- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Quantification of HIV-2 DNA in Whole Blood



Published: Vol 9, Iss 20, Oct 20, 2019 DOI: 10.21769/BioProtoc.3404 Views: 6170
Reviewed by: Alka MehraKirsten A. CoprenAswad Khadilkar
Original research article
The authors used this protocol in:
Nov 2018
Advertisement




Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Time to AIDS infection is longer with HIV-2, compared to HIV-1, but without antiretroviral therapy both infections will cause AIDS-related mortality. In HIV-2 infection, monitoring of antiretroviral treatment (ART) efficacy is challenging since a large proportion of HIV-2-infected individuals displays low or undetectable plasma RNA levels. Hence, quantification of cellular DNA load may constitute an alternative method for monitoring ART efficacy. Moreover, sensitive HIV-2 DNA quantification protocols are also important for the characterization of the HIV-2 reservoirs, and ultimately for the development of HIV-2 cure strategies. We have developed a sensitive and robust HIV-2 DNA quantification protocol based on whole blood as DNA source, including normalization of leukocyte cell numbers using parallel quantification of the single copy porphobilinogen deaminase gene. The specificity and sensitivity of the assay was 100%. The limit of detection was 1 copy and limit of quantification was 5 copies. When applying this protocol to HIV-2 infected, it was found that HIV-2 viral DNA was detectable in individuals in whom viral RNA was undetectable or under quantification level. Thus, this method provides a sensitive approach to HIV-2 DNA viral quantification from whole blood of HIV-2 infected patients.
Keywords: HIV-2Background
Acquired immunodeficiency syndrome (AIDS) can be caused by two viruses, either human immunodeficiency virus (HIV) type 1 (HIV-1) or type 2 (HIV-2) (Azevedo-Pereira and Santos-Costa, 2016). Although these two retroviruses have similar life cycles, they differ in that HIV-2 displays lower pathogenicity, translating into longer progression time to AIDS and reduced transmissibility (Marvin et al.,2015; Azevedo-Pereira and Santos-Costa, 2016). Another distinct feature of HIV-2 is that the plasma viral RNA load of infected individuals is significantly lower, compared with those infected with HIV-1 (Berry et al., 1998; Popper et al., 1999; Andersson et al., 2000). The cellular viral DNA load in peripheral blood has, instead, been suggested to be similar between HIV-1 and HIV-2 infections (Ariyoshi et al., 1996; Popper et al., 2000; Damond et al., 2001). However, others have indicated that also the DNA load is lower in HIV-2, compared with HIV-1, infection (Gueudin et al., 2008). Overall, HIV-2 DNA load quantification studies are relatively few, and most studies have not considered a protocol for a house-keeping gene to quantify the number of cells used in the reaction (Ariyoshi et al., 1996; Gomes et al., 1999; Popper et al., 2000; Damond et al., 2001; Soares et al., 2006; Gueudin et al., 2008; Bertine et al., 2017).
Despite the fact that the estimated time to AIDS development in HIV-2 infected individuals is longer than for individuals with HIV-1, it is clear that HIV-2 will cause AIDS-related mortality without antiretroviral therapy (ART) in infected individuals with long follow-up (Esbjornsson et al., 2018). The monitoring of treatment efficacy is, however, challenging since a large proportion of ART-naïve HIV-2 infected individuals display low or undetectable plasma RNA levels (Berry et al., 1998; Popper et al., 1999; Andersson et al., 2000; Buggert et al., 2016; Honge et al., 2018). Hence, quantification of cellular DNA load may constitute an alternative method for monitoring of ART efficacy.
For simplicity, robustness and the requirement of small volumes of blood, viral DNA quantification protocols based on whole blood, as the source of DNA, are desirable. Currently there are, however, a limited number of publications where HIV-2 DNA quantification has been based on whole blood (Bertine et al., 2017), and the need for protocols combining sensitivity with robustness is still important.
In a recent study (Esbjornsson et al., 2018), we showed that both HIV-1-infected and HIV-2-infected individuals have a high probability of developing AIDS without antiretroviral treatment.
In HIV-1 infection it has been shown that viral DNA load is an independent marker of disease progression and is well correlated with the number of latently HIV-1 infected cells, that comprises the viral reservoir (Parisi et al., 2012; Rouzioux and Avettand-Fenoel, 2018). Comprehensive studies on the HIV-2 reservoirs are, however, lacking. Thus, sensitive HIV-2 DNA quantifications protocols are also important for the characterization of the HIV-2 reservoirs, and ultimately for the development of HIV-2 cure strategies. We therefore decided to develop a sensitive and robust cellular HIV-2 DNA quantification method based on whole blood as DNA source, including normalization of leukocyte numbers using parallel quantification of the single copy porphobilinogen deaminase gene (PBDG) (Raich et al., 1986; Mbisa et al., 2009).
Materials and Reagents
- DNA extraction
- Filter tips (1,000 μl filter tip: Sarstedt, catalog number: 70.762.211; 200 μl filter tip: Sarstedt, catalog number: 70.760.211; 20 μl filter tip: Sarstedt, catalog number: 70.760.213)
- Low Binding Eppendorf tubes (DNA LoBind Snap Cap PCR Tube, 1.5 ml) (Eppendorf, catalog number: 022431021)
- PM1 cells (Lusso et al., 1995)
- Whole Blood specimen of HIV-2 infected individuals, storage in -70 °C freezer. Collect samples in EDTA tubes (BD Life Sciences, Vacutainer spray-coated K2EDTA tubes, catalog number: 367841)
- Whole Blood specimen of uninfected individuals, storage in -70 °C freezer. Collect samples in EDTA tubes (BD Life Sciences, Vacutainer spray-coated K2EDTA tubes, catalog number: 67841)
- Carrier RNA (poly A) (1350 μg) (Qiagen, catalog number: 1017647) store at room temperature before use, then store at -20 °C
- GibcoTM RPMI 1640 Medium (Thermo Fisher Scientific, Fisher Scientific, catalog number: 11875093), store at 4 °C before opening the bottle
- GibcoTM Fetal Bovine Serum (FBS) (Thermo Fisher Scientific, Fisher Scientific, catalog number: 26140079), storage in -20 °C freezer before heat inactivation, then portion it and store at -20 °C
- GibcoTM Penicillin-Streptomycin (10,000 U/ml) (Thermo Fisher Scientific, Fisher Scientific, catalog number: 15140122), store at -20 °C
- QIAamp DNA Mini Kit (Qiagen, catalog number: 51304), store at room temperature
- QIAamp Blood DNA Mini Kit (Qiagen, catalog number: 51104), store at room temperature
- GibcoTM Phosphate buffered saline (PBS, pH = 7.4) (Thermo Scientific, Fisher Scientific catalog number: 10010023), store at room temperature
- Complete RPMI-1640 medium (see Recipes)
- Preparation of Standards
- Filter tips (1,000 μl filter tip: Sarstedt, catalog number: 70.762.211; 200 μl filter tip: Sarstedt, catalog number: 70.760.211; 20 μl filter tip: Sarstedt, catalog number: 70.760.213)
