- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

ATAC-seq on Sorted Adult Mouse Neurons
Published: Vol 9, Iss 19, Oct 5, 2019 DOI: 10.21769/BioProtoc.3382 Views: 8059
Reviewed by: Chiara AmbrogioEnrico PatruccoShibali Das
Original research article
The authors used this protocol in:

Apr 2019
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Transcription regulation is a key aspect of cellular identity established during development and maintained into adulthood. Molecular and biochemical assays that probe the genome are critical tools in exploring mechanisms of transcription regulation and cell type identity. The mammalian brain is composed of a huge diversity of cell types with distinct properties and functions. To understand these specific roles, it is necessary to selectively target cell populations for study. However, the need to selectively study restricted cell populations poses a challenge in neurobiology. It is often difficult to collect sufficient cellular input for many standard biochemical and molecular assays. Recently, important advances have been made to scale assays down, opening up new frontiers to explore molecular mechanisms in neurons. Concurrently, methodologies for preparing neurons for such assays has advanced taking into consideration specific methods to preserve the cell biology meant to be assayed. Here we describe a method for preparing live neurons from adult brain tissue for the Assay for Transposase Accessible Chromatin (ATAC).
Keywords: ATACBackground
The Assay for Transposase Accessible Chromatin coupled with next generation sequencing (ATAC-seq) is becoming a widely used method to assess genome-wide chromatin accessibility (Buenrostro et al., 2013; Corces et al., 2017). A major advantage of ATAC-seq over older methods such as DNase I hypersensitivity mapping, is that it can produce high-quality data from significantly smaller input. This allows chromatin and transcriptional regulatory mechanisms to be explored in previously inaccessible populations of rare or sparse cell types. This is particularly useful in the nervous system where cellular diversity is incredibly high and cells of interest are post-mitotic and often sparse. While single cell methods play an important role in identifying heterogeneity and defining certain characteristics of cell types, they are often not suitable for deeper, reproducible characterization, or targeted manipulation and comparison of cell types. The growing number of marker genes and other genetic strategies to label restricted neuronal populations (Shima et al., 2016) is increasing the depth and breadth of molecular and biochemical assays, including ATAC-seq, used to study diverse types of neurons. However, isolation of healthy, high-quality neurons from live tissue requires special considerations. Here we adapt an earlier protocol for isolating neurons from live, adult brain tissue (Hempel et al., 2007) to generate ATAC-seq profiles from sorted populations of 20,000-50,000 cells expressing a fluorescent marker. We successfully used this method to generate ATAC-seq profiles from a diverse range of adult neuronal types (Sugino et al., 2019).
Materials and Reagents
Materials
- Tube with cell strainer cap (35 µm mesh, Thermo Fisher Scientific, catalog number: 08-771-23)
- Cell strainer (nylon mesh with 40, 70, and 100 µm micron pores, Thermo Fisher Scientific, catalog number: 08-771-19)
- Filter-Steriflip, 50 ml, membrane tube device (Thermo Fisher Scientific, catalog number: SCGP00525)
- Tools and supplies for animal dissection and incubation of brain slices (Hempel et al., 2007)
- 100-mm glass Petri dish
- Glass Pasteur pipettes with tips fire-polished to diameters of ~600, 400-300 and, ~200 µm (measured under the microscope with a fine-grade ruler or stage micrometer)
- Gas Diffuser (VWR, catalog number: 80073-710)
Reagents
For Cutting Solution (CS) and modified ACSF (mACSF)
- 95% O2/5% CO2 compressed gas (for solution oxygenation)
- N-Methyl-D-glucamine (NMDG) (Sigma, catalog number: M2004-500G)
- D-Trehalose Dihydrate (Sigma-Aldrich, catalog number: T5251)
- Sodium Hydroxide (NaOH) (Thermo Fisher Scientific, catalog number: S318-1)
- Choline chloride (Thermo Fisher, catalog number: AC110295000)
