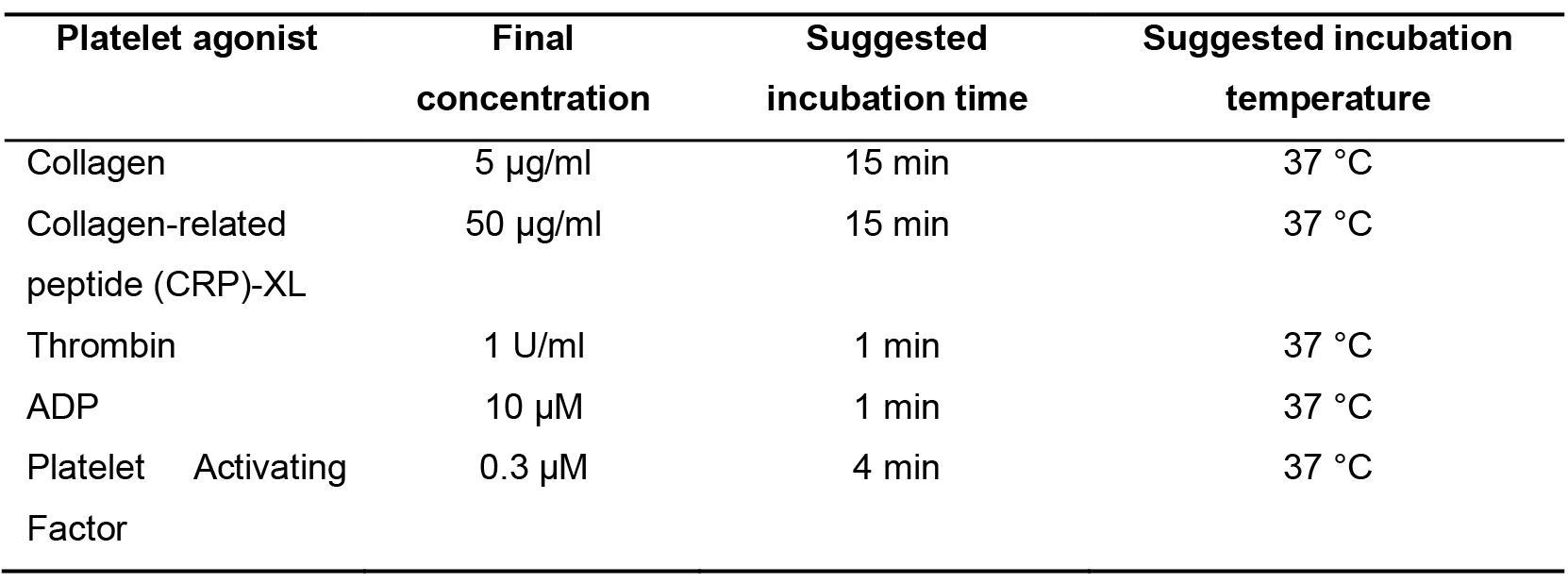
- Generation of platelet lysates for ELISA
- Following platelet activation, resuspend 8 x 108 platelets in 400 μl of serum-free DMEM. Adjust volumes according to platelet number. Use protease, phosphatase, etc, inhibitors if required.
- Freeze-thaw tubes three times using liquid nitrogen. Submerse the tube in liquid nitrogen to freeze completely, before removing and allowing to defrost completely at room temperature.
- Sonicate lysates three times for 30 s on ice.
- Centrifuge for 3 min at 13,000 x g, RT to pellet debris. Transfer supernatant to a clean microcentrifuge tube.
- Store at -20 °C until use.
- Making platelet lysates for Western Blot
- Following platelet activation, resuspend 3 x 108 platelets directly into 100 μl of 1x Laemmli buffer.
- Heat at 95 °C for 5 min, then cool on ice.
- Load 10 μl for 3 x 107 platelets per lane of an SDS-PAGE mini gel.
- Platelet staining for flow cytometry
- Stain platelet suspensions for surface proteins of interest using any standard staining protocol for flow cytometry. Keep platelets at room temperature at all times. Handling a platelet pellet from small volumes is difficult, so protocols that avoid washing steps are preferable to avoid losing platelets during supernatant aspiration.
- Co-stain 10 µl of control or activated platelets with a platelet surface marker and platelet activation marker of interest (see Tables 2 and 3 for examples).
