- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Measuring Small-molecule Inhibition of Protein Interactions in Live Cells Using FLIM-FRET
Published: Vol 9, Iss 20, Oct 20, 2019 DOI: 10.21769/BioProtoc.3401 Views: 7269
Reviewed by: Chiara AmbrogioMuhammad AslamAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Mar 2019


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
This protocol was designed to quantitatively measure small-molecule displacement of proteins in live mammalian cells using fluorescence lifetime imaging microscopy–Förster resonance energy transfer (FLIM-FRET). Tumour cell survival is often dependent on anti-apoptotic proteins, which bind to and inhibit pro-apoptotic proteins, thus preventing apoptosis. Small-molecule inhibitors that selectively target these proteins (termed BH3-mimetics) are therefore a promising avenue for the treatment of several cancers. Previous techniques used to study the efficacy of these drugs often use truncated versions of both pro- and anti-apoptotic proteins, as they are membrane bound and hydrophobic in nature. As a result, the true efficacy of these drugs to displace full-length pro-apoptotic proteins in their native environment within a cell is poorly understood. This protocol describes FLIM-FRET methods to directly measure the displacement (or lack of displacement) of full-length Bcl-2 family proteins in live mammalian cells.
Keywords: FLIMBackground
Tumour cell survival is dependent on anti-apoptotic proteins such as Bcl-2 and Bcl-XL, which maintain the integrity of the mitochondrial outer-membrane, thus preventing programmed cell death. Small-molecule inhibitors that selectively target these anti-apoptotic proteins (termed BH3-mimetics) are therefore a promising avenue for the treatment of several cancers. However, in order to study the efficacy of BH3-mimetics, tools to measure Bcl-2 family interactions are required. Previous techniques used to study the efficacy of these drugs often used truncated versions of Bcl-2 and Bcl-XL, as they are membrane bound and contain hydrophobic sequences that make purification difficult. However, the use of truncated proteins in vitro fails to recapitulate the whole story, since Bcl-2 family proteins often behave differently on membrane surfaces (Lovell et al., 2008; Pécot et al., 2016). Moreover, the use of detergents in immunoprecipitation of endogenous proteins can artificially promote or dissociate Bcl-2 family protein interactions (Hsu and Youle, 1997; Billen et al., 2008). Our approach is to use fluorescence lifetime imaging microscopy–Förster resonance energy transfer (FLIM-FRET), to measure full-length protein interactions in living cells (Osterlund et al., 2015; Liu et al., 2019). This is accomplished by expressing two proteins of interest in a desired cell type, with each protein fused to appropriate fluorescent proteins. As a donor fluorescent protein, we use mCerulean3 (mCer3) and as the acceptor, Venus. During a protein-protein interaction, the fluorescent proteins are in close proximity, enabling the donor fluorescent protein to non-radiatively transfer energy to the acceptor. Small molecule inhibitors that inhibit the studied protein interaction, result in a decrease in energy transfer due to a reduction in protein binding. The advantage of this technique is that it enables the use of full-length proteins interacting in their native environment within the cell. Additionally, with the use of proper controls, FLIM-FRET can distinguish true protein interactions from random collisions that occur on confined organelle-membrane surfaces (Osterlund et al., 2015). Here we describe a detailed protocol on how to collect and analyze FLIM-FRET data, as in our most recent publication (Liu et al., 2019).
Materials and Reagents
- 10 cm culture dish (Sarstedt, catalog number: 83.3902)
- 1.7 ml graduated microcentrifuge tubes, autoclaved/sterile (FroggaBio Scientific Solutions, catalog number: LMCT1.7B)
- CellCarrier-384 Ultra microplate, tissue culture treated (PerkinElmer, catalog number: 6057300)
- Mirus TransIT-X2® (Mirus, catalog number: MIR6003, store at -20 °C)
- Opti-MEMTM (Thermo Fisher Scientific, catalog number: 51985091)
- DMEM (Gibco, 12800-082)
- Fetal Bovine Serum (FBS) (Sigma-Aldrich, catalog number: 17B500)
- Non-essential amino acids (NEAA) (Thermo Fisher Scientific, catalog number: 11140050)
- Phosphate Buffered Saline (PBS) (Thermo Fisher Scientific, catalog number: 10010031)
- Trypsin (10x) (Gibco, catalog number: 15400-054)
- ABT-263 (Navitoclax) (Selleck Chemicals, catalog number: S1001. Dissolve in DMSO, store at -20 °C)
- DMSO (Thermo Fisher Scientific, catalog number: 85190)
- NaOH (BioShop, catalog number: SHY700.1)
- BMK Bax-/-Bak-/- cells expressing mCer3-Bcl-XL (Liu et al., 2019)
- EGFP constructs for mCer3-Bcl-XL, Venus-Bad, Venus-Bad(4E) (Liu et al., 2012; Osterlund et al., 2015; Liu et al., 2019). Please contact the corresponding author for constructs
- Fluorescein (Thermo Fisher Scientific, catalog number: F1300)
- 10 nM Fluorescein standard in NaOH (see Recipes)
- Media for cells (see Recipes)
Equipment
- Biological safety cabinet (Thermo Fisher Scientific, 1300 Series A2, catalog number: 1323TS)
- Tissue culture incubator (Thermo Fisher Scientific, HeracellTM 150i CO2 incubator, catalog number: 512026282)
- Vortex (Scientific Industries, Vortex Genie 2, catalog number: SI-0236)
- Hemocytometer (Millipore Sigma, Bright-LineTM, catalog number: Z359629-1EA)
- ISS-Alba FLIM/FCS Confocal Microscope (ISS)
Note: This microscope is custom built from ISS. For full specifications, please refer to (Osterlund et al., 2015).