- Sodium chloride (Thermo Fisher, catalog number: S271-500)
- Potassium chloride (Thermo Fisher, catalog number: P217-500)
- NaH2PO4·H2O (Thermo Fisher, catalog number: S369-500)
- NaHCO3 (Thermo Fisher, catalog number: S233-500)
- HEPES (Thermo Fisher, catalog number: BP310-500)
- Na ascorbate (AppliChem, catalog number: A5048)
- Thiourea (Sigma-Aldrich, catalog number: T8656-500G)
- Na pyruvate (AppliChem, catalog number: A4859)
- N-acetyl-L-cystein (Sigma-Aldrich, catalog number: A7250-100G)
- MgSO4·7H2O (Thermo Fisher, catalog number: M65-500)
- CaCl2·2H2O (Thermo Fisher, catalog number: BP510-250)
- Tris-HCl (Sigma-Aldrich, catalog number: T2819)
- EDTA (Sigma-Aldrich, catalog number: E5134-500G)
- Ketamine 100 mg/ml (Webster Vet Supply, catalog number: 07-803-6637)
- Kynurenic acid (Sigma-Aldrich, catalog number: K3375-1G)
- Tetrodotoxin (TTX) (Tocris, catalog number: 1069)
- TTX 0.1 mM (see Recipes)
- Ketamine 500 mM (see Recipes)
- Cutting Solution (CS) (see Recipes)
- Modified Artificial Cerebral Spinal Fluid (mACSF) (see Recipes)
For tissue preparation and single cell suspension preparation
- Loctite 40104 (Construction Equipment Plus, catalog number: 82525940)
- Pronase (Sigma-Aldrich, catalog number: P5147-1G)
- Fetal bovine serum (FBS) (Atlanta Biological, catalog number: S11550)
- DNase I (Thermo Fisher, catalog number: AM2222)
- Percoll (Sigma-Aldrich, catalog number: P1644-100ML)
For nuclei preparation and tagmentation
- Digitonin (Millipore, catalog number: 300410-250MG)
- Tween-20 (Thermo Fisher, catalog number: BP337-100)
- Dimethyl Formamide (Sigma-Aldrich, catalog number: 270547-100ML)
- 100% acetic acid (Thermo Fisher, catalog number: A38500)
- Tn5
Note: We used a discontinued kit (Illumina, catalog number: FC-121-1030). However, Tn5 can be purchased from several manufactures specifically for ATAC-seq and there are published protocols for producing and purifying Tn5 in house (Picelli et al., 2014). - IGEPAL CA-630 (Sigma-Aldrich, catalog number: I8896-50ML)
- 1% (100x) Digitonin (see Recipes)
- 10% (100x) Tween-20 (see Recipes)
- 2x TD buffer (see Recipes)
- Lysis buffer (see Recipes)
For library preparation
- Zymo DNA Clean and Concentrator-5 Kit (Zymo, catalog number: D4014)
- 1x Low TE buffer (see Recipes)
- NEBNext High-Fidelity 2x PCR Master Mix (New England Labs, catalog number: M0541)
- SYBR Green 10,000x (Thermo Fisher, catalog number: S7567)
- DMSO (Sigma-Aldrich D8418-50ML)
- Primers (Tables 1 and 2)
Table 1. Library amplification primers (HPLC purified) (24097267)name sequence Ad1_noMX AATGATACGGCGACCACCGAGATCTACACTCGTCGGCAGCGTCAGATGTG Ad2.1_TAAGGCGA CAAGCAGAAGACGGCATACGAGATTCGCCTTAGTCTCGTGGGCTCGGAGATGT Ad2.2_CGTACTAG CAAGCAGAAGACGGCATACGAGATCTAGTACGGTCTCGTGGGCTCGGAGATGT Ad2.3_AGGCAGAA CAAGCAGAAGACGGCATACGAGATTTCTGCCTGTCTCGTGGGCTCGGAGATGT Ad2.4_TCCTGAGC CAAGCAGAAGACGGCATACGAGATGCTCAGGAGTCTCGTGGGCTCGGAGATGT Ad2.5_GGACTCCT CAAGCAGAAGACGGCATACGAGATAGGAGTCCGTCTCGTGGGCTCGGAGATGT Ad2.6_TAGGCATG CAAGCAGAAGACGGCATACGAGATCATGCCTAGTCTCGTGGGCTCGGAGATGT Ad2.7_CTCTCTAC CAAGCAGAAGACGGCATACGAGATGTAGAGAGGTCTCGTGGGCTCGGAGATGT Ad2.8_CAGAGAGG CAAGCAGAAGACGGCATACGAGATCCTCTCTGGTCTCGTGGGCTCGGAGATGT Ad2.9_GCTACGCT CAAGCAGAAGACGGCATACGAGATAGCGTAGCGTCTCGTGGGCTCGGAGATGT Ad2.10_CGAGGCTG CAAGCAGAAGACGGCATACGAGATCAGCCTCGGTCTCGTGGGCTCGGAGATGT Ad2.11_AAGAGGCA CAAGCAGAAGACGGCATACGAGATTGCCTCTTGTCTCGTGGGCTCGGAGATGT Ad2.12_GTAGAGGA CAAGCAGAAGACGGCATACGAGATTCCTCTACGTCTCGTGGGCTCGGAGATGT Ad2.13_GTCGTGAT CAAGCAGAAGACGGCATACGAGATATCACGACGTCTCGTGGGCTCGGAGATGT Ad2.14_ACCACTGT CAAGCAGAAGACGGCATACGAGATACAGTGGTGTCTCGTGGGCTCGGAGATGT Ad2.15_TGGATCTG CAAGCAGAAGACGGCATACGAGATCAGATCCAGTCTCGTGGGCTCGGAGATGT Ad2.16_CCGTTTGT CAAGCAGAAGACGGCATACGAGATACAAACGGGTCTCGTGGGCTCGGAGATGT Ad2.17_TGCTGGGT CAAGCAGAAGACGGCATACGAGATACCCAGCAGTCTCGTGGGCTCGGAGATGT Ad2.18_GAGGGGTT CAAGCAGAAGACGGCATACGAGATAACCCCTCGTCTCGTGGGCTCGGAGATGT Ad2.19_AGGTTGGG CAAGCAGAAGACGGCATACGAGATCCCAACCTGTCTCGTGGGCTCGGAGATGT Ad2.20_GTGTGGTG CAAGCAGAAGACGGCATACGAGATCACCACACGTCTCGTGGGCTCGGAGATGT Ad2.21_TGGGTTTC CAAGCAGAAGACGGCATACGAGATGAAACCCAGTCTCGTGGGCTCGGAGATGT Ad2.22_TGGTCACA CAAGCAGAAGACGGCATACGAGATTGTGACCAGTCTCGTGGGCTCGGAGATGT Ad2.23_TTGACCCT CAAGCAGAAGACGGCATACGAGATAGGGTCAAGTCTCGTGGGCTCGGAGATGT Ad2.24_CCACTCCT CAAGCAGAAGACGGCATACGAGATAGGAGTGGGTCTCGTGGGCTCGGAGATGT