- Pipette Tips (1,000 μl filter tips: Sarstedt, catalog number: 70.762.100; 200 μl filter tips: Sarstedt, catalog number: 70.760.502; 10 μl tips: Sarstedt, catalog number: 70.1130)
- Low Binding Eppendorf tubes (DNA LoBind Snap Cap PCR Tube, 1.5 ml) (Eppendorf, catalog number: 022431021)
- MicroAmpTM Optical 8-Tube Strip, 0.2 ml (Thermo Scientific, Applied BiosystemsTM, catalog number: 4316567)
- MicroAmpTM Optical 8-Cap Strips (Thermo Scientific, Applied BiosystemsTM, catalog number: 4323032)
- HIV-2 (NIH-Z Strain) Purified Virus and electron microscopy counted (Advanced Biotechnologies Inc, catalog number: 10-127-000), storage in -70 °C freezer
- Qiagen miRNeasy micro Kit (Qiagen, catalog number: 217084), store at 4 °C
- DNA oligonucleotides primers were obtained from Invitrogen and listed in Table 1, store oligonucleotides at -20 °C
- SuperScriptTM III One-Step RT-PCR System with PlatinumTM Taq DNA Polymerase (Thermo Fisher Scientific, Invitrogen, catalog number: 12574018), storage in -20 °C freezer. SuperScriptTM III One-Step RT-PCR System is supplied with the following:
- SuperScript® III RT/Platinum® Taq Mix (50 μl)
- 2x Reaction Mix (containing 0.4 mM of each dNTP, 3.2 mM MgSO4) (1 ml)
- 5 mM Magnesium Sulfate (500 μl)
- PlatinumTM Taq DNA Polymerase High Fidelity (Thermo Fisher Scientific, Invitrogen, catalog number: 11304011), storage in -20 °C freezer. Platinum® Taq DNA Polymerase High Fidelity (20 μl, at 5 U/μl) is supplied with the following:
- 10x High Fidelity Buffer [600 mM Tris-SO4 (pH 8.9), 180 mM (NH4)2SO4] (1.25 ml)
- 50 mM MgSO4 (1 ml)
- Agarose (Thermo Fisher Scientific, Invitrogen, catalog number: 16500500), store at room temperature
- Milli-Q quality water (RNase, DNase free water [dH2O]) (VWR, catalog number: SH30538.02), store at room temperature
- AccuGENETM 50x TAE Buffer (Lonza, catalog number: 51216), store at room temperature
- Gel Red (VWR International, catalog number: 41003), store at room temperature
- QIAquick PCR purification Kit (Qiagen, catalog number: 28104), store at room temperature
Table 1. Primers used for HIV and PBDG standard preparation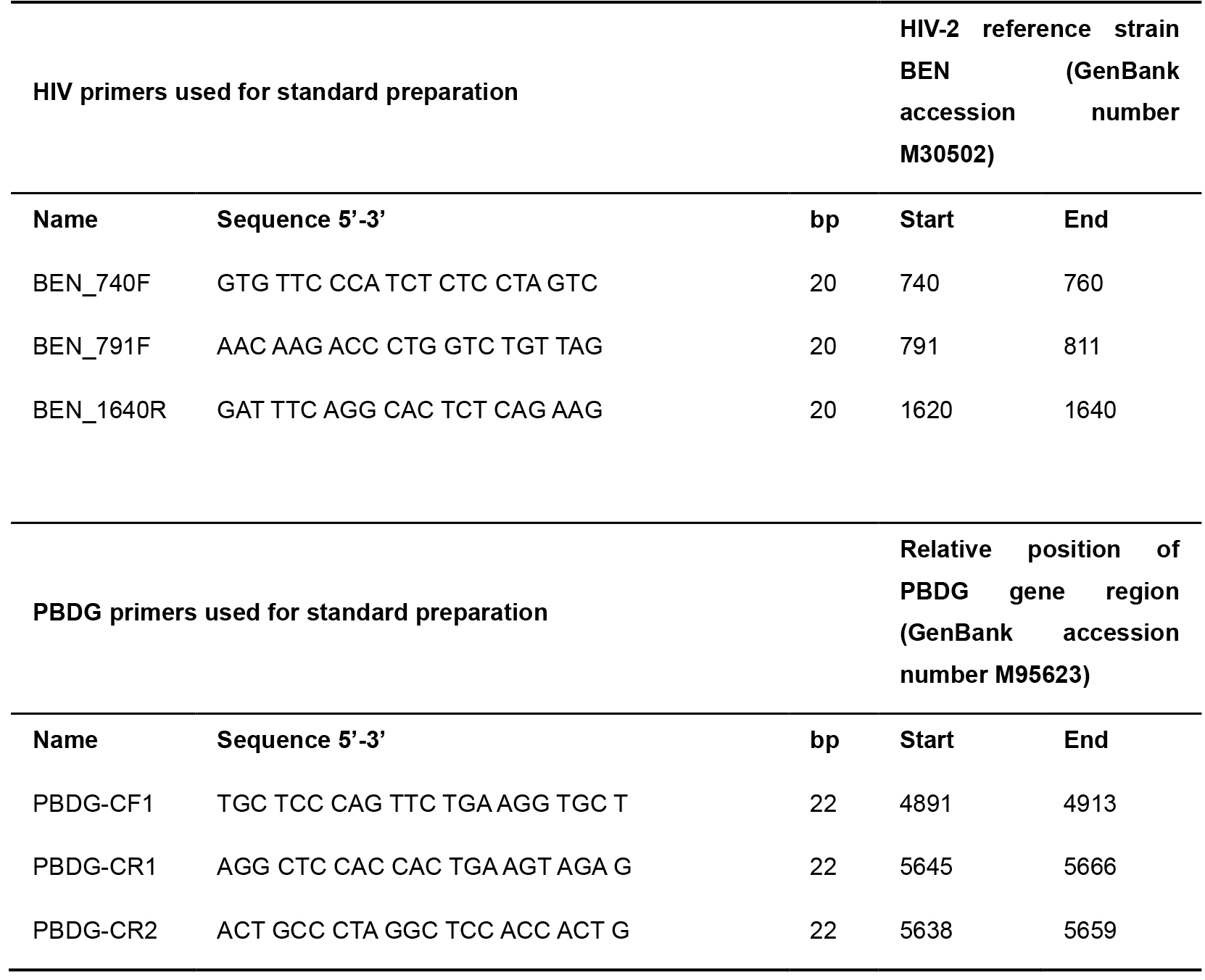
- qPCR for HIV-2 DNA detection
- Pipette tips (1,000 μl filter tips: Sarstedt, catalog number: 70.762.100; 200 μl filter tips: Sarstedt, catalog number: 70.760.502)
- 96-well qPCR plates (Applied BiosystemsTM, catalog number: 4346906)
- Optical Adhesive Film (Applied BiosystemsTM, catalog number: 4360954)
- HyCloneTM Water, Molecular Biology Grade (RNase, DNase, proteinase free) (Thermo Fisher Scientific, Fisher Scientific, GE Healthcare HyClone SH30538.FS, catalog number: 10247783), store at room temperature
- HIV-1 infected U937 Cells (U1) (Folks et al., 1987)
- Maxima probe qPCR Master Mix (2x) (Thermo Scientific, catalog number: K0231), storage in -20 °C freezer. qPCR Master Mix contains the following:
- 2x Maxima Probe/ROX qPCR Master Mix containing Maxima Hot Start Taq DNA Polymerase and dNTPs (1.25 ml)
- Nuclease-Free Water (1.25 ml)
- Primer and probe sequences for HIV-2 was obtained from Damond et al. (2002) (PMID: 12354861). Storage in -20 °C freezer
- Primer and probe sequences of the housekeeping human gene PBGD was obtained from Mbisa et al. (2009) (PMID 19020818). Storage in -20 °C freezer
- Agarose (Thermo Fisher Scientific, Invitrogen, catalog number: 16500500), store at room temperature
- Milli-Q quality water (RNase, DNase free water [dH2O]) (VWR, catalog number: SH30538.02), store at room temperature
- AccuGENETM 50x TAE Buffer (Lonza, catalog number: 51216), store at room temperature
- Gel Red (VWR International, catalog number: 41003), store at room temperature
- 1 Kb Plus DNA Ladder (Thermo Fisher Scientific, Invitrogen, catalog number: 10787018), storage in -20 °C freezer
Equipment
- EVETM Automated Cell Counter, NanoEnTek (VWR, Avantor, catalog number: 10027-452)
- EVETM Cell Counting Slides with Trypan Blue (VWR, Avantor, catalog number: 10027-446)
- Eppendorf Microcentrifuge, model: 5415D, with rotor F 45-24-11, AC/DC input 230 V AC, 50-60 Hz (Sigma-Aldrich, catalog number: Z604062)
- Life Eco Thermal Cycler, 96 well gradient, 100-240V, 50-60Hz, 600W, (Bioer Technology Co., catalog number: BTC42096)
- Gilson single-channel/adjustable-volume pipettes: 0.1-2 μl, 2-20 μl, 20-200 μl, 100-1,000 μl (Eppendorf, catalog number: F167380)
- Applied Biosystems StepOnePlus Real-Time PCR System (Thermo Scientific, Applied BiosystemsTM, catalog number: 4376600)
- NanoDrop ND-1000 spectrophotometer (Thermo Scientific, catalog number: ND-2000C)
- Mini ReadySub-CellTM electrophoresis system (Bio-Rad Laboratories, catalog number: 1704467)
- UV-Transparent gel tray (Bio-Rad Laboratories, catalog number: 1704435)
- 8-well Comb (Bio-Rad Laboratories, catalog number: 1704463)
- Gel DocTM XR+ Gel Documentation System (Bio-Rad Laboratories, catalog number: 1708195)
Software
- Image Analysis Software, Image LabTM Software (Life Sciences, Bio-Rad)
- Excel 2016 (Microsoft)
- GraphPad Prism 7.0.
Procedure
Main steps of preparation of HIV-2 qPCR:
- Extract DNA from PM1 cells as outlined below Steps A1 and A2.
- Prepare PBDG, cellular controls as indicated in Procedure B “Preparation of PBDG standard” by using extracted DNA of PM1 cells.
- Extract viral RNA -from HIV-2 isolate- to prepare HIV-2 standards, as mentioned in Procedure B “Preparation of HIV-2 standard”.
- Extract viral DNA from whole blood as indicated in Step A3.
- Set up HIV-2 qPCR as outlined below Procedure C.
- DNA extraction
- Collect 5 x 106 PM1 cells (Lusso et al., 1995) in 1.5 ml Eppendorf tubes and centrifuge for 5 min at 200 x g. Discard supernatant and wash pellets twice in 1 ml of sterile PBS. Store dried pellet at -20 °C until DNA extraction.
Notes:- PM1 cell line is a modified CD4+ T-cell clone, which expresses co-receptors such as CCR5 and CXCR4 (Lusso et al., 1995).
- Maintain PM1 cells in complete RPMI-1640 (Invitrogen) (see Recipes) until passage number 3, after defreezing. Count cells by using Automated Cell Counter.
- U1s are subclones of U937 chronically HIV-1 infected cells (Folks et al., 1987). These cells are carrying integrated HIV-1 in their genome, thus extracted DNA can be used as a negative control, and to detect the specificity.
- For negative control, collect 5 x 106 U1 cells (Folks et al., 1987) as the same way as it is mentioned above for PM1 cells. And count cells by using Automated Cell Counter.
- To extract cellular DNA of PM1 cells use Qiagen QIAamp DNA Mini Kit according to the manufacturer’s instructions. A single elution of 50 μl buffer AE is used.
Notes:- DNA extractions should be performed in a cell biology-grade clean hood equipped with separate air-handling mechanisms or under a DNA-free hood. Add extra centrifugation step before elution to decrease the EtOH contamination: 20,000 x g for 1 min. For elution use prewarmed Buffer AE (56 °C) and incubate it for 2 min at room temperature.