Software
- VistaVision 4.1 (or higher) (ISS, http://www.iss.com/microscopy/software/vistavision.html)
- ImageJ bundled with 64-bit Java 1.8.0 (NIH, https://imagej.nih.gov/ij/)
- ImageJ ROI Macro (Script attached to key resource table of Liu et al., 2019) (https://github.com/DWALab/Liu_et_al_2018_eLife/blob/master/ROILifetimeAnalysis.ijm)
- Excel (Microsoft, https://www.microsoft.com/en-ca/)
- GraphPad Prism 7 (GraphPad, https://www.graphpad.com/)
Procedure
- Reverse transfection and plating of cells
The presented example is an experiment to determine if 10 μM of the BH3-mimetic ABT-263 can displace the BH3-only protein, Bad, from the anti-apoptotic protein Bcl-XL in live cells (Figure 1). The acceptor fluorescent protein is fused to the N-terminus of Bad (Venus-Bad), while the donor fluorescence protein is fused to the N-terminus of Bcl-XL (mCer3-Bcl-XL). Our cell line of choice is baby mouse kidney (BMK) cells genetically deficient for the pro-apoptotic proteins Bax and Bak (BMK-dko cells, Liu et al., 2019) stably expressing mCer3-Bcl-XL. These cells are resistant to apoptotic stimuli, and thus tolerate higher expression of pro-apoptotic proteins and higher concentrations of apoptotic inducing agents like BH3-mimetics. It is worth noting that a wide range of different cell lines and proteins can be used with this protocol. Our laboratory has previously used MCF-7 and HCT-116 cell lines. Additionally, we use a wide range of different proteins, both pro-apoptotic (Bad, Bim, tBid, Puma, Noxa, etc.) and anti-apoptotic (Bcl-XL, Bcl-2, Bcl-w, and Mcl-1).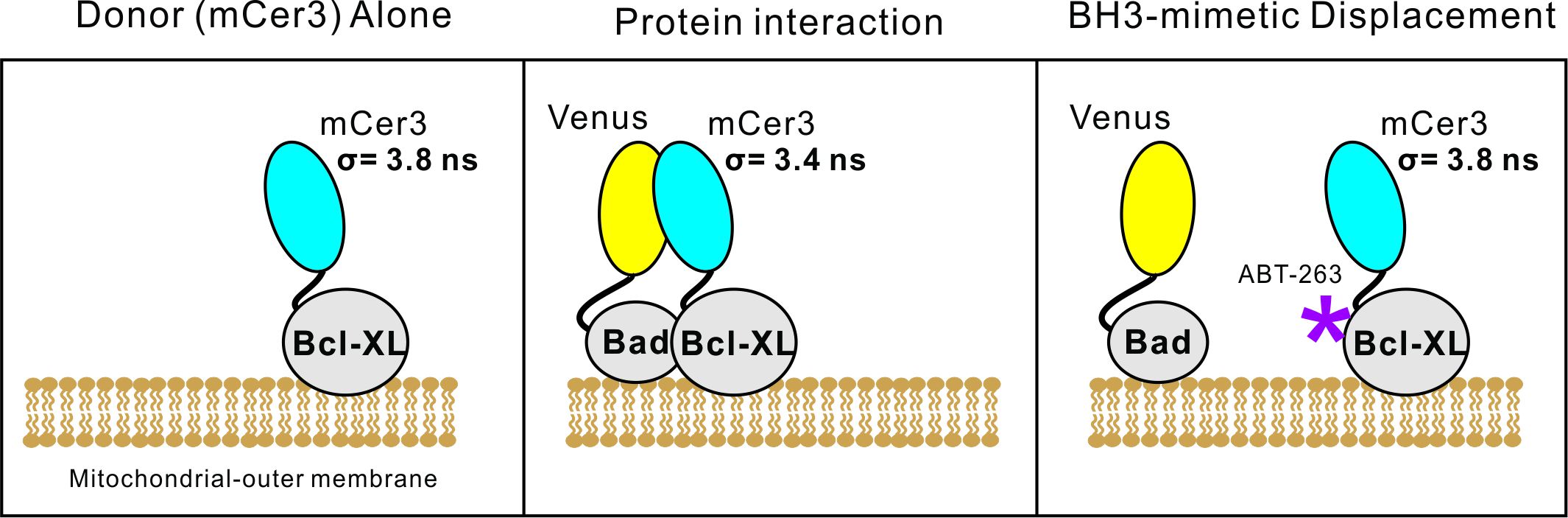
Figure 1. Overview of FLIM-FRET in live cells. The donor fluorescence protein (mCer3) alone has a measured lifetime of 3.8 ns. In close proximity to the acceptor fluorescent protein, Venus, the lifetime for mCer3 decreases, indicative of Förster resonance energy transfer. The BH3-mimetic ABT-263 binds to Bcl-XL, displacing and/or preventing protein interactions. When fully displaced, the fluorescence lifetime for mCer3 returns to 3.8 ns. Partial displacement results in intermediate fluorescence lifetimes enabling estimation of relative binding affinities.- The following conditions are for the transfection of 2 wells in a 384-well format plate (~10,000 cells per well). In a sterile tissue-culture hood, put 10 μl of Opti-MEMTM into a sterile 1.7 ml microcentrifuge tube, then add 200 ng of plasmid DNA with the Venus-Bad gene. Vortex the mixture, ensuring the small volume of 10 μl falls back to the bottom of the tube.
Note: Other plate types can be used, such as 6-well dishes and whole mount slides. However, we prefer using 384-well plates as less reagents are required per experiment. For all plate types, it’s critical that there is no background fluorescence from the plate and that the plate or chamber slide must be compatible with cell culture. - Add 0.2 μl of Mirus TransIT-X2® then immediately vortex the mixture. Ensure a thorough vortex for up to 20 s. After mixing, ensure the small volume falls back to the bottom of the tube. Let the reaction sit at room temperature for approximately 10 min.
- While the transfection reaction sits, we often use this time to split and count the BMK-dko cells stably expressing our donor fluorescent protein (mCer3-Bcl-XL), as follows:
- Remove a 10 cm culture dish containing cells that are ~80% confluent from the incubator, and place it in the sterile tissue culture hood.
- Aspirate the DMEM media in the dish.
- Wash the cells with 10 ml of PBS and then remove the PBS using vacuum aspiration.
- Add 1 ml of 1x-Trypsin. Using your hands, tilt the 10 cm dish to ensure the 1 ml of Trypsin is evenly distributed. Place the 10 cm dish back into the 37 °C incubator, periodically checking on the plate (every few minutes) until it becomes visually apparent that the majority of cells have detached from the dish.
- When the cells are detached from the dish, add 4 ml of fresh DMEM media to the dish and gently pipette the cells up and down (~10 times) to disrupt large cell aggregates. Then place the suspended cells into a 15 ml falcon tube.
- Using a hemocytometer, count the number of suspended cells and express this value as (number of cells) per (1 μl). In our example experiment, we counted a total of 520 cells/μl.
- Since the prepared transfection is for 2-wells (of a 384 well plate), 2 x 104 cells in a total volume of 80 μl are required (this is 1 x 104 cells in a volume of 40 μl per well). Dilute a small fraction of your counted cell suspension to a final concentration of 250 cells/μl (2 x 104 cells divided by 80 μl volume).
- Take 80 μl of your suspended cells (at a concentration of 250 cells/μl) and when the 10 min incubation (Step A2) is complete, mix with the transfection reaction. Add 45 μl of this mixture to a single well of a PerkinElmer cellcarrier ultra 384-well plate (2 wells in total).
- Allow the cells to settle to the bottom of the plate by leaving the plate to rest flat in the tissue culture hood for approximately 10 min. Afterward, the plate should be placed in the 37 °C incubator.
- Approximately four hours after Step 6, remove the plate from the incubator and place in the sterile tissue culture hood.
- Remove all media from the wells, and replace with desired media containing the treatment of choice. In our example experiment, either media containing DMSO, or media containing 10 μM ABT-263 is added back to the wells of the 384-well plate.
- Return the plate back to the incubator, and wait 20 h.