Table 2. QC qPCR primers
Note: These primers are used to screen libraries post-amplification but prior to sequencing. Primers should amplify 3-4 regions at the TSS of genes that are highly accessible in your samples and 3-4 background regions that are typically not accessible in your samples. The primers listed above worked well for mouse neurons isolated from multiple different tissues.Uvrag_F CTAAACCTGGGGCGTAACTT (TSS primer) Uvrag_R CTAGAGCAATAGCCCACGAG Usp5_F ATGGAAGGAAGTCGTGGTCG (TSS primer) Usp5_R CCGGGTCCATAAAGACGAGT Ap2a1_F AGTACCGGGCTATTTCCGTT (TSS primer) Ap2a1_R GGAGCTGGTAGTCCATCCAA ATACbg1_F CTAGTGGGGTAAATTGGCAGG (background primer) ATACbg1_R AACGGCACACAAATAGGCTC ATACbg2_F ATATGCCCAGAACTACCGAC (background primer) ATACbg2_R TTCTTTTCAGAAACAGGCGG ATACbg3_F TTCTGCACTTTTCATGGCAG (background primer) ATACbg3_R ACAGATGCAGGACCCATAAG
- 100x SYBR Green (see Recipes)
For library quality assessment
- KAPA Library Quantification kit (Roche, catalog number: KK4854)
- High Sensitivity DNA kit (Agilent, catalog number: 5067-4626)
- SSO Evagreen Master mix (Bio-Rad, catalog number: 1725203)
Equipment
- Vibratome (such as Leica VT1000S)
- Flow Cytometer (such as BD FACSAria)
- Dissection stereomicroscope, such as Leica MZ16FA (Leica) equipped with fluorescence and transmitted light (such as Leica MZ16FA).
- Osmometer (such as Advanced Instruments MicroOmometer 3300)
- Real-time PCR thermocycler (such as Corbett Research RotorGene 6000)
- Bioanalyzer (such as Agilent 2100)
- Centrifuges (such as Sorvall Legend RT+ Centrifuge and Eppendorf Centrifuge, model: 5415 D)
- Laboratory water bath (such as Fisher Scientific Isotemp 110 Water Bath)
Software
- FastQC (Andrews, 2010)
- bowtie2 (Langmead and Salzberg, 2012)
- picard-tools-1.123 (Broad Institute, 2019)
Procedure
Procedure Overview
- Harvest and section brain tissue (1-2 h depending on the number of mice).
- Micro dissection and tissue digestion (1-1.5 h).
- Trituration and Percoll separation (1-1.5 h depending on the amount of tissue).
- Sorting (variable depending on machine and sample size, typically more than 2 h).
- Preparing nuclei and tagmentation (~1 h).
Pause point: Tagmented nuclei can be stored at -20 °C for up to two weeks. - Library preparation (2-3 h depending on the number of samples).
Pause point: Amplified DNA can be stored at -20 °C. - Quality assessment (2-3 h).
Harvest and Section Brain Tissue
- Add the following reagents to Cutting Solution (CS) and Modified Artificial Cerebral Spinal Fluid (mACSF):
Ketamine (0.05 mM final concentration) or 0.05 mM APV
TTX (100 nM final concentration)
Kynurenic acid (0.8 mM final concentration) or 0.02 mM DNQX
Notes:- These inhibitors are important for high quality ATAC from neurons. They prevent excitotoxicity as tissue is sliced and dissociated.
- The amount of CS and mACSF needed will depend on the number and size of animals perfused and the set-up used. For reference, when perfusing four young adult mice typically weighing ~20 mg, 300 ml of CS and 100 ml of mACSF are prepared. If working with a cell type that requires multiple rounds of Percoll centrifugation (Step 26), extra mACSF may be needed. Prior to attempting the protocol, we recommend calculating the volume needed by measuring and summing the volumes of liquid needed for each step. To allow for the possibility of poor perfusions requiring additional volume (Step 4d), prepare 50 ml extra of CS.
- To mACSF, add 5% (w/v) Trehalose.
Note: This is a protectant for tissue dissociation (Saxena et al., 2012). The effects of Trehalose are a bit controversial (Lee et al., 2018), but we found it increases the number of intact neurons after trituration. - Stir at room temp (RT) and oxygenate using gas diffuser until Kynurenic acid dissolves.
Note: mACSF can remain at room temp.- Move enough CS to a new container to oxygenate and suspend one or more cell strainers.
- Place container in a water bath at 34 °C.
- After sectioning, brain slices will recover at 34 °C (Ting et al., 2014).
- Move remaining CS to 4 °C and oxygenate.
- Move enough CS to a new container to oxygenate and suspend one or more cell strainers.
- Harvest brain.
- Make an ice bath around the vibratome buffer tray.