- For negative control, extract DNA from collected U1 cells (Folks et al., 1987) by using Qiagen QIAamp DNA Mini Kit according to the manufacturer’s instructions, then elute in 50 μl buffer AE.
- Extract viral DNA from 200 μl of whole blood in the presence of 5 μg carrier RNA using Qiagen QIAamp Blood DNA Mini Kit according to the manufacturer’s instruction. Load DNA to spin column and elute in 50 μl elution buffer.
Notes- DNA extractions should be performed in a biosafety cabinet.
- Whole Blood sample should be collected in EDTA tubes and stored at -70 °C until DNA extraction.
- To reduce degradation of DNA, avoid freezing/thawing of the whole blood samples.
- Extraction should be performed by using low-binding Eppendorf tubes.
- Store DNA extracts at -20 °C for up to a few weeks.
- Collect 5 x 106 PM1 cells (Lusso et al., 1995) in 1.5 ml Eppendorf tubes and centrifuge for 5 min at 200 x g. Discard supernatant and wash pellets twice in 1 ml of sterile PBS. Store dried pellet at -20 °C until DNA extraction.
- Preparation of Standards
Preparation of HIV-2 standard- Prepare the HIV-2 standard by extracting RNA from 1.44 x 1011 RNA-copies/ml of electron microscopy counted HIV-2 particles using Qiagen miRNeasy micro Kit according to the manufacturer’s instructions with minor modifications.
Notes:- HIV-2 control stock solution has a concentration of 7.2 x 1010 viral particles/ml. As two RNA genomes are packed in each viral particle the final stock contains: 1.44 x 1011 RNA-copies/ml = 1.44 x 108 copies/μl (7.2 x 1010multiply with two). Extract a sample (HIV-2) with 5 μl from the stock + 95 μl PBS. Extracted RNA should be eluted in 30 μl. In this case, the concentration of sample is 2.4 x 107 copies/μl ((1.44 x 108 x 5)/30). Use 10-fold dilution series to reach 5,000 copies/μl RNA concentration.
- By using miRNeasy micro Kit purification gives a better yield and allows elution in smaller volume.
- To increase the yield of extracted RNA: increase the amount or qiazol reagent from 700 μl to 1000 μl. Double incubation time (10 min instead of 5 min). Use 200 μl of chloroform instead of 140 μl during extraction.
- To improve the yield of extraction add 5 μl carrier RNA (1 μg/ μl) before and after transferring the aqueous phase.
- Wash it twice by using 350 μl RWT buffer and between washing steps add DNase to decrease the amount of DNA contamination.
- Elute RNA in 30 μl of RNase-free water.
- Perform cDNA synthesis directly after RNA extraction by using SuperScript III One-Step RT-PCR System and Platinum Taq DNA polymerase. Prepare cDNA synthesis in 25 μl reaction volume containing 2x reverse transcription buffer (0.4 mM of each dNTP, 3.2 mM MgSO4), 1 μl of eluted total RNA, 100 ng forward and reverse primers, SuperScriptTM III One-Step RT-PCR System with PlatinumTM Taq DNA Polymerase and RNase/DNase–free sterile water up to the final volume (see Table 2).
Table 2. Preparation of cDNA synthesis by reverse transcription and first round nested PCR
- Quantify the HIV-2 gag fragment of RT-PCR (850 base pairs) using the following program: initial cDNA synthesis for 30 min at 50 °C, followed by amplification: initial denaturation for 2 min at 94 °C, 35 cycles of 15 s at 94 °C, 30 s at 50 °C, 45 s at 68 °C and a final elongation step for 5 min at 68 °C.
Note: By using primer pair BEN_740F and BEN_1640R plus strand of HIV-2 gag can be amplified. The estimated size of the amplified fragment is 850 base pairs. - Visualize PCR products on 1% agarose gels stained with Gel Red, run at 110 V for 45 min in 1x TAE buffer.
- Purify PCR Product by using QIAquick PCR purification Kit according to the manufacturer’s instruction. Load DNA to spin column and elute in 50 μl sterile water. Quantify the concentration of PCR product by using NanoDrop ND-1000 spectrophotometer and visualize it on 1% agarose gel with the same running conditions (see Figure 1).
- Purified HIV-2 gag should be stored at -20 °C until needed.
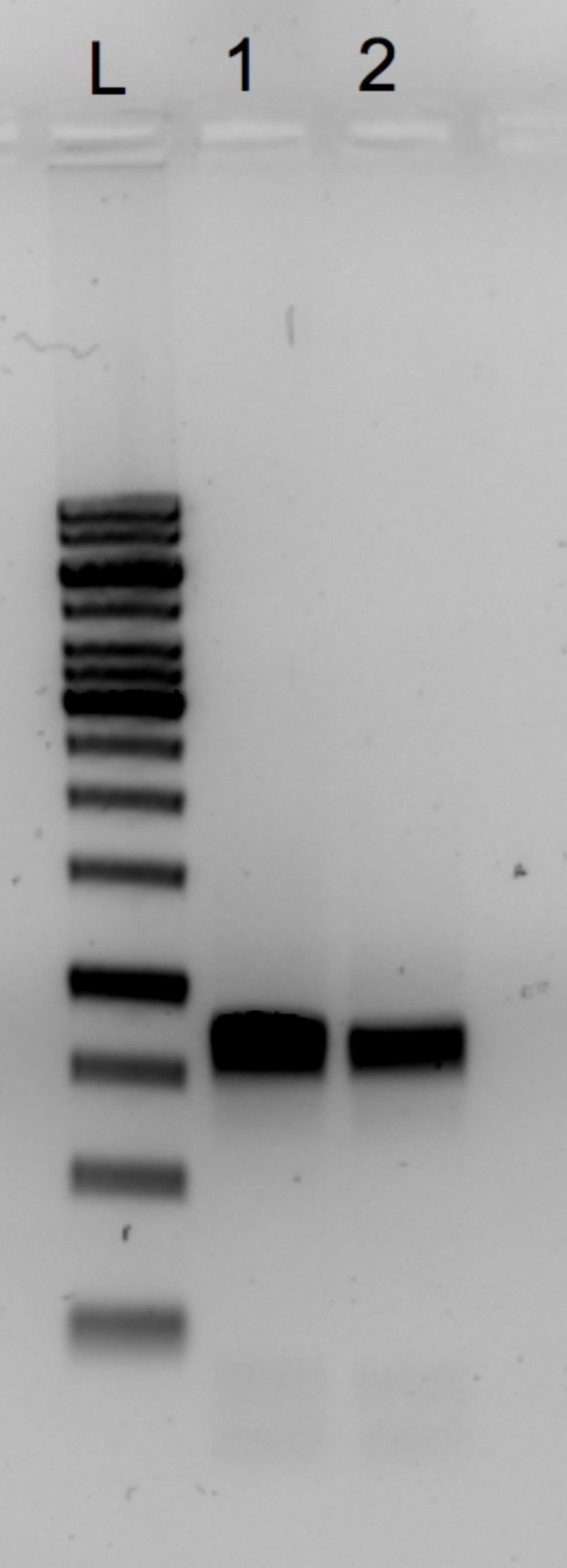
Figure 1. Detection of cleaned PCR product of HIV-2 gag PCR. An example of successful of PCR purification is visualized on 1% Agarose gel stained with Gel red. L = 1 KB DNA ladder, 1 and 2 = HIV-2 PCR products, with the estimated size of 850 bp.
- Perform host gene standard (PBDG fragment) amplification in two steps (outer followed by nested PCR). Prepare outer PCR in 25 μl solution containing: 10 ng eluted DNA of PM1 cells, Dream Taq DNA polymerase, 5 mM dNTP mix and 100 ng/μl primers (PBGD-CF1 and PBGD-CR2) (see Table 3). Visualize PCR product on 1% agarose gel stained with Gel Red, run at 110 V for 45 min in 1x TAE buffer.
- Reaction of nested PCR solution should contain: 1 μl of outer PCR product, Dream Taq DNA polymerase, 5 mM dNTP mix and 100 ng/μl primers (PBGD-CF1 and PBGD-CR1). Detect PCR product on 1% agarose gel stained with Gel Red at the same conditions as aforementioned.