- The following conditions are for the transfection of 2 wells in a 384-well format plate (~10,000 cells per well). In a sterile tissue-culture hood, put 10 μl of Opti-MEMTM into a sterile 1.7 ml microcentrifuge tube, then add 200 ng of plasmid DNA with the Venus-Bad gene. Vortex the mixture, ensuring the small volume of 10 μl falls back to the bottom of the tube.
- Calibrating the ISS-Alba system
- Turn the ISS-Alba instrument on in the following order: computer, climate control unit (set at 25 °C), TI-PS100W power supply, Nikon TI-S-CON motorized stage control, Nikon Intensilight C-HGFIE, excitation boxes for 445 and 514 nm lasers (Ext. 20 MHz), Detector box, Nikon Eclipse Ti microscope, A320 FastFLIM (91070 2012), on the computer load VistaVision Suite software, A512 3-Axis DAC controller, A403 scanning mirror driver (91032 1009), signal conditioning unit (98018 3001). Allow the entire apparatus to warm up for 30 min.
Note: This protocol is not limited to the ISS-Alba microscope. Several other commercial and custom-built instruments can be used, but with differences in downstream image processing (Osterlund et al., 2019; Hirmiz et al., 2019). We recommend using confocal microscopy as many Bcl-2 family interactions occur on mitochondrial membranes, confocal microscopy allows you to achieve sub-cellular resolution interaction maps. For more accurate estimation of fluorescence lifetime, we also recommend single photon counting detectors. Single photon avalanche photo-diodes (SPADs) with single photon time correlated or time-gating capabilities are ideal. Frequency domain FLIM measurements can also be utilized but the data interpretation is less straight forward (Sun et al., 2012). - Take the plate out of the incubator and add to an empty well ~60 μl of fluorescein at a concentration of 10 nM in 0.1 M NaOH (see Recipe below).
Note: We often find day to day variability in the photon count, despite not making any changes to laser power or filters. As a result, we use this fluorescein sample as a reference point to ensure we consistently achieve the same photon counts as previous experiments. Additionally, we measure the mono-exponential lifetime of fluorescein as a control for the instrument. - Lower the objective, and apply a small bead of water. Ensure the water resupply tube is in close proximity to the droplet. Place the 384-well plate into the holder on the microscope, and begin raising the objective until the water droplet makes contact with the bottom of the plate.
- Move the stage to a well containing cells, and use the brightfield to focus on the cells.
- Once focused on the cells, select “z-reset” on the microscope, and move 100 μm above the cells to ensure the focal plane in within the solution. Move the stage to the well containing fluorescein, turn off brightfield, cover the stage with a black dark fabric, and select L100 on the microscope to connect the light path to the detectors.
- Within the VistaVision software, select “TD-FLIM” under “Measurement” on the left hand side, this is time-domain FLIM using the method of time-correlated single photon counting (Osterlund et al., 2015).
Note: Use Figure 2 as a reference for the correct buttons or location of certain settings within VistaVision. The red numbers indicate which step it’s referring to.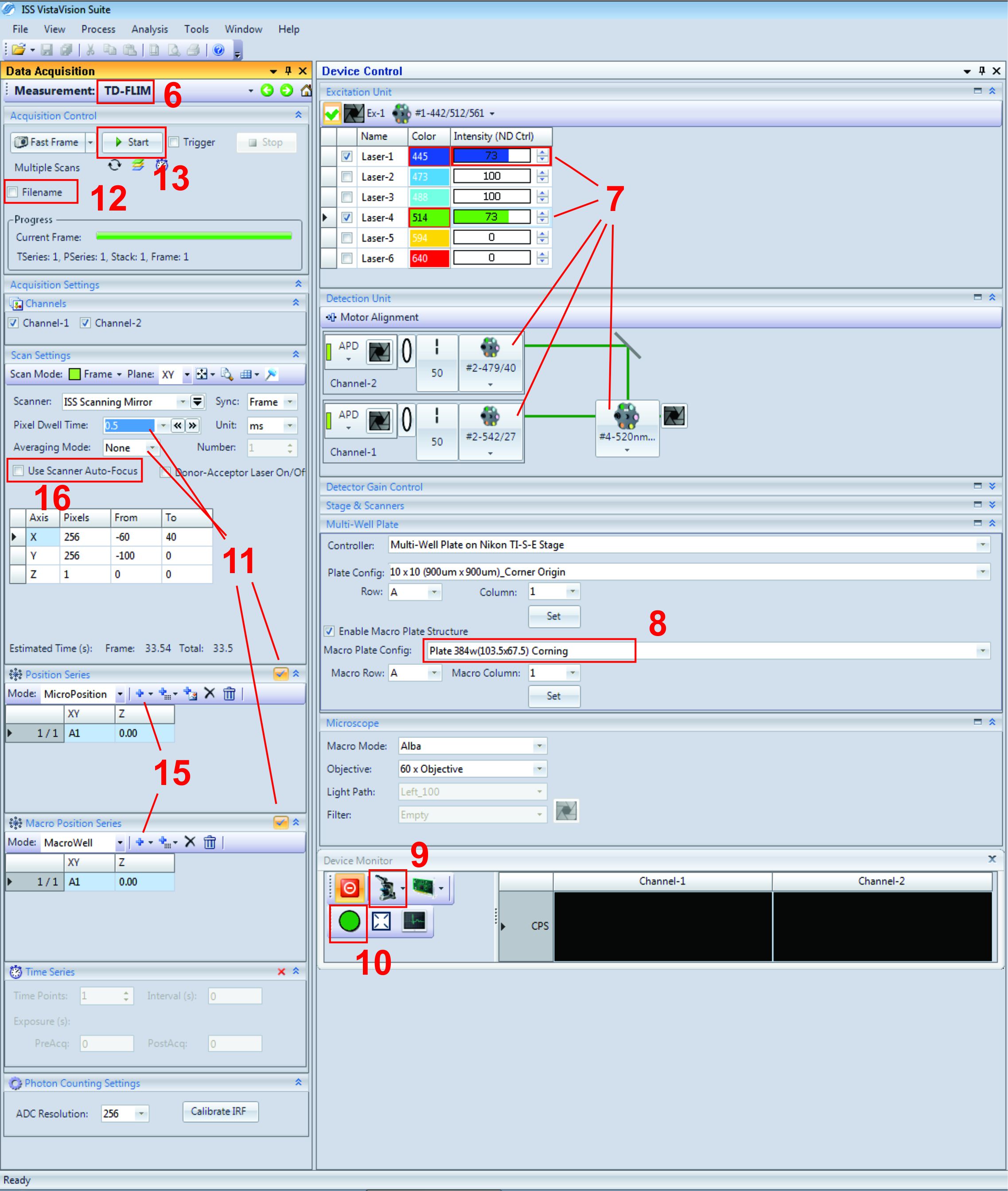
Figure 2. Screenshot of VistaVision software for acquiring FLIM-FRET data. Note that red numbers within the figure refer to the corresponding step in the protocol. - Next, set the 445 nm and 514 nm lasers to ~70%. We often cycle through the filters under “Motor Alignment” to ensure they are in their correct positions. Set Channel-2 to the 479/40 nm filter and Channel-1 to the 542/27 nm filter. Filter #1 is a multi-band filter which allows the light of three different wavelengths to pass, enabling the examination of the three most common fluorescent proteins (cerulean, green and red fluorescent protein). Filter #4 will pass the emission of Venus and reflect the light of mCer3, resulting in the signal of Venus for Channel-1 (Ch1) and mCer3 for Channel-2 (Ch2).