- Dilute platelet suspensions with FACs buffer to 120 µl (see Recipes).
- Analyze platelets by flow cytometry ensuring the FSC acquisition threshold is sufficiently low to recognize the whole platelet population, whilst excluding noise (see Notes).
Table 2. Examples of antibodies for platelet marker staining
Table 3. Examples of antibodies used to quantify platelet activation markers
- Platelet depletion in mice
Note: Platelets may be depleted in vivo by administration of anti-CD42b, which targets platelets for clearance.- Determine the baseline platelet count in mice by collecting blood (~20 µl) from the pedal vein (see Section B), and platelet enumeration by flow cytometry (see Section C).
- Weigh mice using scales.
- Prepare anti-CD42b so that each mouse receives 1.8 µg/g body weight in 100-200 µl of sterile PBS. For example, for a single 25 g mouse, add 75 µl of PBS to 125 µl of 0.5 mg/ml anti-CD42b stock to achieve a working solution of 0.31 mg/ml and administer 147 µl. Use sterile PBS only as a vehicle control.
- Place mice into a warming box for rodents at 37 °C for 15 min to facilitate dilation of veins.
- Place mouse into a restrainer and administer anti-CD42b or vehicle control by intravenous tail vein injection with a 27 G needle. Hold the injection site under pressure until bleeding subsides.
- Repeat pedal vein blood sampling and platelet counts at the frequency required.
Note: Bleeding at sampling sites will be prolonged while the platelet count is low, so apply pressure for at least ten minutes and monitor hemostasis carefully.
Data analysis
- Collect events on plot using FSC-A and SSC-A parameters (Figure 4).
- Plot PE fluorescence intensity (platelet marker +ve events) on a logarithmic scale against FITC fluorescence intensity (platelet activation marker +ve events).
- Use quadrant gating on the control samples to restrict the CD41-positive control platelet population to the upper left quadrant.
- PE+/FITC+ populations will appear in the upper right quadrant for any conditions resulting in double positive staining.
- Calculate median fluorescence intensity (MFI) of upper left and upper right quadrants. To remove background, subtract the MFI of unstained cells from the stained cells.
- Calculate the mean MFI from duplicates of each condition.
- Data can also be analyzed as percentage of total cell population. For this, calculate percentage population of interest by dividing PE+/FITC+ events (upper right gate) by PE+ events (upper left and upper right gates) and multiply by 100.

Figure 4. Example of changes in activation state of platelets following stimulation with collagen. Platelets were prepared as above and stimulated with collagen prior to co-labeling with a PE-conjugated platelet marker (in this case CD41), and a FITC-conjugated platelet activation marker/protein of interest (in this case cell-surface IL-1α). Treatment with collagen results in a rightward shift in FITC-positive platelets.
Notes
- Platelets are sensitive and should be handled gently to avoid activation. Whole blood, platelet rich plasma, and washed platelets should be kept at room temperature at all times unless indicated (i.e., 37 °C).
- Sodium citrate is the recommended anticoagulant for these protocols, as its chelation of divalent cations is reversible with the addition of CaCl2, allowing for the easy production of serum. These protocols can also be performed using other non-reversible anticoagulants if necessary. If serum is required, the addition of atroxin (Bothrops atrox venom) to a final concentration of 750 ng/ml will cleave fibrinogen to fibrin and remove it from the plasma (Wentzensen et al., 2011; Karapetian, 2013).
- The detection of small particles by flow cytometry (including platelets) depends on accurately differentiating them from debris and electronic noise. For the Accuri C6, an FSC-H threshold of ~10,000 and a medium flow rate was suitable to detect the platelet population.