Table 3. Host gene performed by 2-step PCR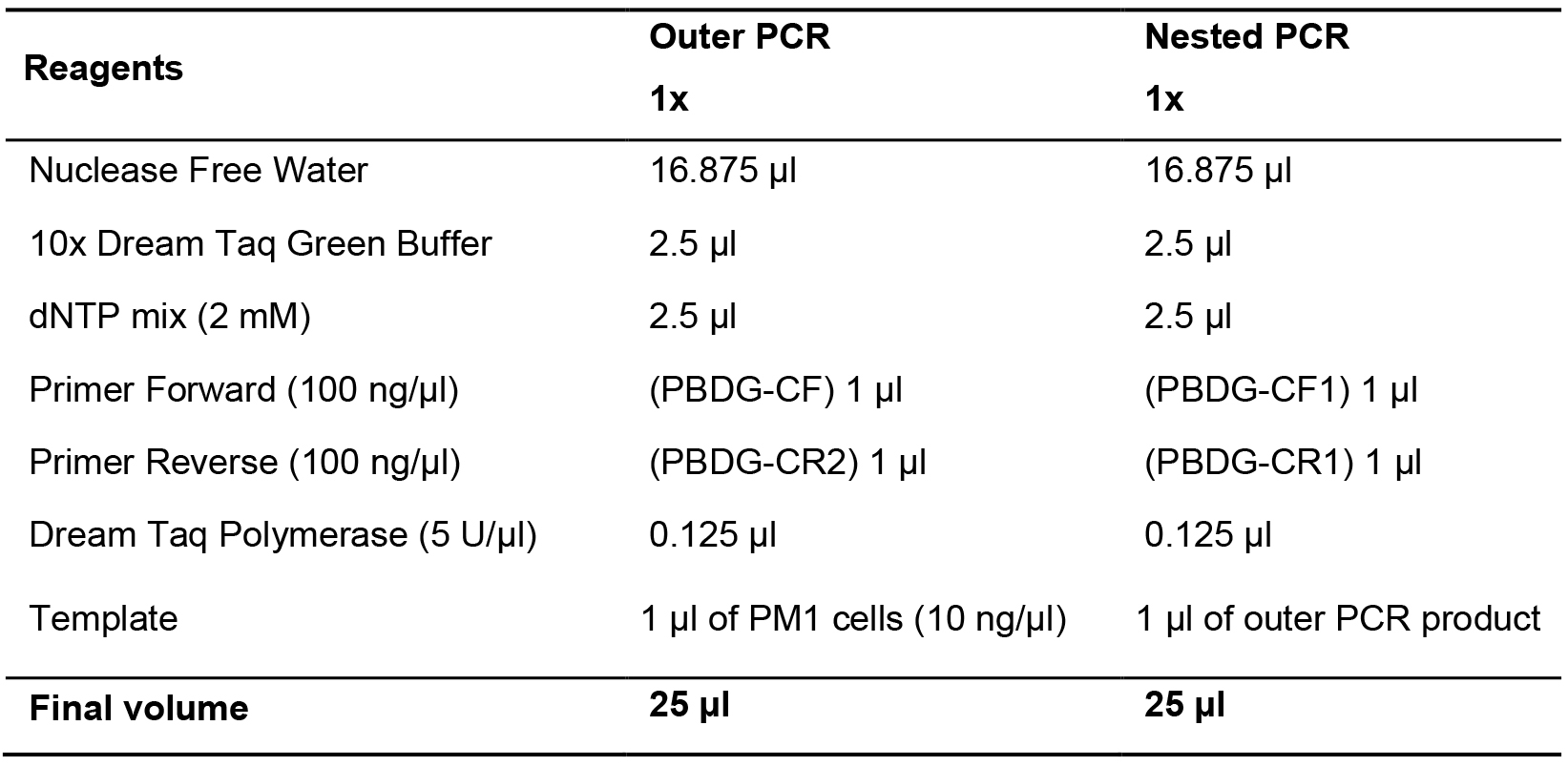
- Outer and Nested PCR cycling parameters: an initial denaturation 2 min at 94 °C followed by 35 cycles of 30 s at 94 °C, 30 s at 55 °C, 45 s at 72 °C and a final elongation step for 5 min at 72 °C.
- Purify PCR product by using QIAquick PCR purification Kit according to the manufacturer’s instruction. Load DNA to spin column, elute in 50 μl sterile water and quantify the concentration of PCR product by using NanoDrop ND-1000 spectrophotometer and detect the product on 1% agarose gel with the same running conditions as mentioned before.
Notes:- For RNA elution use RNase/ DNase/ proteinase free water.
- cDNA synthesis and One-Step PCR reactions are conducted in 0.2 ml, MicroAmpTM Optical 8-Tubes.
- Stock and dilutions must be kept on ice after thawing and mixing, and frozen at -20 °C when not in use. Pipette mix or flick mix samples and dilutions, do not vortex to mix.
- RNA extractions, dilution of RNA and DNA, PCR setups must be done in the amplicon-free room and should be performed using filtered pipette tips.
- Modified PCR protocol of Damond et al. (2002) is used for HIV-2 qPCR.
- Prepare the HIV-2 standard by extracting RNA from 1.44 x 1011 RNA-copies/ml of electron microscopy counted HIV-2 particles using Qiagen miRNeasy micro Kit according to the manufacturer’s instructions with minor modifications.
- qPCR for HIV-2 DNA detection
Main steps of the qPCR (see Figure 2 for an example of experiment layout): 1) Prepare Real-Time PCR reaction of HIV-2 from extracted DNA of infected and uninfected individuals and with sterile water instead of DNA, as mentioned in Steps C1, C2 and C3. 2) Prepare PCR reaction for HIV-2 standard from HIV-2 gag fragment corresponding to 106 to 1 DNA copies as mentioned in Step C5. 3) Prepare Real-Time PCR reaction of PBDG from extracted DNA of infected and uninfected individuals and with sterile water instead of DNA as mentioned in Step C6. 4) Prepare PCR reaction for PBDG standard from purified housekeeping gene fragment corresponding to 106 to 100 DNA copies as mentioned in Step C7. 5) All reactions need to be performed using a 96-well plate. 6) Carry out qPCR reaction in the conditions mentioned in Step C10. 7) Calculate the concentration of samples by using the formula in Step C11.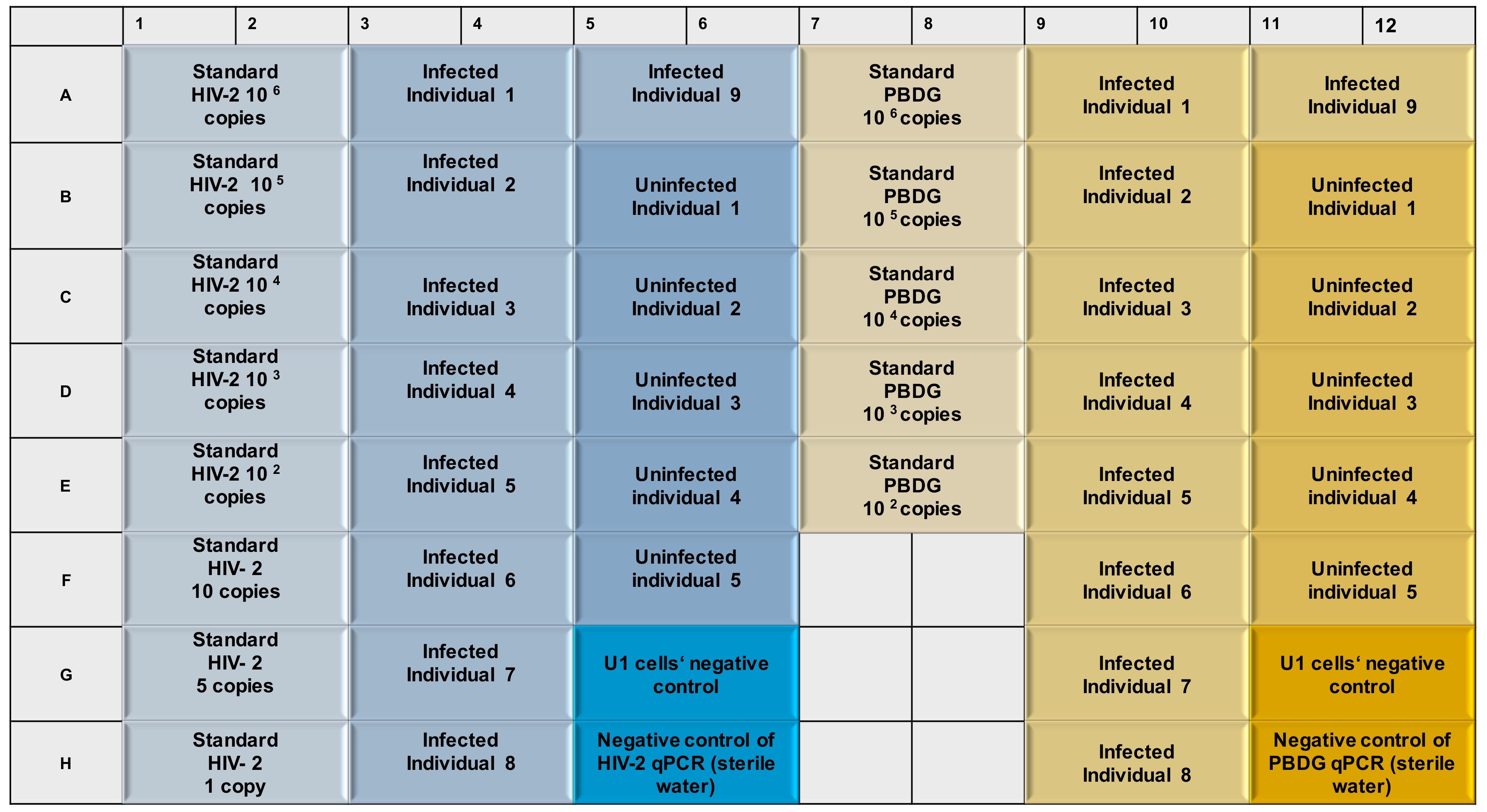
Figure 2. Example of the experimental layout. Blue colors indicate the qPCR of HIV-2 gag, while yellow colors stand for Real-Time PCR of PBDG gene. Standard curves are generated from eight (for HIV-2) (106 to 1 copies) and from five dilutions (for PBDG) (106 to 100 copies) dilutions. Each sample and control are prepared in duplicates on a 96-well qPCR plate. DNA of U1 cells stands for negative and specificity controls (as these cells are carrying an integrated HIV-1 genome, but no HIV-2 genome).- Prepare the following Real-Time PCR reactions for HIV-2 in 30 μl solution (quantities per sample) containing 15 μl Thermo Fisher Maxima probe qPCR Master Mix, 4 μM of Taqman Probe, 100 ng/μl of each primer and 8 μl of template DNA.