- Under the “Multi-well Plate” tab, select the correct plate-map configuration as your experiment. For ours, we are using a 384 well plate from Corning as it works for the PerkinElmer plate.
- Under “Device Monitor”, select the small microscope icon, and select Alba to direct light from the microscope to the instrument.
- Select the green circle directly below to open the shutters. You should now be able to see the photon counts from Ch1 and Ch2 directly below. We often strive to achieve photon counts between 1 x 105 – 1.5 x 105 for both channels by adjusting the laser power or using the “Motor Alignment” to adjust the pin hole location. Using either one of these tools to achieve consistent photon counts between experiments means the data collected from multiple experiments can be directly comparable. Record the photon counts for Ch1 and Ch2 then uncheck the green circle to close the shutters.
- The next step is to ensure we are measuring the correct lifetime for fluorescein. Under the “Scan Settings” tab, change the pixel dwell time to 0.5 (ms), and averaging mode set to “none”. Also, uncheck both “Position Series” and “Macro Position series” directly below.
- Click “filename” and enter the name of “reference”.
- Finally click “Start”. A new window should appear on the right-hand side. After image acquisition, please follow Steps A3-A6 under Data Analysis, A. VistaVision Analysis, to check if the instrument can correctly measure fluorescein’s lifetime (4 +/- 0.1 ns). In addition to lifetime, we also use this fluorescein image to check for homogeneity of intensity. Being a pure solution of fluorescein, we expect the intensity to be homogenous, or at the very least, well centered. If it’s not, you may consider changing the X and Y position for the field of view as seen in step 16 of Figure 1.
- Turn the ISS-Alba instrument on in the following order: computer, climate control unit (set at 25 °C), TI-PS100W power supply, Nikon TI-S-CON motorized stage control, Nikon Intensilight C-HGFIE, excitation boxes for 445 and 514 nm lasers (Ext. 20 MHz), Detector box, Nikon Eclipse Ti microscope, A320 FastFLIM (91070 2012), on the computer load VistaVision Suite software, A512 3-Axis DAC controller, A403 scanning mirror driver (91032 1009), signal conditioning unit (98018 3001). Allow the entire apparatus to warm up for 30 min.
- Live cell image acquisition on ISS
Note: From this step we enter the process to collect data for cells and regions of interest.- Next, on the microscope turn on the brightfield, reset the stage from 100 back to 0 (focal height of cells), and move to your most top-left well containing your cells (Since the stage moves left to right, up to down to collect data). Confirm you are focused on your cells in the most top-left view within the most top-left well using the eyepiece and brightfield. Press “Zero reset” on the microscope, select “Scanner Auto-Focus” to lock the focal plane. Finally, press “L100” to switch from the brightfield to the detector.
- Within VistaVision, select your wells of interest under “Macro Position Series” (always refer your starting well as A1) then your fields of view within each well under “Position series”.
- Select Z-reset under the “Stage and scanners” tab, then check “Use Scanner Auto-Focus”.
- Finally, change the file name to a name that makes sense to store your data (for example the name of the cells and experiment) and click “start”. By the end of the experiments, a “. iss” file will be automatically stored in your selected path.
- Be sure to check the water supply has enough reservoir for the time of image acquisition.
Data analysis
- VistaVision analysis
- The three files we need for downstream analysis are the intensities from Ch1 (Venus), Ch2 (mCer3), and the lifetime for mCer3 (Tau). Ch1 and Ch2 tiff files have been automatically extracted and saved with image acquisition. The Tau file can be extracted using VistaVision offline version (which means not necessarily connected with the microscope). Open VistaVision software, and make sure to select the offline mode under “Device Configuration” if you are using a separate computer. Click continue.
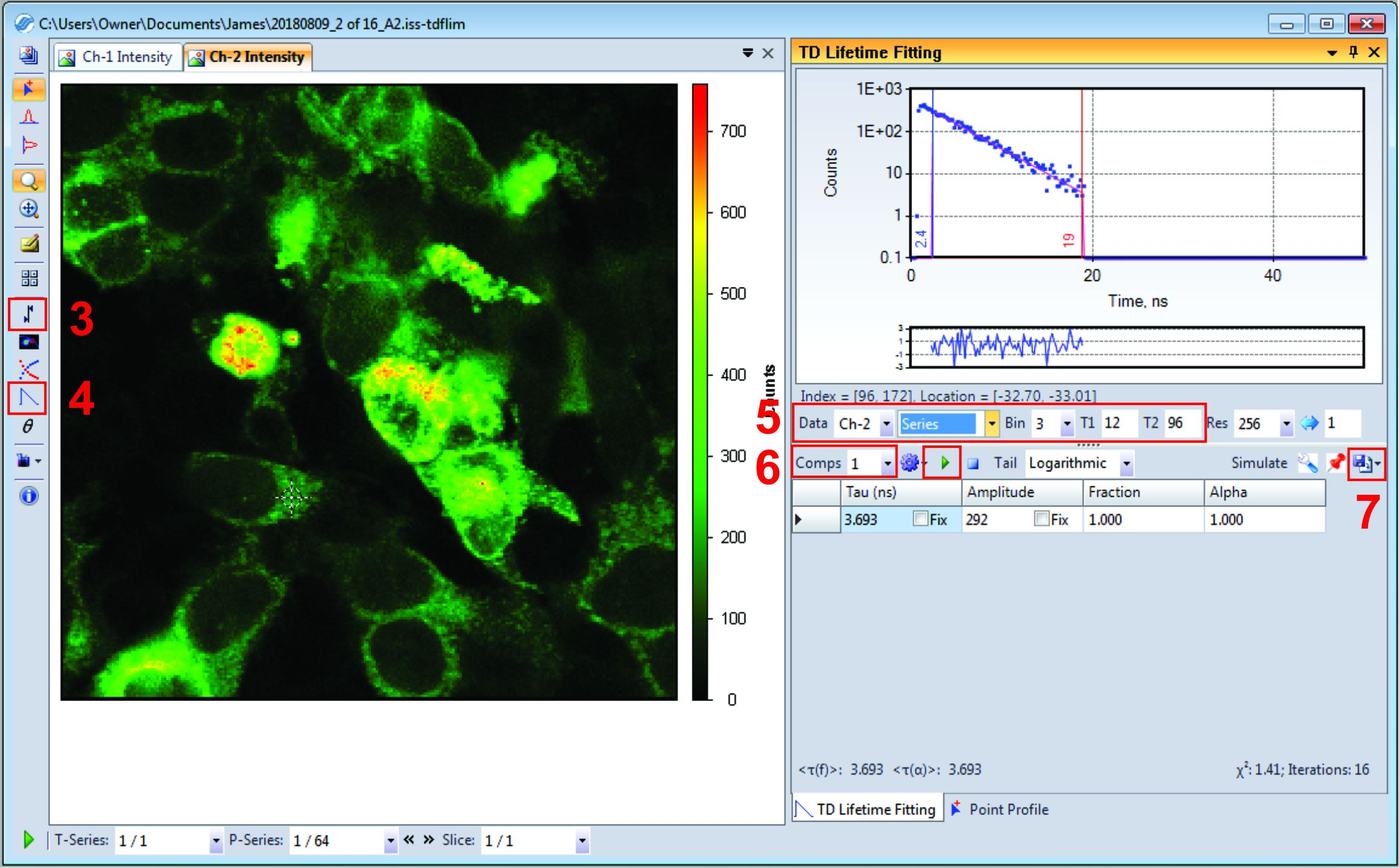
Figure 3. Screenshot of VistaVision software for the analysis of Tau files. The Red numbers within the figure refer to the corresponding step in the protocol. - Next, drag and drop the saved ISS file into VistaVision. Under file type, it should say (ISS Fluorescence Lifetime Image Package).