Recipes
- Sodium citrate for anticoagulation of blood
- Prepare a 3.8% solution of sodium citrate by dissolving 380 mg sodium citrate in 7 ml water
- Adjust pH to 7 with concentrated hydrochloric acid
- Add water to a final volume of 10 ml
- Store solution at 2-8 °C for 6 months. Allow solution to reach room temperature before use
- To anticoagulate blood with 0.38% sodium citrate final concentration, add 1 ml of 3.8% sodium citrate solution to 9 ml of whole blood. Adjust the volume of sodium citrate according to final blood volume
- Hank’s Balanced Salt Solution (HBSS) with EDTA
- Add 250 µl of 1 M EDTA to 50 ml of HBSS and mix well
- Adjust pH to 6.4 with concentrated hydrochloric acid
- Store solution at 2-8 °C for 2 weeks. Allow solution to reach room temperature before use
- FACs buffer
- Dissolve 0.5 g of BSA in 50 ml of PBS
- Add 250 µl of 10% Sodium Azide solution (0.5 g Sodium Azide dissolved in 5 ml deionized water)
- Filter through a 0.22 μm syringe filter prior to use
- Store solution at -20 °C for 6 months. Allow solution to reach room temperature before use
- 1x Laemmli buffer
To a 15 ml tube, add:
0.8 ml Tris 1 M, pH adjusted to 6.8
1 ml 20% SDS in deionized water
1 ml glycerol
0.53 ml β-mercaptoethanol
5 mg bromophenol blue
Adjust to 10 ml with deionized water
Store solution at 2-8 °C for 3 months
Acknowledgments
This work was funded by British Heart Foundation Grants FS/13/3/30038, FS/18/19/33371 and RG/16/8/32388 to MCHC, PG/18/64/33922 to NP, the BHF Cambridge Centre for Research Excellence RE/13/6/30180, and the Cambridge NIHR Biomedical Research Centre. The protocols described here are adapted from previous work (Burzynski et al., 2019).
Competing interests
The authors declare that they have no conflicts of interest, financial or otherwise.
References
- Bennett, J. S. (1996). Structural biology of glycoprotein IIb-IIIa. Trends Cardiovasc Med 6(1): 31-36.
- Boilard, E., Nigrovic, P. A., Larabee, K., Watts, G. F., Coblyn, J. S., Weinblatt, M. E., Massarotti, E. M., Remold-O'Donnell, E., Farndale, R. W., Ware, J. and Lee, D. M. (2010). Platelets amplify inflammation in arthritis via collagen-dependent microparticle production. Science 327(5965): 580-583.
- Burzynski, L. C., Humphry, M., Pyrillou, K., Wiggins, K. A., Chan, J. N. E., Figg, N., Kitt, L. L., Summers, C., Tatham, K. C., Martin, P. B., Bennett, M. R. and Clarke, M. C. H. (2019). The coagulation and immune systems are directly linked through the activation of interleukin-1α by thrombin. Immunity 50(4): 1033-1042.e1036.
- Karapetian, H. (2013). Reptilase time (RT). In: Methods in Molecular Biology. Humana Press 992: 273-277.
- Lane, D. A., Philippou, H. and Huntington, J. A. (2005). Directing thrombin. Blood 106(8): 2605-2612.
- Li, Z., Delaney, M. K., O’Brien, K. A. and Du, X. (2010). Signaling during platelet adhesion and activation. Arterioscler Thromb Vasc Biol 30(12): 2341-2349.
- Monroe, D. M., Hoffman, M. and Roberts, H. R. (2002). Platelets and thrombin generation. Arterioscler Thromb Vasc Biol 22(9): 1381-1389.
- Thornton, P., McColl, B. W., Greenhalgh, A., Denes, A., Allan, S. M. and Rothwell, N. J. (2010). Platelet interleukin-1α drives cerebrovascular inflammation. Blood 115(17): 3632-3639.
- Wentzensen, N., Rodriguez, A. C., Viscidi, R., Herrero, R., Hildesheim, A., Ghosh, A., Morales, J., Wacholder, S., Guillen, D., Alfaro, M., Safaeian, M., Burk, R. D. and Schiffman, M. (2011). A competitive serological assay shows naturally acquired immunity to human papillomavirus infections in the guanacaste natural history study. J Infect Dis 204(1): 94-102.
- Yun, S. H., Sim, E. H., Goh, R. Y., Park, J. I. and Han, J. Y. (2016). Platelet activation: the mechanisms and potential biomarkers. Biomed Res Int 2016: 9060143.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Burzynski, L. C., Pugh, N. and Clarke, M. C. (2019). Platelet Isolation and Activation Assays. Bio-protocol 9(20): e3405. DOI: 10.21769/BioProtoc.3405.
Category
Molecular Biology > Protein > Detection
Immunology > Host defense > General
Cell Biology > Cell isolation and culture > Cell isolation > Flow cytometry
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.