Notes:- Sequences of primers and probe were obtained from Damond et al. (2002) (PMID:12354861).
- Extract sample DNA from 200 μl of whole blood as mentioned in Step A3. To improve the yield of extraction use carrier RNA during viral DNA extraction.
- Since carrier RNA was used in the extraction, it is not possible to quantify the DNA reliably since spectrophotometric quantification will detect both RNA and DNA in the solution.
- As a negative control, add 8 μl of sterile water instead of DNA.
- Set up the same reaction by using DNA from uninfected individuals to ensure that the primers are specific for viral DNA detection.
Note: Extract DNA from 200 μl of whole blood of uninfected individuals in the same way as it was performed for infected samples (see Step A3). - Also, prepare the reaction in the presence of 1 μg PM-1 cell’s DNA to exclude whether or not human DNA contamination has an effect on the sensitivity of real-time PCR.
- For quantification, use a series of ten-fold dilutions of the HIV-2 gag fragment corresponding to 106 to 1 DNA copies of the standard, and include it in each experiment in order to generate an external standard curve. Each sample and standard should be quantified in duplicate (see Figures 3 and 4).
Note: The most critical step of HIV-2 qPCR is the preparation of standards. Prepare the dilutions by using 1.5 ml low-binding Eppendorf tubes. Mix every tube by pipetting up and down, then centrifuge it before preparing the next dilution.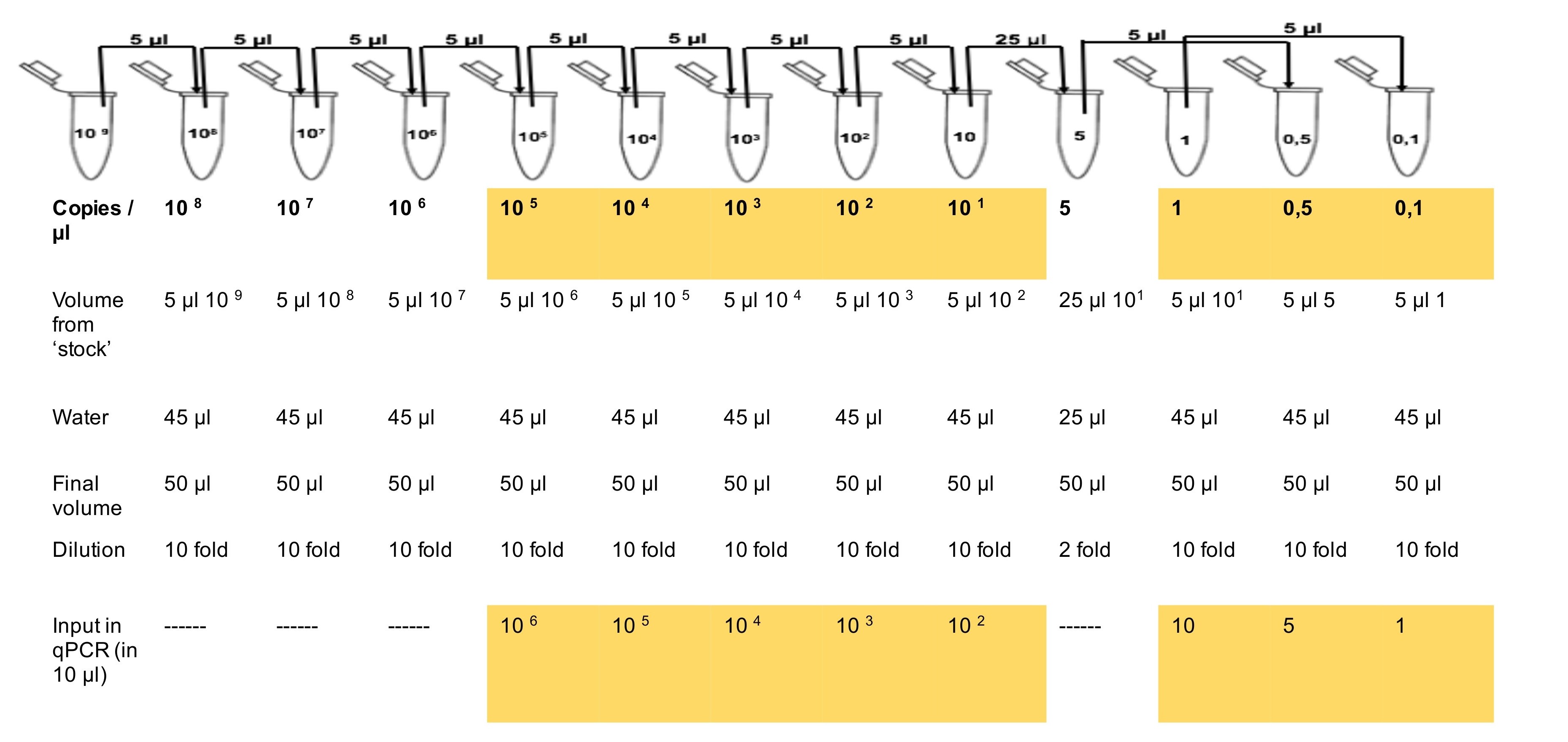
Figure 3. The serial dilution scheme for HIV-2 standard. Prepare serial dilution in 1.5 ml Eppendorf tubes by pipetting 5 μl from 109 ‘stock’ to 108 copies/μl tube and add 45 μl water. Pipette up and down to mix and shortly centrifuge it. Using a new tip, pipette 5 μl from tube 108 to tube 107. Pipette up and down to mix and centrifuge it for a few seconds. Repeat the same process for the other tubes. Yellow highlighted shows dilutions used to perform HIV-2 standard curve.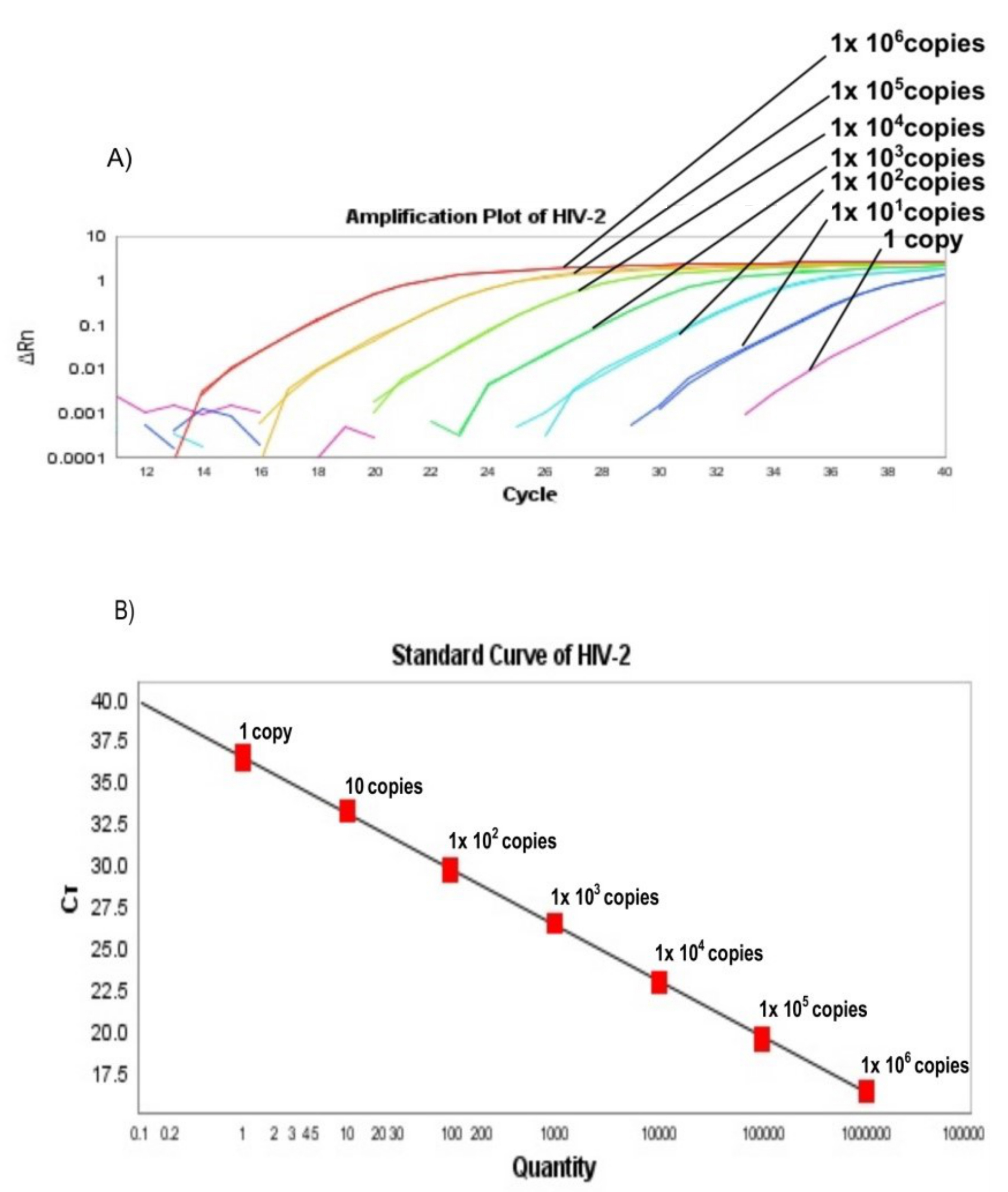
Figure 4. Standard curve of HIV-2 gag fragment. A. Sensitivity test for TaqMan Probe for gene of interest. Amplification plot showed the testing results of the tenfold dilution series of host gene (106, 105, 104, 103, 100, 10 and 1 DNA copies per reaction in duplicate). B. The linear regression analysis between the quantities of host gene (106, 105, 104, 103, 100, 10 and 1 DNA copies per reaction in duplicate) and Ct values. Regression equations were calculated with y = -3.305 + 38.692, R2 = 0.999. The red box represents Ct values. - Normalize HIV-2 DNA to expression of the housekeeping human gene, PBGD. Prepare the following reaction: 12.5 μl Thermo Fisher Maxima probe qPCR Master Mix, 50 ng/μl primers, 2 μM of probe and 3 μl of template DNA.