- Your ISS data should look very similar to Figure 3. Click the “Set intensity threshold for lifetime analysis” button on the left hand side, and input a minimum of 80 for Ch-2. Click okay. Pixels with a Ch2 intensity less than 80 will be discarded from the lifetime data (this variable may change based on your own fluorescence data).
Note: Use Figure 3 as a reference for the following steps. The red number indicates which step it refers to. - Next, click the “lifetime fit” button found on the left column.
- On the right-hand side under the lifetime fitting select the following from their respective dropdown menus; select Ch2 since we are concerned with mCer3 lifetime, select series to apply settings to all images in this stack, select Bin of 3 (this means photons from neighbor pixels will be binned together to fit lifetime). Finally, input T1 and T2 gates as 12 and 96 respectively.
Note: T1 and T2 gates are correlated with the PIE time setting for this microscope. We limit the gate scope after mCer3 excitation and before the disappearance of the emission. - Directly below, set Comps to 1, and click the green “play” button.
Note: If measuring your fluorescein reference, select “Frame” instead of “series”, the lifetime should appear directly below under “Tau”. Fluorescein lifetime should be 4ns (+/- 0.1 ns) for both Ch1 and Ch2. - Once finished, click “Export Data”, select “Export Frame Analysis Data”, and select “Comp-1 Tau” and “Chi-square”. Click the save button, and select the same folder where Ch1 and Ch2 tiff files are currently saved. Click okay.
Note: The “Chi-square” file is our quality record, which could be useful to troubleshoot if we notice abnormal data in later steps.
- The three files we need for downstream analysis are the intensities from Ch1 (Venus), Ch2 (mCer3), and the lifetime for mCer3 (Tau). Ch1 and Ch2 tiff files have been automatically extracted and saved with image acquisition. The Tau file can be extracted using VistaVision offline version (which means not necessarily connected with the microscope). Open VistaVision software, and make sure to select the offline mode under “Device Configuration” if you are using a separate computer. Click continue.
- ImageJ Macro
- For file organization, each transfection/treatment group should have its own designated folder. In this example, and as seen in Figure 4, all data is stored under the file named “VBad”. Within this folder, you need to make four additional ones named “analysis”, “Ch1”, “Ch2”, “tau”. Within each folder place the respective data renamed as the following; For Ch1 and Ch2 tiff image stacks, name them as “1_ch1” and “1_ch2”. If you have additional files from additional wells, they can be named as “2_ch1” and “2_ch2”. The lifetime tiff should be renamed to “1t”, again additional files from additional wells can be renamed to “2t”.
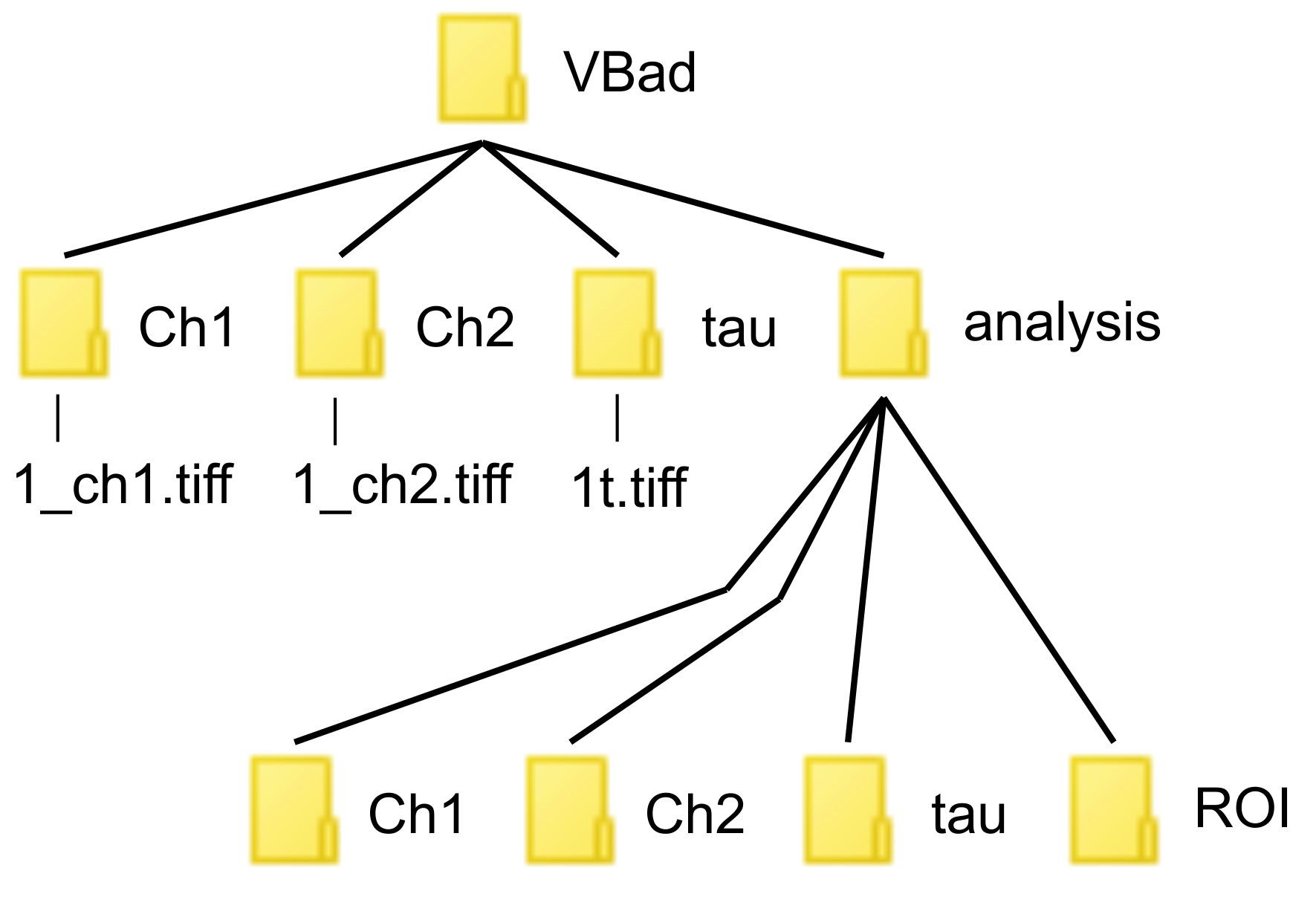
Figure 4. This diagram illustrates how to store your files for the ImageJ script. Please note the correct spelling and capitalization of characters. - Next, open ImageJ software. Under “Plugins” select “New” and then “Macro”.