- Fill vibratome buffer tray with cold CS and oxygenate.
- Administer lethal dose of appropriate anesthetic.
- Ideally before heart stops beating, perfuse cold CS via trans-cardiac perfusion through the left ventricle exiting the right atrium until blood has been completely replaced with CS and tissues such as the liver and lungs are no longer red (Typically 20-30 ml depending on mouse size). If the liver has red patches this indicates poor perfusion of tissues. Continue perfusing until the liver color is evenly white/tan and only clear CS is seen exiting the heart. For a detailed example of trans-cardiac perfusion technique see Gage et al. (2012).
- Quickly remove brain and place in oxygenating CS filled vibratome buffer tray.
Note: For dissection details, see Papouin and Haydon (2018). - If slicing multiple brains repeat (c)-(f) for each animal.
- Make sure brain(s) in tray do not come in direct contact with O2 bubbles.
- Allow the last brain to equilibrate for 5 min.
- Mount brains for slicing.
- Place wet blot paper with oxygenated cold CS inside a 100-mm petri dish.
- Place brain(s) on wetted paper.
- Remove unwanted tissue and cut to create a flat surface to affix the brain.
- Examples of two options for mounting the brain are shown in Video 1. The first brain is mounted by removing the cerebellum so that rostral structures will be sliced first. The second brain is oriented in the opposite direction so that caudal structures may be sliced. Both orientations will produce coronal slices.
- Use a fine scalpel to minimize tissue distortion that can damage neurons.Video 1. Mounting brains on vibratome platform. (Animal experimentation: All experiments were conducted in accordance with the requirements of the Institutional Animal Care and Use Committees at Brandeis University (protocol #17001).)
- Attach brains to platform with Loctite.
Affix the brain in an orientation such that cells of interest are away from the Loctite. - Move platform back to oxygenated CS.

Micro Dissection and Tissue Digestion
- Slice mounted brain(s) into 300 µm sections through the region of interest.
- Set speed to 3 (on a scale of 0-10) and frequency between 6 and 8 (on a scale of 0-10).
Note: Settings should be optimized on individual equipment to minimize tissue distortion/compression. If brains tend to come loose during slicing, then the speed may be too fast or the frequency too low. - Reducing tissue deformation/compression will increase yield. We recommend always using a fresh blade.
- Set speed to 3 (on a scale of 0-10) and frequency between 6 and 8 (on a scale of 0-10).
- Using a wide mouth pipette or a plastic transfer pipette that has been cut to make the opening larger gently move slices to cell strainers in oxygenated CS at 34 °C
- The strainer protects slices from coming into direct contact with oxygenation bubbles.
- If working with cells expressing a fluorescent marker, protect samples from light as much as possible from here on to minimize photobleaching.
- Incubate for 10-12 min.
- Move strainer containing slices to oxygenated mACSF at RT.
- Ensure slices are submerged.
- Allow equilibrating for 5-10 min.
- Dissolve pronase (10% w/v) in RT oxygenated mACSF (20-40 ml) and pour into a 100 mm petri dish.
- Transfer the strainer with slices to Petri dish with pronase solution. Use a wide mouth pipette or cut plastic transfer pipette to move slices from the strainer and into the solution. Incubate for 1-1.5 h at RT.
- While slices digest, micro-dissect region(s) of interest under a stereomicroscope.
- If dissecting a large number of slices, it is ideal to work in batches using several dishes so that the tissue waiting to be dissected or post-dissection can be oxygenated in the pronase solution.
- When finished dissection, oxygenate tissue pieces in pronase solution for any remaining time on the digest incubation.
- This step should be optimized to reduce non-target tissue. Enriching the cells of interest will reduce flow cytometry sort times.
- Prepare 30-50 ml of oxygenated mACSF + 1% FBS (mACSF + FBS), filter with Steriflip and keep on ice
- Add 1 U/ml DNase to 5-10 ml of filtered mACSF + FBS (mACSF + FBS + DNase), keep on ice.
- Move tissue pieces to 1 ml of mACSF + FBS.
Wash tissue 2-3 x with mACSF + FBS to quench and remove protease by allowing tissue to settle to the bottom of the tube and removing most of the supernatant. Add fresh mACSF + FBS and repeat. - Suspend tissue pieces in mACSF + FBS + DNase and divide between Eppendorf tubes such that the volume of tissue does not exceed 0.1% of the volume (Figure 1).
Notes:- DNase prevents genomic DNA released by lysed cells from clumping and blocking the pipette opening during trituration.
- Trituration with too dense cell solution results in low yield of live cells.

Figure 1. Example of tissue amount for trituration in 1 ml
- Keep tissue on ice.
- With the 600 μm fire-polished Pasteur pipette, gently triturate the sample ten times.
- Pre-wet the pipette to prevent tissue from sticking to the inside walls.
- Avoid making bubbles as this can damage cells.
- The pipette mouth should not be held against the wall or floor of the tube.
- The solution should turn cloudy with large chunks remaining.
- Incubate tube(s) on ice allowing chunks to settle for a few minutes.
- Filter supernatant using tube with cell strainer cap.
Note: You may need to prewet the strainer with mACSF + FBS to start gravity flow. - Add 1 ml fresh mACSF + FBS + DNase I to remaining tissue chunks.