Notes:- Primer and probe sequences of PBGD gene were obtained from Mbisa et al. (2009) (PMID 19020818).
- Use extracted DNA of uninfected and infected individuals as a template DNA. For preparation of DNA, see Step A3.
- For each run, a standard curve should be generated from purified PBDG fragment ranging from 106 to 100 DNA copies (see Figure 5).
Notes:- Prepare the same reaction for PBDG standard as mentioned in Step C6.
- Similar to HIV-2 standard preparation, dilutions of PBDG should be performed by using 1.5 ml low-binding tubes. Mixing by pipetting up and down, then centrifugation is necessary before every step of dilution.
- Distribute the reaction mix in the wells of the 96-well qPCR plate and avoid air bubbles. An example of a plate layout is provided in Figure 2.
- Cover the qPCR plate with an adhesive plastic film, centrifuge it briefly.
- Carry out qPCR with an initial denaturation step at 95 °C for 10 min, followed by 40 cycles of denaturation at 95 °C for 15 s, annealing at 60 °C for 30 s and final extension at 72 °C for 30 s.
- HIV-2 DNA concentrations should be first reported as HIV-2 DNA copies/μl obtained from the qPCR reaction. The numbers of copies of HIV-2 DNA are then normalized to the host gene. The final results are reported as the number of copies/105 cells. The formula to convert the results is: (HIV-2 DNA copies per μl)/(PBDG copies per μl/2 chromosomes per cell) x 105 cells = HIV-2 DNA copies per 105 cells.
Note: Perform quantitative real-time PCR (qPCR) by using the Applied Biosystems StepOnePlus Real-Time PCR System. Extracted DNA from U1 cells, HIV negative and HIV-1 infected individuals can be used as controls.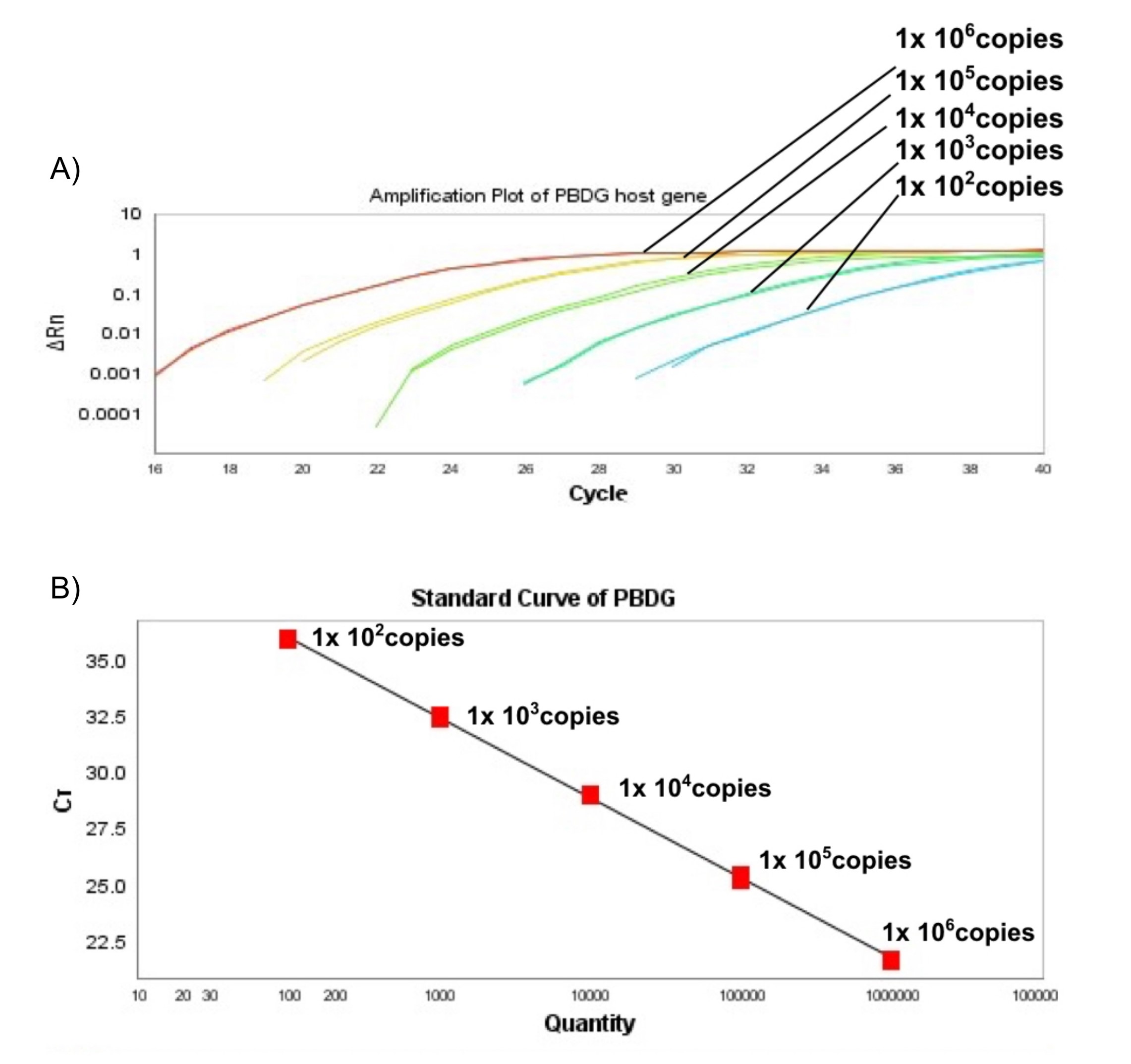
Figure 5. Standard curve for PBDG (host gene). A. Sensitivity test for TaqMan Probe for PBDG host gene. Amplification plot showed the testing results of the tenfold dilution series of host gene (106, 105, 104, 103 and 100 DNA copies per reaction in duplicate). B. The linear regression analysis between the quantities of host gene (106, 105, 104, 103 and 100 DNA copies per reaction in duplicate) and Ct values. Regression equations were calculated with y = -3.483 + 39.893, R2 = 0.999. The red box represents Ct values.
- Prepare the following Real-Time PCR reactions for HIV-2 in 30 μl solution (quantities per sample) containing 15 μl Thermo Fisher Maxima probe qPCR Master Mix, 4 μM of Taqman Probe, 100 ng/μl of each primer and 8 μl of template DNA.
Data analysis
- First export the obtained copy numbers from AB StepOnePlus Real-Time PCR System. Calculate for each sample the average of copy number of duplicate measurements.
Note: To determine HIV-2 DNA load in whole blood, express HIV-2 DNA copies per cell number using normalization to the porphobilinogen deaminase (PBDG) gene copy number. - First present DNA levels as copies per μl obtained in the PCR reaction, after that normalize the level of HIV-2 DNA to the PBDG copies per μl obtained in the PCR reaction. Set up duplicates of samples and standards in each experiment. Use the following formula to convert the results: (HIV-2 DNA copies per μl)/(PBDG copies per μl)/2 chromosomes per cell) x 105 cells = HIV-2 DNA copies per 105 cells. An example of calculation is provided in Figure 6.