- Copy the ImageJ macro found in the key resource table of Liu et al. (2019)., or found at this link: https://github.com/DWALab/Liu_et_al_2018_eLife/blob/master/ROILifetimeAnalysis.ijm.
Note: This ImageJ macro selects ROI (regions of interest) based on Ch2. As a result, the ROIs selected have different ratios of Ch1 to Ch2 intensities. ROIs with intensities too high or low are excluded from the downstream analysis as they may lead to inaccurate lifetime fitting. - Within the open Macro window, click “Macro” and then “Run Macro”.
- The macro will prompt you to “Choose Source Directory”. Select your “VBad” folder (or whichever folder contains your data).
- Input the number of images contained within your tiff stacks. Once you input this value, the script will automatically start to run. When the analysis is done, a new window will appear stating “Analysis complete”. Click okay and close ImageJ.
Note: To figure out the number of images per stack, open either 1_ch1 or 1_ch2 within ImageJ, the number of images should be displayed on top of the image stack as 1/(number of images in stack), this is also the number of fields you imaged per well).
- For file organization, each transfection/treatment group should have its own designated folder. In this example, and as seen in Figure 4, all data is stored under the file named “VBad”. Within this folder, you need to make four additional ones named “analysis”, “Ch1”, “Ch2”, “tau”. Within each folder place the respective data renamed as the following; For Ch1 and Ch2 tiff image stacks, name them as “1_ch1” and “1_ch2”. If you have additional files from additional wells, they can be named as “2_ch1” and “2_ch2”. The lifetime tiff should be renamed to “1t”, again additional files from additional wells can be renamed to “2t”.
- Organizing/binning data in Microsoft Excel
- Within the folder “VBad”, open the folder called “analysis”. The ImageJ script should have automatically saved your data into three separate excel files named; “Ch1”, “Ch2” and “tau”. Copy the mean (column C) from each excel file, and paste into a new Excel workbook called “Bin_analysis”. Paste Ch1 data into Column A, Ch2 data into Column B, and tau data into Column C. Use row 1 for each column as a space to label the data. For each of these columns of data, each row represents the average values for a ROI.
- Create a new Column D named “A/D”, which represents the acceptor to donor ratio. More specifically, the value obtained from taking the mean intensity for Venus divided by the mean intensity of mCer3 for each ROI. The acceptor/donor ratio acts as a surrogate for the relative amount of protein concentrations used to plot binding curves. In column D (“A/D”) take Ch1 data and divide it by Ch2 (i.e., In cell D2, type: = A2/B2), and extend this to the bottom of the Excel workbook.
- Now, in cell E1, assign the name “Bin”. This column “Bin” assigns each ROI to a different acceptor/donor ratio bin for averaging and plotting data. To do this, copy and paste the following IF statement into cell E2 and extend it to the bottom of the data [= IF(D2 > 0.01,IF(D2 > 0.05,IF(D2 > 0.1,IF(D2 > 0.2,IF(D2 > 0.3,IF(D2 > 0.4,IF(D2 > 0.5,IF(D2 > 0.75,IF(D2 > 1,IF(D2 > 1.5,IF(D2 > 2,IF(D2 > 2.5,3,2.5),2),1.5),1),0.75),0.5),0.4),0.3),0.2),0.1),0.05),0.01)].
- Finally, in cell F1, name this column “FRET” as it will contain the calculated FLIM-FRET, the average value of energy transfer that occurred for each ROI. This value is calculated by the following formula:
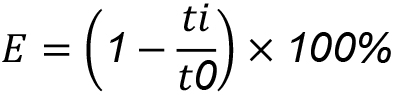
where,
E is FLIM-FRET efficiency, ti is the mean lifetime for the ROI, and t0 is the average lifetime of mCer3 in all ROIs not expressing Venus (acceptor protein). In cell F2, copy and paste the following formula: [= (1-(C2/3.8)) x 100], and extend this to the bottom of the data. - In a new sheet (sheet 2), select well A1 and insert “Pivot table”. Select all the data from the first sheet (Columns A-F). Under Rows select “Bins”. Drag “Fret” to the Values box, select the dropdown tab, and Value field settings. From here you can select the “Average” value. This will then display the average FRET value for each bin.
- Repeat step 5 but this time select “Count” instead of “average”, to have an idea how many ROIs contribute to each Bin.
- Repeat step 5 again, but this time select “StdDev”, to know the standard deviation of FRET values in each bin.
- Finally, in cell E1 (of sheet 2 beside the pivot table), label this column as “SEM”. We plot our FLIM-FRET data using standard error of the mean (SEM), a value not available in Excel’s pivot tables. In cell E2 type the following formula: [= D2/(SQRT(C2))]. Extend this down to the bottom of the pivot table.
- Plotting data with GraphPad Prism
- Open GraphPad Prism version 7.04. Select “XY” under "New table & graph”. Under “Enter/import data” select “X: numbers” and “Y: enter and plot values calculated elsewhere; mean, SEM, N”. Click “create”.
- On the left hand side, Data table 1 is automatically selected for you. Input your Acceptor/donor ratio bin data (Column A on your Excel pivot table) under column “X” in Graphpad. Copy and paste mean FLIM-FRET efficiency (column B of your pivot table) into “Mean” column next to “X” in GraphPad. Column E in Excel is your calculated standard error of the mean “SEM” for FRET values. Copy Column E and paste it under the “SEM” labeled column in GraphPad. Finally, copy Column C, the number of ROIs per bin, and paste this data under the column labeled “N” in GraphPad.