Note: If working with precious small amounts of tissues, rinse the cell strainer cap with the 1 ml and transfer any chunks left in the top back to the Eppendorf for the next trituration step. - Triturate ten times with the 400 μm fire-polished Pasteur pipette as described in Step 17.
Note: The solution should become cloudy again, and remaining chunks be smaller. - Filter supernatants (as in step 19) into the same tube and add fresh mACSF + FBS + DNase (as in Step 20).
Trituration and Percoll Separation
- Triturate ten times with the 200 μm fire-polished Pasteur pipette. Again, filter supernatant into the same tube.
- If chunks remain you may repeat the 200 μm trituration on leftover chunks.
- Transfer the combined, filtered cell suspension to a 15 ml falcon tube.
- Centrifuge at 4 °C through a Percoll solution. Always load ~25 μl of 100% Percoll at the bottom of the tube underneath the Percoll solution to act as a cushion for your cell pellet.
Note: This step should be optimized for the cell type of interest. The goal is to separate cell bodies from smaller debris to make sorting more efficient. We proved example conditions from three types of neurons that are small, medium, and large in size for guidance in optimizing.- Example conditions
Percoll solution- For cerebellar Purkinje neurons, which are large, the cell suspension was brought to a final concentration of 5% (v/v) Percoll in 5-10 ml.
- For cortical pyramidal neurons, the cell suspension was brought to a final concentration of 10% (v/v) Percoll in 5-10 ml.
- For cerebellar granule neurons, which are small, the cell suspension was brought to a final concentration of 20% (v/v) Percoll in 5-10 ml.
Centrifugation- For cerebellar Purkinje neurons in 10 ml, 54 x g for 15 min is used.
- For cortical pyramidal neurons in 10 ml, 54 x g for 15 min is used.
- For cerebellar granule neurons in 10 ml, 91 x g for 20 min is used.
Note: The first cerebellar granule cell pellet typically contains too much debris. To clean it up further the pellet is resuspended in another 5 ml of 20% Percoll, and centrifuged again.
- When optimizing, move the supernatant to a new tube and keep on ice.
- ~25 μl of the supernatant can be placed on slide or small dish. Once cells have settled for 1-2 min the droplet can be examined under a fluorescent microscope to estimate the number of cell bodies that failed to pellet.
- If needed, you may add a Percoll cushion to the bottom of the tube containing the supernatant and centrifuge again. This typically results in a pellet with additional cells, but often contains more debris than the first pellet.
- Considerations
- Pelleting cell bodies through the Percoll solution is a balance between speed, time, the concentration of Percoll (and volume, to lesser degree). Less force and less time spent centrifuging typically produced higher quality samples. Optimize for the minimal Percoll concentration that removes enough debris to significantly shorten sorting on the flow cytometer. It is not necessary to fully fractionate the sample.
- An additional option is to layer the cell suspension on top of the Percoll solution. This has the disadvantage of making it difficult to do additional spins on the supernatant but may allow better fractionation of some samples.
- Example conditions
- Suspend cell pellet in 1-3 ml cold mACSF + FBS immediately after removing supernatant.
Note: Avoid leaving cells pelleted for long periods of time. - Filter through a clean tube with cell strainer cap.
Sorting
- Sort on a flow cytometer
During optimization we recommend sorting ~100 cells onto a slide or small dish and examine the droplet under a fluorescent microscope to confirm cells of interest were isolated and estimate debris contamination using fluorescent illumination and brightfield.- Gating strategies are highly cell type and flow cytometer dependent. The goal is to capture individual healthy cells containing high levels of fluorescence signal. Our gates typically capture the top 1-10% of fluorescent events and subset to capture events with side scatter and forward scatter indicative of single cells and excluding doublets/clumps and debris.
- Adding a small amount of DNA dye such as Hoechst be useful in distinguishing small cell bodies containing a nucleus from debris when working with small neurons such as granule cells.
Notes:- Hoechst will penetrate live cells. If using other dyes such as DAPI or propidium iodide Triton x 100 will need to be added to permeabilize cells.
- With the bright mCitrine+ cells from Nelson enhancer trap lines (Shima et al., 2016) we often observe cell lysis under the microscope. A cell becomes dim and the fluorescent protein can be seen diffusing away. Therefore, simply estimating the number of cells that lack fluorescence by eye as a measure of contamination would likely give an overestimate and should not be used for this purpose.
- Sort bulk samples into a tube containing cold mACSF + FBS.
- Optional: to increase the quality of difficult samples tagment the sample in ~10,000 cell batches.
- For this, collect ~10,000 cells and follow Steps 29-39 while the next batch of cells is collected. Scale the tagmentation reaction in Step 36 by 1/5.
- Batching the sample reduces the overall time early sorted cells sit in suspension. Batches can be pooled later for library prep as needed, prioritizing batches collected earlier in the sort as these tended to give higher quality ATAC.
- Pellet sorted cells at 4 °C.
- Centrifuge sorted cells at 500 x g for 2 min.
- Rotate tube 180° in holder, centrifuge again at 700 x g for 1 min.
- Rotate tube 180° again and centrifuge at 500 x g for 2 min.
Note: Alternating the force angle helps collect cells off the tube wall and reduces cell loss when removing the supernatant. - The pellet is often not visible for samples < 20,000 cells.