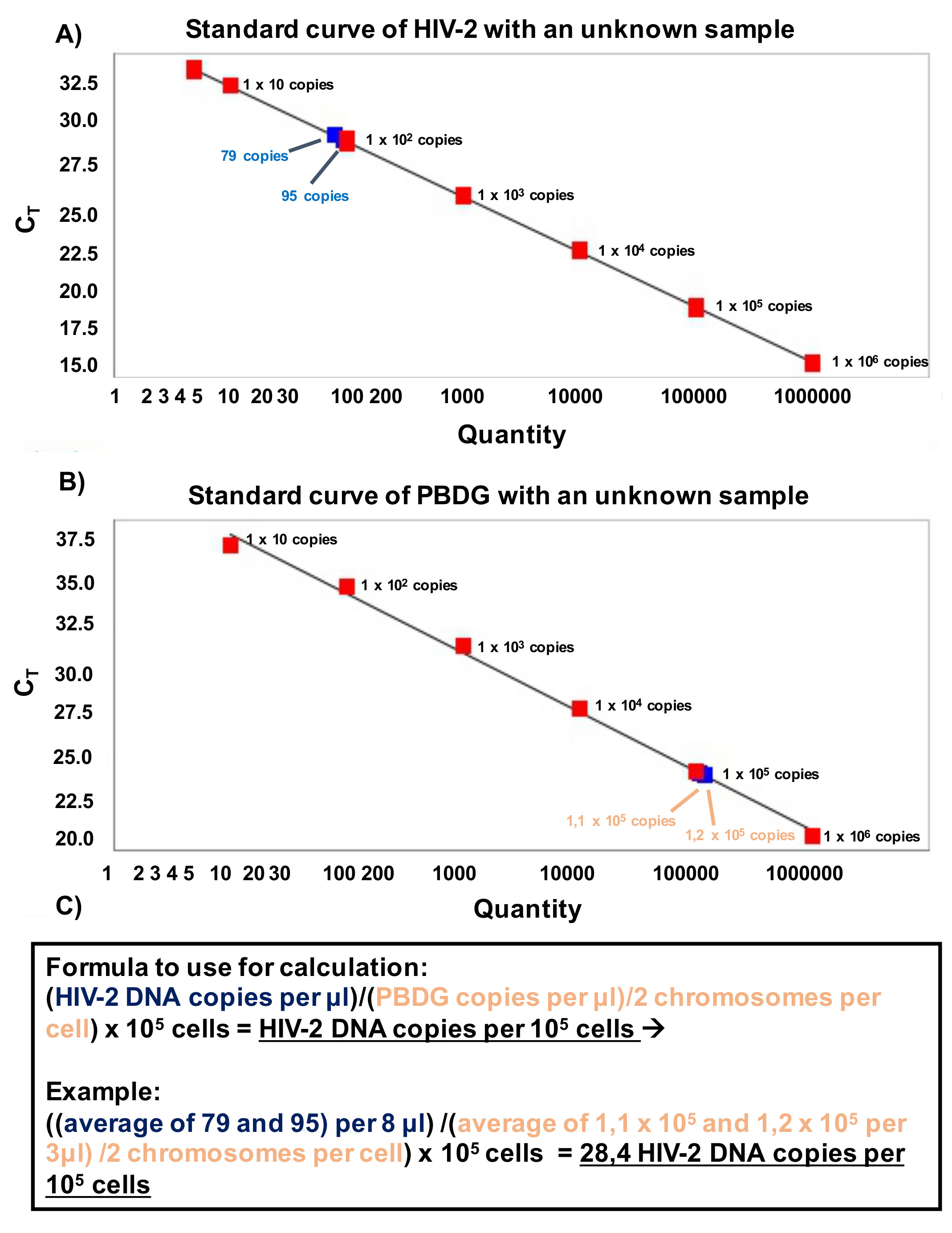
Figure 6. Calculation of HIV-2 DNA copies of an unknown sample. A. Standard curve of HIV-2 gag fragment with the copy numbers of unknown samples. The red box represents Ct values of HIV-2 gag fragment. Linear regression analysis between the quantities of HIV-2 gag gene 106, 105, 104, 103, 100, 10 and 5 DNA copies per reaction in duplicate) and Ct values. The blue box represents HIV-2 gag Ct values of unknown samples. B. Standard curve for PBDG (housekeeping gene) and unknown sample. The linear regression analysis between the quantities of PBDG gene (106, 105, 104, 103, 100 and 1 DNA copies per reaction in duplicate) and Ct values. The red box represents Ct values of PBDG and blue box represents Ct values of PBDG of unknown sample. C. Formula used to convert the results to HIV-2 DNA copies per 105 cells. - For quantification, the standard with dilutions (see Figure 3) was analyzed in the presence or absence of 1 μg of HIV-2 negative PM1 cell DNA (background DNA). The two curves revealed no differences in their linear range of quantification or PCR efficiency. Moreover, in the presence of 1 μg background DNA, no inhibitory effect was detected (see Figure 7).
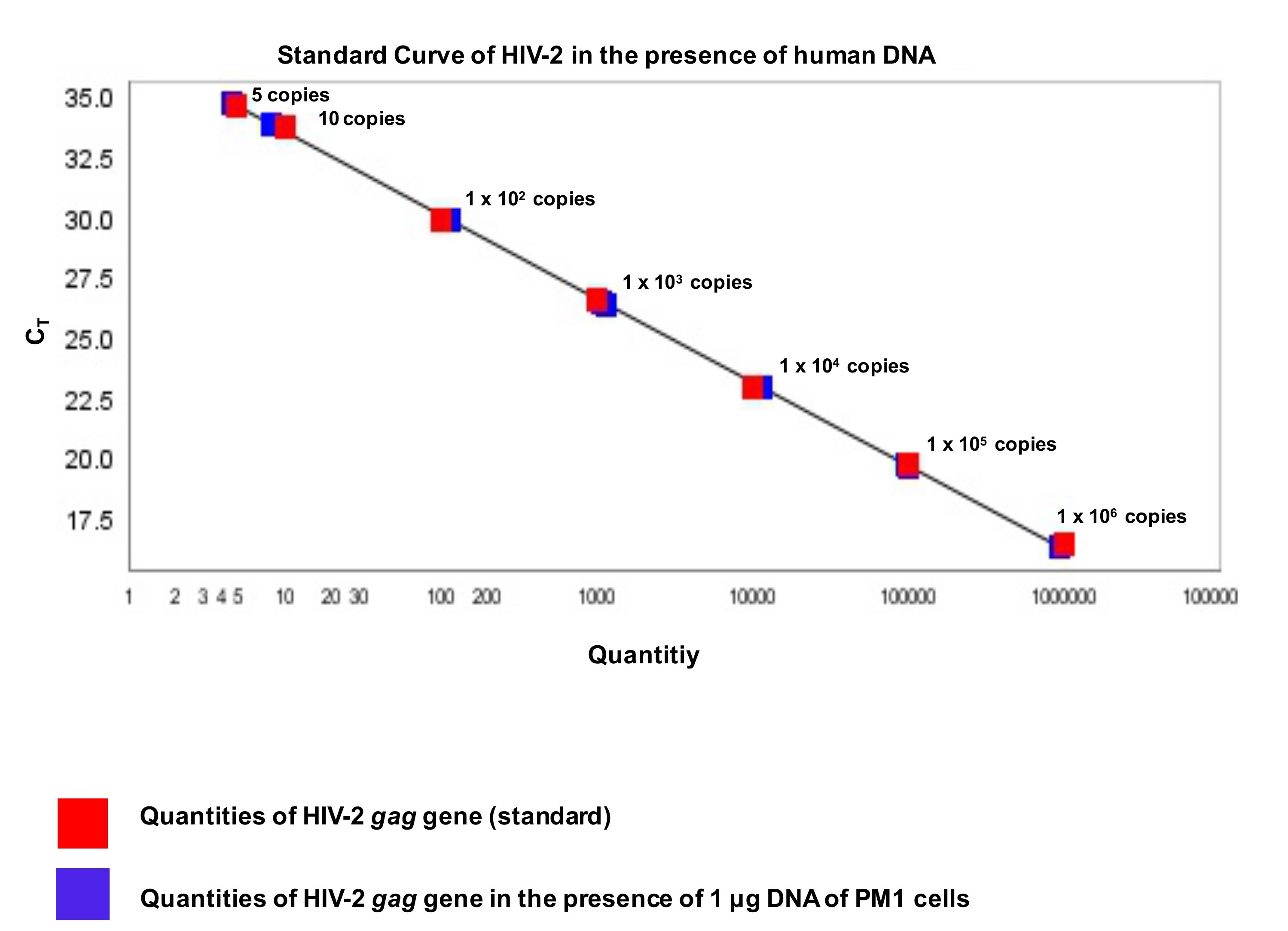
Figure 7. Standard of HIV-2 gag fragment in the presence of 1 μg human DNA. To exclude that background human DNA has an inhibitory effect or not, qPCR was performed in the presence of 1 μg DNA of PM1 cells. The linear regression analysis between the quantities of HIV-2 gag gene (106, 105, 104, 103, 100, 10 and 5 DNA copies per reaction in duplicate) and Ct values in the presence of DNA of PM1 cells. Regression equations were calculated with y = -3.305 + 38.692, R2 = 0.999. The red box represents Ct values of standard without human DNA. The blue box represents Ct values in the presence of PM1 cells’ DNA. - Sensitivity of the assay was 100% at five copies/reaction (detected in 14/14 runs, 0.69 log10 with SD 0.44), while 78% at one copy/reaction (11/14 runs). Limit of detection was 1 copies/reaction (11/14 runs), the median correlation coefficient was 0.999 (range: 0.998-1.000) and limit of quantification was 5 copies/reaction at CT 33 (SD: 0.9). Median slope was -3.15 (range: -3.00--3.37) and median efficiency was 97.0% (range: 98.4% to 98.5%).
- LOD and LOQ was calculated by using a formula described by Schwarz et al. (2004). Detection limit of proviral DNA was 0.5 copies/105 leukocytes.
- To exclude that this method is specific only for HIV-2, run qPCR on extracted DNA of U1 (subclone of U937, which has been chronically infected with HIV-1) cells (Folks et al., 1987), and negative controls, such as extracted DNA from whole blood of HIV negative individuals and extracted DNA from HIV-1 infected patients. As none of the HIV-negative samples, nor HIV-1 infected samples gave positive results, specificity was 100%.