- Within GraphPad on the left hand side, select Data1 under the “Graphs” folder. Your data should appear on a simple XY-plot with unlabeled axes. Label the X-axis by selecting it and typing “Ratio of Venus to mCer3”. Label the Y-axis by selecting it and typing “FLIM-FRET Efficiency %”.
- The next step is to fit the data. Under the “Analysis” tab at the top, select “Fit a curve with nonlinear regression”. The new window that appears is called “Parameters: Nonlinear Regression”. Within this new window select the tab called “Fit” and select “One site-Specific binding with Hill slope”.
- Next, select the tab called “Compare” and select the question “Do the best-fit values of selected unshared parameters differ between data sets?”. Below a new box should appear labeled “Choose one or more parameters”. Select the “Bmax” box.
Note. This step should only be used when all data sets are from the same protein [or slightly changed mutants like VBad-2A which has 2 amino acids changed via site-directed mutagenesis (Liu et al., 2019), since we expect the Bmax value to be the same for all. - Under the “Confidence” tab, select “Calculate CI of parameters” and select the confidence interval of 95%. Below under “Confidence or prediction bands”, check the box to “Plot confidence/prediction bands”, and again select the 95% value. Also ensure the box for “Show SE of parameters” is checked. Finally, click okay to fit your data.
- Your data should now be fitted with the Hill slope equation and display the 95% confidence interval for this fit. A new sheet is created on the left hand side under “Results” and should be labeled as “Nonlin fit of Data1”. If you go back to the “Data table” and input the value of 0.5 under the “X” column, an additional folder will appear under “Results” and be labeled as “Interpolated Y values”.
- Repeat all the above steps under “Data Analysis” for your other FLIM-FRET groups, plotting their data within the same GraphPad file. In our example, our groups were Venus-Bad (VBad), VBad + 10 μM ABT-263, and VBad-4E. Mutating the BH3 domain of VBad (specifically the key hydrophobic residues h1-h4) has previously been reported to significantly inhibit the binding with Bcl-XL and Bcl-2 in live cells (Aranovich et al., 2012; Liu et al., 2019). Importantly, this mutation does not disrupt the membrane binding domain of Bad, making VBad-4E an excellent control to account for the FLIM-FRET efficiency measured due to random collisions on subcellular membranes (Wilfling et al., 2012). As seen in Figure 5, VBad-4E has significantly reduced FLIM-FRET efficiency compared to VBad (Green line). The addition of 10 μM ABT-263 to VBad (Blue line) reduces the FLIM-FRET efficiency, however, it remains higher than VBad-4E. A reduction in FLIM-FRET efficiency from the addition of BH3-mimetic suggests a reduction in protein binding with mCer3-Bcl-XL. Since the FLIM-FRET efficiency remains higher than the non-binding mutant (VBad-4E), we define the remaining percentage of FLIM-FRET efficiency as resistance to displacement from the specified 10 μM of ABT-263.
Note: For FLIM-FRET experiments, it’s imperative to have negative controls. Bcl-2 family proteins localize to mitochondrial membranes, and as a result, have locally high concentrations. This increases the frequency of random collisions that can contribute to the measured FLIM-FRET efficiency. Because VBad-4E binds to subcellular membranes, but no longer binds to Bcl-XL, it is an excellent negative control.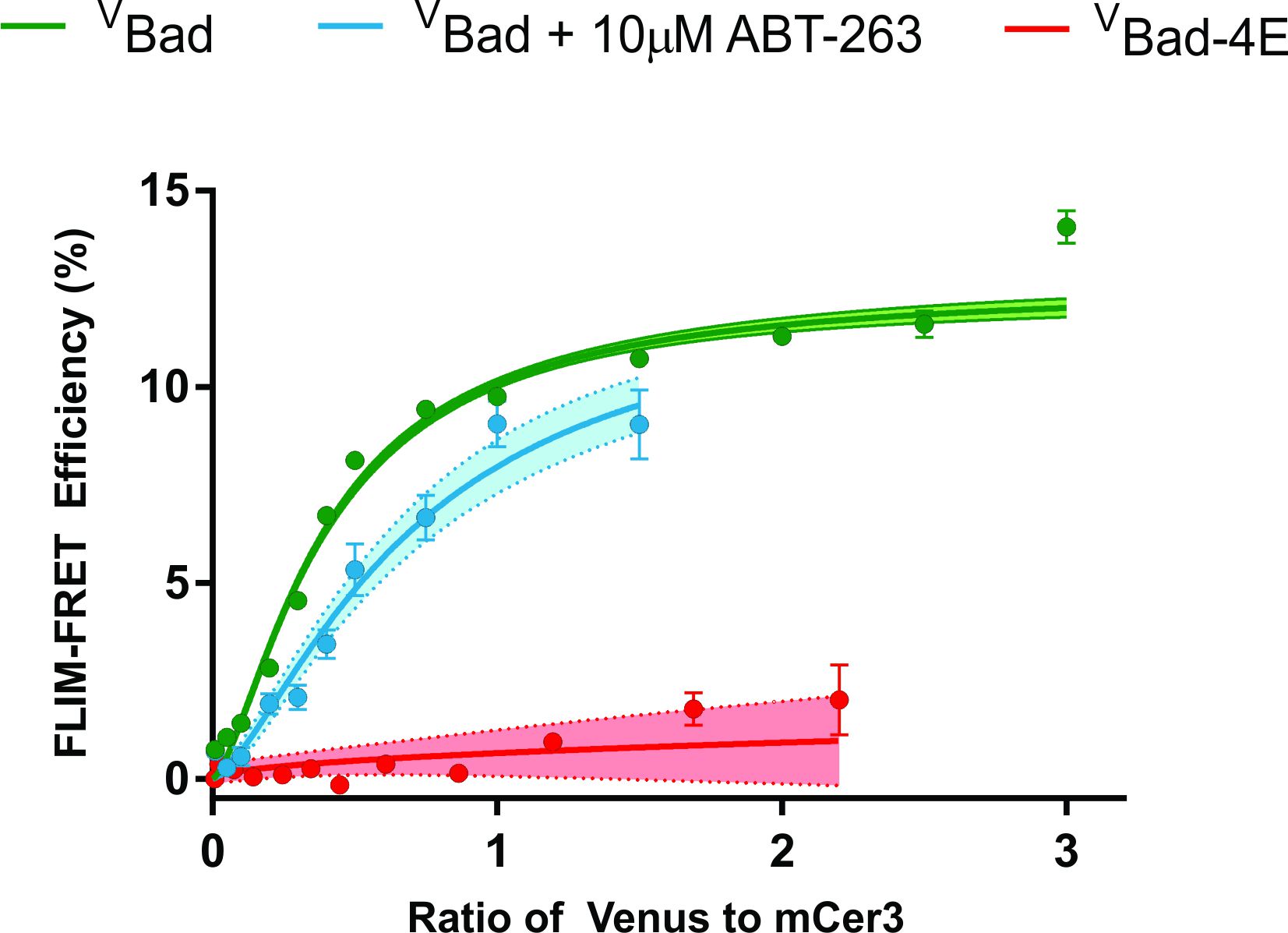
Figure 5. Final result plotting data in GraphPad Prism. Note that the addition of 10 μM ABT-263 reduces the FLIM-FRET efficiency (%) (compare green to blue lines). In addition, introducing four glutamic residues to the BH3-domain of Bad (VBad-4E) completely abrogates all binding with mCer3-Bcl-XL (Red line) as shown in Liu et al. (2019). - To quantify resistance to displacement (Resistance (%)), we compare the FLIM-FRET efficiency (%) between VBad, and VBad + 10 μM ABT-263 at the ratio of Venus to mCer3 of 0.5. To find the FLIM-FRET efficiency (%) value at this acceptor/donor ratio, input the value of 0.5 in the “data table” (in the ratio column below all raw data), then select the tab under “Results” called “Interpolate Y values”. The FLIM-FRET efficiency (%) value will be calculated automatically according to the fit model.