Preparing Nuclei and Tagmentation Solution
- Remove the supernatant leaving ~2 μl and immediately resuspend the cell pellet by gentle flicking.
- Add 100 μl of Lysis Buffer and mix gently.
- Incubate on ice for 2-5 min.
Note: The goal is to lyse the cell membrane while leaving the nucleus mostly intact. - Quench lysis by diluting with 1 ml of mACSF+FBS.
This reduces the concentration of Triton X-100 allowing 2-3 μl to be left behind at Step 37 without inhibiting tagmentation later on. - Pellet nuclei at 4 °C.
- Centrifuge nuclei at 700 x g for 2 min.
- Rotate tube 180°, centrifuge at 1,000 x g for 1 min.
- Rotate tube 180°, centrifuge at 700 x g for 2 min.
- The pellet is often not visible for samples < 40,000 nuclei.
- Prepare 1x tagmentation solution:
For 50,000 cells:
Notes:25 μl 2x TD 0.5 μl 1% digitonin 0.5 μl 10% Tween-20 (variable) μl PBS for final volume of 50 μl* (variable) μl Tn5 transposase (100 nM final)** - *Accounting for the 2-3 μl of supernatant left in Step 37 becomes more important with smaller tagmentation reactions where all volumes are scaled down except Step 37)
- **The volume of Tn5 will vary depending on the enzyme source. 100 nM is recommended as a starting point for further optimization. This concentration worked well for most samples but had to be doubled for cerebellar granule neurons, likely due to significant debris contamination.
- Remove most of the supernatant from pelleted nuclei.
Note: It can be difficult to remove all of the volume without disturbing the pellet. Leave 2-3 μl and resuspend nuclei by gentle flicking in this volume. - Add 1x tagmentation solutionand flick tube gently to mix.
- Incubate at 37 °C with agitation for 30 min.
- Agitation option 1: Incubate stationary and gently flick tube every ~5 min.
- Agitation option 2: Incubate in a large bacterial flask incubator shaker set to ~250 RPM.
- Add 5 volumes of DNA Binding Buffer from Zymo DNA Clean and Concentrator-5 Kit.
Pause point: Sample can be stored at -20 °C for up to two weeks.
Library Preparation
- If starting from frozen samples, allow samples to warm to room temp.
- Purify tagmented DNA.
- Load and wash columns according to Zymo kit instructions.
- Elute in a volume to obtain ~2,500 cells worth of material/μl.
Note: ~1 μl is typically not recovered from the column. Therefore, an extra 1 μl can be added when calculating volumes. - To increase recovery of DNA from the column the volume can be split between two elutions.
- For example, 40,000 cells worth of tagmented material can be eluted in 8.5 μl twice to give ~16 μl total.
- The kit recommends using a minimum volume of 6 μl for elution. For samples where two 6 μl elutions would dilute the sample below 2,500 cells/μl, elute 2 times with 6 μl using H2O or diluted EB buffer* and rotovap to concentrate to ~2,500 cells worth of material/μl.
- *1x EB buffer comes with the Zymo DNA Clean kit. Dilute with H2O such that it will be 1x in the target final volume after rotovap concentration. For example, if the target final volume is 6 μl, rotovaping will concentrate the sample that was eluted twice with 6 μl by 2-fold, so a 1:2 dilution is needed.
- Prepare PCR Amplification Mix.
For 50,000 cells worth of material :
Note: If final libraries contain significant primer dimer contamination, the primer concentration can be cut in half.25 μl NEBNext Master Mix 2.5 μl 25 μM Primer Ad1 2.5 μl 25 μM Primer Ad2 (barcode) 0.5 μl 100x SYBR Green (in DMSO) - Add PCR Amplification Mix to the tube containing eluted DNA, mix by pipetting up and down 5-10 times then transfer to appropriate PCR tube(s) for amplification.
Notes:- The original ATAC and Omni-ATAC protocols suggest amplifying a small fraction of the sample in a qPCR thermocycler to determine the optimal number of PCR cycles and then amplify the rest of the sample in a different thermocycler appropriate for the larger volume.
- We found the optimal number of cycles varied for a single master mix aliquoted in different volumes as well as between different thermocyclers. To ensure the whole sample was amplified optimally and consistently, the entire sample was amplified in a qPCR thermocycler and monitored in real time to observe which cycle the fluorescent signal began to increase. Therefore, depending on the machine and sample volume, it may be necessary to divide the sample between multiple tubes. Aliquot the sample into the minimum number of PCR tubes (to reduce sample loss) without exceeding the recommended maximum volume for the qPCR machine.
- Amplify samples on a real-time PCR thermocycler with the cycling protocol indicated below:
Cycling ProtocolStep Temp (°C) Time 1 72 5 min 2 98 30 s 3 98 10 s 4 63 30 s 5 repeat steps 3-4 for < 12 cycles - Stop the amplification 2-3 cycles after the fluorescent signal begins to rise.
Notes:- Good quality libraries typically show an increase in fluorescent signal in < 6 cycles.
- Libraries that require > 12 cycles typically have low complexity.