Recipes
- Complete RPMI-1640 medium (pre-warmed at 37 °C before use)
RPMI-1640 medium
2.0 mM L-glutamine
10% FBS (Heat inactivate at 56 ± 2 °C for 30 ± 2 min in a water-bath, and portion at 50 ml/centrifuge tubes, and store at -20 °C until use)
1% penicillin/streptomycin solution
Acknowledgments
This work was supported by Physicians Against AIDS Research Foundation, Sweden, and the Swedish Research Council, Sweden (no. 321-2012-3274 for P.M., no. 2016-02285 for M.J.), Crafoord Foundation, Österlund Foundation and by EFOP-3.6.3-VEKOP-16-2017-00009 for ZsSz.
We thank the members of the Sweden Guinea-Bissau Cohort Research (SWEGUB CORE) group, including: Babetida N’Buna, Antonio Biague, Ansu Biai, Cidia Camara, Joakim Esbjörnsson, Marianne Jansson, Sara Karlson, Jacob Lopatko Lindman, Patrik Medstrand, Fredrik Månsson, Hans Norrgren, Angelica A. Palm, Gülsen Özkaya Sahin, and Zacarias Jose da Silva.
The following reagents were obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: PM1 from Dr. Marvin Reitz and HIV-1 infected U937 Cells (U1) from Dr. Thomas Folks.
Competing interests
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. Authors have no conflict of interest.
Ethics
The study was approved by the ethical committees of the National Health Ethics Committee in Guinea-Bissau (Ref 038/CNES/INASA/2016) and the Regional Ethical Review Board, Lund University, Sweden (Dnr 2016/426). All participants received information about the study before inclusion and provided oral and written informed consent. To ensure confidentiality, all study data was managed under code.
References
- Andersson, S., Norrgren, H., da Silva, Z., Biague, A., Bamba, S., Kwok, S., Christopherson, C., Biberfeld, G. and Albert, J. (2000). Plasma viral load in HIV-1 and HIV-2 singly and dually infected individuals in Guinea-Bissau, West Africa: significantly lower plasma virus set point in HIV-2 infection than in HIV-1 infection. Arch Intern Med 160(21): 3286-3293.
- Ariyoshi, K., Berry, N., Wilkins, A., Ricard, D., Aaby, P., Naucler, A., Ngom, P. T., Jobe, O., Jaffar, S., Dias, F., Tedder, R. S. and Whittle, H. (1996). A community-based study of human immunodeficiency virus type 2 provirus load in rural village in West Africa. J Infect Dis 173(1): 245-248.
- Azevedo-Pereira, J. M. and Santos-Costa, Q. (2016). HIV Interaction With Human Host: HIV-2 As a Model of a Less Virulent Infection. AIDS Rev 18(1): 44-53.
- Berry, N., Ariyoshi, K., Jaffar, S., Sabally, S., Corrah, T., Tedder, R. and Whittle, H. (1998). Low peripheral blood viral HIV-2 RNA in individuals with high CD4 percentage differentiates HIV-2 from HIV-1 infection. J Hum Virol 1(7): 457-468.
- Bertine, M., Gueudin, M., Melard, A., Damond, F., Descamps, D., Matheron, S., Collin, F., Rouzioux, C., Plantier, J. C. and Avettand-Fenoel, V. (2017). New Highly Sensitive Real-Time PCR Assay for HIV-2 Group A and Group B DNA Quantification. J Clin Microbiol 55(9): 2850-2857.
- Buggert, M., Frederiksen, J., Lund, O., Betts, M. R., Biague, A., Nielsen, M., Tauriainen, J., Norrgren, H., Medstrand, P., Karlsson, A. C., Jansson, M. and group, S. C. (2016). CD4+ T cells with an activated and exhausted phenotype distinguish immunodeficiency during aviremic HIV-2 infection. AIDS 30(16): 2415-2426.
- Damond, F., Descamps, D., Farfara, I., Telles, J. N., Puyeo, S., Campa, P., Lepretre, A., Matheron, S., Brun-Vezinet, F. and Simon, F. (2001). Quantification of proviral load of human immunodeficiency virus type 2 subtypes A and B using real-time PCR. J Clin Microbiol 39(12): 4264-4268.
- Damond, F., Gueudin, M., Pueyo, S., Farfara, I., Robertson, D. L., Descamps, D., Chene, G., Matheron, S., Campa, P., Brun-Vezinet, F. and Simon, F. (2002). Plasma RNA viral load in human immunodeficiency virus type 2 subtype A and subtype B infections. J Clin Microbiol 40(10): 3654-3659.
- Esbjornsson, J., Mansson, F., Kvist, A., da Silva, Z. J., Andersson, S., Fenyo, E. M., Isberg, P. E., Biague, A. J., Lindman, J., Palm, A. A., Rowland-Jones, S. L., Jansson, M., Medstrand, P., Norrgren, H., Sweden and Guinea-Bissau Cohort Research, G. (2018). Long-term follow-up of HIV-2-related AIDS and mortality in Guinea-Bissau: a prospective open cohort study. Lancet HIV. doi: 10.1016/S2352-3018(18)30254-6.
- Folks, T. M., Justement, J., Kinter, A., Dinarello, C. A. and Fauci, A. S. (1987). Cytokine-induced expression of HIV-1 in a chronically infected promonocyte cell line. Science 238(4828): 800-802.
- Gomes, P., Taveira, N. C., Pereira, J. M., Antunes, F., Ferreira, M. O. and Lourenco, M. H. (1999). Quantitation of human immunodeficiency virus type 2 DNA in peripheral blood mononuclear cells by using a quantitative-competitive PCR assay. J Clin Microbiol 37(2): 453-456.
- Gueudin, M., Damond, F., Braun, J., Taieb, A., Lemee, V., Plantier, J. C., Chene, G., Matheron, S., Brun-Vezinet, F. and Simon, F. (2008). Differences in proviral DNA load between HIV-1- and HIV-2-infected patients. AIDS 22(2): 211-215.
- Honge, B. L., Jespersen, S., Medina, C., Te, D. S., da Silva, Z. J., Christiansen, M., Kjerulff, B., Laursen, A. L., Wejse, C., Krarup, H., Erikstrup, C. and Bissau, H. I. V. C. s. g. (2018). The challenge of discriminating between HIV-1, HIV-2 and HIV-1/2 dual infections. HIV Med 19(6): 403-410.
- Lusso, P., Cocchi, F., Balotta, C., Markham, P. D., Louie, A., Farci, P., Pal, R., Gallo, R. C. and Reitz, M. S., Jr. (1995). Growth of macrophage-tropic and primary human immunodeficiency virus type 1 (HIV-1) isolates in a unique CD4+ T-cell clone (PM1): failure to downregulate CD4 and to interfere with cell-line-tropic HIV-1. J Virol 69(6): 3712-3720.
- Marvin, S., Reitz, Jr. and Robert, C. G. (2015). Human Immunodeficiency Viruses. In: Mandell, Douglas, and Bennett's Principles and Practice of Infectious Diseases (Eighth Edition). Elsevier 2: 2054-2065.
- Mbisa, J. L., Delviks-Frankenberry, K. A., Thomas, J. A., Gorelick, R. J. and Pathak, V. K. (2009). Real-time PCR analysis of HIV-1 replication post-entry events. Methods Mol Biol 485: 55-72.
- Parisi, S. G., Andreis, S., Mengoli, C., Scaggiante, R., Ferretto, R., Manfrin, V., Cruciani, M., Giobbia, M., Boldrin, C., Basso, M., Andreoni, M., Palu, G. and Sarmati, L. (2012). Baseline cellular HIV DNA load predicts HIV DNA decline and residual HIV plasma levels during effective antiretroviral therapy. J Clin Microbiol 50(2): 258-263.
- Popper, S. J., Sarr, A. D., Gueye-Ndiaye, A., Mboup, S., Essex, M. E. and Kanki, P. J. (2000). Low plasma human immunodeficiency virus type 2 viral load is independent of proviral load: low virus production in vivo. J Virol 74(3): 1554-1557.
- Popper, S. J., Sarr, A. D., Travers, K. U., Gueye-Ndiaye, A., Mboup, S., Essex, M. E. and Kanki, P. J. (1999). Lower human immunodeficiency virus (HIV) type 2 viral load reflects the difference in pathogenicity of HIV-1 and HIV-2. J Infect Dis 180(4): 1116-1121.
- Raich, N., Romeo, P. H., Dubart, A., Beaupain, D., Cohen-Solal, M. and Goossens, M. (1986). Molecular cloning and complete primary sequence of human erythrocyte porphobilinogen deaminase. Nucleic Acids Res 14(15): 5955-5968.
- Rouzioux, C. and Avettand-Fenoel, V. (2018). Total HIV DNA: a global marker of HIV persistence. Retrovirology 15(1): 30.
- Schwarz, G., Baumler, S., Block, A., Felsenstein, F. G. and Wenzel, G. (2004). Determination of detection and quantification limits for SNP allele frequency estimation in DNA pools using real time PCR. Nucleic Acids Res 32(3): e24.
- Soares, R., Foxall, R., Albuquerque, A., Cortesao, C., Garcia, M., Victorino, R. M. and Sousa, A. E. (2006). Increased frequency of circulating CCR5+ CD4+ T cells in human immunodeficiency virus type 2 infection. J Virol 80(24): 12425-12429.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Szojka, Z., Karlson, S., Jansson, M. and Medstrand, P. (2019). Quantification of HIV-2 DNA in Whole Blood. Bio-protocol 9(20): e3404. DOI: 10.21769/BioProtoc.3404.
Category
Microbiology > Microbial genetics > DNA > DNA quantification
Molecular Biology > DNA > DNA quantification
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.