Note: The standard error of the mean describes the accuracy of our measurement while the 95% confidence interval describes how well our data fits the model. - To calculate Resistance (%), we use the following formula:
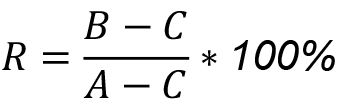
where, R denotes Resistance (%), A is the interpolated mean FLIM-FRET efficiency value for VBad + DMSO, B is the interpolated mean FLIM-FRET efficiency value for VBad + 10 μM ABT-263, and C is the interpolated mean FLIM-FRET efficiency value for VBad-4E. - In our example, we calculated a resistance of 63% at the dose used of 10 μM ABT-263.
Recipes
- 10 nM Fluorescein standard in NaOH
- Dissolve 0.3765 g fluorescein disodium salt in 0.1 M NaOH up to 5 ml (0.2 μM fluorescein stock)
- Next, dilute this stock 1/20 for imaging. i.e., Take 1 ml of this 0.2 μM stock and mix with 19 ml 0.1 M NaOH to achieve 10 nM solution
- Store these stocks at 4 °C, wrapped in tinfoil or tape to avoid light. Stocks can be stored up to 6 months
- Media for cells
- Follow the manufacturer’s instructions for resuspending DMEM powder, then autoclave the solution
- Next, to 450 ml of autoclaved DMEM media add 50 ml of Fetal Bovine Serum (FBS), achieving a final concentration of 10%
- Finally, add 5 ml of non-essential amino acids (NEAA). Mix well, and store at 4 °C
Acknowledgments
Research was funded by Canadian Institutes of Health Foundation grant FRN 12517 to DWA who also holds a Tier 1 Canada Research Chair in membrane biogenesis. The protocol described here is based on research previously published in Osterlund et al. (2015) and Liu et al. (2019).
Competing interests
The authors declare that they have no financial or non-financial competing interests.
References
- Aranovich, A., Liu, Q., Collins, T., Geng, F., Dixit, S., Leber, B. and Andrews, D. W. (2012). Differences in the mechanisms of proapoptotic BH3 proteins binding to Bcl-XL and Bcl-2 quantified in live MCF-7 cells. Mol Cell 45(6): 754-763.
- Billen, L. P., Kokoski, C. L., Lovell, J. F., Leber, B. and Andrews, D. W. (2008). Bcl-XL inhibits membrane permeabilization by competing with Bax. PLoS Biol 6(6): e147.
- Hirmiz, N., Tsikouras, A., Osterlund, E. J., Richards, M., Kun, J., Andrews, D. W. and Fang, Q.(2019). Investigating Bcl-2 family protein-protein interactions using a high-speed multiplexing confocal FLIM microscope. In Proc.SPIE. Vol. 11076.
- Hsu, Y. T. and Youle, R. J.( 1997). Nonionic detergents induce dimerization among members of the Bcl-2 family. J Biol Chem 272 (21): 13829-13834.
- Liu, Q., Leber, B. and Andrews, D. W. (2012). Interactions of pro-apoptotic BH3 proteins with anti-apoptotic Bcl-2 family proteins measured in live MCF-7 cells using FLIM FRET. Cell Cycle 11(19): 3536-3542.
- Liu, Q., Oesterlund, E. J., Chi, X., Pogmore, J., Leber, B. and Andrews, D. W. (2019). Bim escapes displacement by BH3-Mimetic anti-cancer drugs by double-bolt locking both Bcl-XL and Bcl-2. ELife 8:e37689.
- Lovell, J. F., Billen, L. P., Bindner, S., Shamas-Din, A., Fradin, C., Leber, B. and Andrews, D. W. (2008). Membrane binding by tBid initiates an ordered series of events culminating in membrane permeabilization by Bax. Cell 135(6): 1074-1084.
- Osterlund, E. J., Hirmiz, N, Tardif, C. and Andrews, D. W. (2019). Rapid imaging of BCL-2 family interactions in live cells using FLIM-FRET. In Methods in Molecular Biology. Vol. 1877: 305-335.
- Osterlund, E., J., Liu, Q. and Andrews, D. W. (2015). The Use of Flim-Fret for the detection of mitochondrial protein-protein interactions. Methods Mol Biol 1264: 395-419.
- Pécot, J., Maillet, L., Le Pen, J., Vuillier, C., Trécesson, S. C., Fétiveau, A., Sarosiek, K. A., Bock, F. J., Braun, F., Letai, A., Tait S. W. G. ,Gautier, F. and Juin, P. P. (2016). Tight sequestration of BH3 proteins by BCL-XL at subcellular membranes contributes to apoptotic resistance. Cell Rep 17 (12): 3347-3358.
- Sun, Y., Hays, N. M., Periasamy, A., Davidson, M. W. and Day, R. N. (2012). Monitoring protein interactions in living cells with fluorescence lifetime imaging microscopy. Methods Enzymol 504: 371-391.
- Wilfling, F., Weber, A., Potthoff, S., Vögtle, F. N., Meisinger, C., Paschen, S. A. and Häcker., G. (2012). BH3-only proteins are tail-anchored in the outer mitochondrial membrane and can initiate the activation of Bax. Cell Death Differ19 (8): 1328-1336.
Article Information
Copyright
![]() Pemberton et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Pemberton et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Pemberton, J. M., Liu, Q. and Andrews, D. L. (2019). Measuring Small-molecule Inhibition of Protein Interactions in Live Cells Using FLIM-FRET. Bio-protocol 9(20): e3401. DOI: 10.21769/BioProtoc.3401.
- Liu, Q., Oesterlund, E. J., Chi, X., Pogmore, J., Leber, B. and Andrews, D. W. (2019). Bim escapes displacement by BH3-Mimetic anti-cancer drugs by double-bolt locking both Bcl-XL and Bcl-2. ELife 8:e37689.
Category
Biochemistry > Protein > Interaction > Protein-protein interaction
Cancer Biology > Cancer biochemistry > Drug resistance
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.