Note: The 2x NEBNext Master Mix is sensitive to freeze thaw cycles. Best practice is to aliquot, store in an enzyme box, and protect from temperature swings while in the freezer. We recommend tracking the number of cycles need for libraries over time. If cycles creep up over batches, it may indicate the need to replace 2x Master Mix and further optimize storage. - If obtaining ATAC libraries with a large fraction of PCR duplicates (e.g., > 50%), reduce the number of cycles by 1.
- Purify amplified libraries using Zymo DNA Clean and Concentrator-5 Kit.
- Pool replicate tubes from the same sample.
- Optional: If concerned about yield, rinse PCR tubes with DNA Binding Buffer.
- Elute in 6 μl of low TE twice (total volume will be ~11 μl).
Quality Assessment
- Move 1 μl to a new tube and dilute 1:2 with low TE. Use this aliquot for quality control testing and quantification.
- Run 1 μl on a High Sensitivity Bioanalyzer Chip to examine the size distribution of fragments
Note: Figure 2A shows examples of the size distribution diversity from libraries that gave high quality sequencing data. While nucleosome laddering can indicate a high-quality library, good libraries do not always have this pattern. - Use 0.5 μl for qPCR with primers against genomic regions as in Table 2.
Notes:- Libraries with low complexity may show strong signal from 1-2 TSS regions but lack signal at 1-2 other TSS regions.
- High quality libraries typically showed strong signal from at least 3 different TSS regions.
- Libraries with good signal to noise at open regions typically gave a mean (TSS): mean (background) ratio > 50. This ratio may need to be established empirically if using other regions, species or tissue types.
- Use 0.5 μl to quantify using Kapa Library Quantification Kit according to manufacturer’s instructions. Samples are typically 5-50 nM.
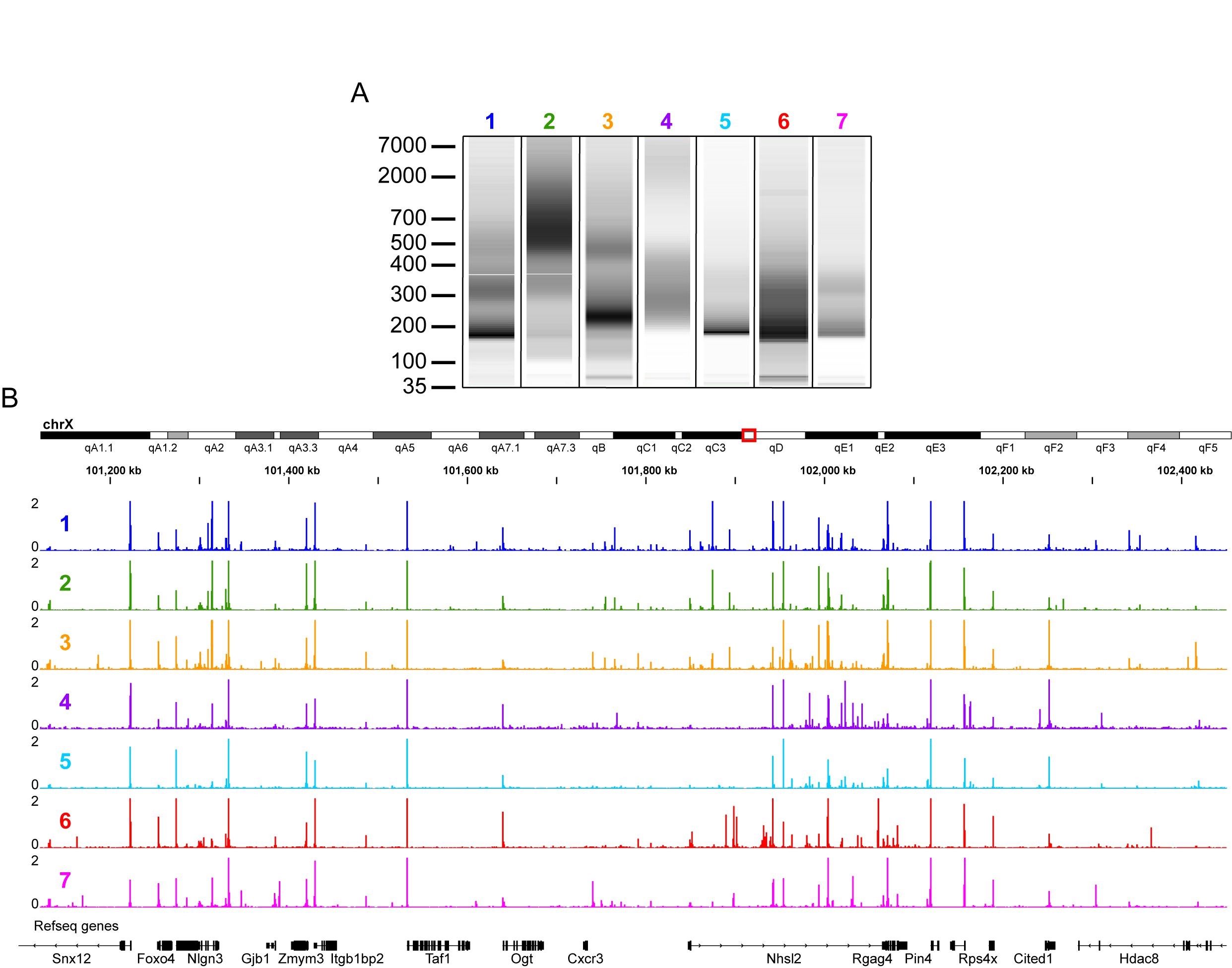
Figure 2. Examples of fragment size distributions from high quality ATAC libraries that deviated from standard nucleosome laddering. A. 0.5 μl of sample was run on a High Sensitivity DNA chip. B. Unique non-mitochondrial reads per million from the samples in A. Transcription start site (TSS) enrichment calculated following ENCODE guidelines for all seven samples was ≥ 20. Examples are from multiple different cell types.
- Run 1 μl on a High Sensitivity Bioanalyzer Chip to examine the size distribution of fragments
- Store the remaining 10 μl at -20 °C until sequenced.
- Sequence libraries.
- The protocol used for sequencing depends on the project goals. Paired-end sequencing is most commonly used with ATAC. This provides data on the total length of the fragment generated by transposition. Filtering data for certain sized fragments can be useful for certain analyses.
- When considering fragments versus reads it is important to keep in mind that the ends of fragments or one end of a read represents the location of Tn5 access to the genome while the fragment itself is the intervening DNA between two transposition events.
Data analysis
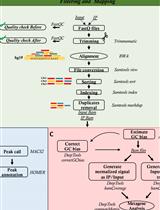
There are numerous options for analysis of ATAC-seq data that depend on the project goals. As with any project involving bioinformatic analysis it is strongly advised to consult with a statistician or bioinformatician to assess the data needed to achieve project goals. This section is meant to provide basic examples of early analytical steps to aid in visualizing data quality, as in Figure 2B. The parameters of each step should be assessed for their appropriateness to individual datasets. A useful resource is the ENCODE project pipeline (https://www.encodeproject.org/pipelines/ENCPL792NWO/).
- Check sequence quality with FastQC (Andrews, 2010). Assess the read quality by importing data from FastQ files.
- Trim adaptor sequences using cutadapt (version 1.9.1) (Martin, 2015). Using cutadapt (version 1.9.1) on paired end reads:
>cutadapt -a CTGTCTCTTATACACATCT -A CTGTCTCTTATACACATCT -q 30 --minimum-length 36 -o trimmed_output_1.fastq -p trimmed_output_2.fastq input_file_name_1.fastq.gz input_file_name_2.fastq.gz - Map reads to genome allowing only paired and concordant alignments with a maximum fragment length of 2,000 bp using bowtie2 (Langmead and Salzberg, 2012). Reference genomes can be obtained from iGenome.
> bowtie2 -t -X2000 --no-mix --no-discordant -x igenome/UCSC_mm10/Sequence/Bowtie2Index/genome -1 trimmed_output_1.fastq -2 trimmed_output_2.fastq -S mapped _output.sam
> samtools view -bS mapped_output.sam > mapped_output.bam
> samtools sort mapped_output.bam -o sorted_output.bam
> samtools index sorted_output.bam - Remove mitochondrial reads using samtools (Li et al., 2009). Mitochondrial reads can be large contaminant in ATAC data but varies between cell types.
> samtools idxstats sorted_output.bam | cut -f 1 | grep -v chrM | xargs samtools view -b sorted_output.bam > noMT_output.bam - Remove duplicate reads using picard-tools-1.123 (Broad Institute, 2019).
> java -jar -Xmx20g MarkDuplicates.jar METRICS_FILE=metrics.txt INPUT= noMT_output.bam OUTPUT=noDupM.bam REMOVE_DUPLICATES=TRUE - Shift unique non-mitochondrial reads to account for Tn5 protein offset using BEDTools-Version-2.16.2 (Quinlan and Hall, 2010).
> bamToBed -i noDupMT.bam > noDupMT.bed
> cat noDupMT.bed | awk -F $'\t' 'BEGIN {OFS = FS}{if ($6 == "+") {$2 = $2 + 4} else if ($6 == "-") {$3 = $3 - 5}print $0}' > shifted_output.bed - Make coverage and bigwig files for visualization using BEDTools-Version-2.16.2 and UCSC tools (Kent et al., 2010).
> genomeCoverageBed -g igenome/UCSC_mm10/genome_size.tab.txt -i shifted_output.bed -bg output_cov.bed
> wigToBigWig output_cov.bed igenome/UCSC_mm10/genome_size.tab.txt output.bw
Notes
- We recommend optimizing the steps of Percoll separation (Step 26), flow cytometry sorting (Step 29), and tagmentation (Steps 36 and 39) for each cell type. We provide examples of three specific cell types throughout the protocol for reference in optimizing.
- For optimizing Steps 26 and 29, the goal of centrifuging the cell suspension through a Percoll solution is to separate cell bodies from debris by density. Removing debris can make sorting by flow cytometry more efficient, yielding a larger number and higher quality nuclei. This is especially true if using one of the Nelson Lab enhancer trap lines (Shima et al., 2016) where mCitrine is expressed at high levels including in neuronal projections. MCitrine+ debris from these cells is often too bright to remove by simple intensity gaiting on the flow cytometer. In particular, small cells such as cerebellar granule cells pose a significant challenge to distinguish from debris. While projections from the cells of interest may not be problematic for assays such as RNA-seq, it is a problem for tagmentation where debris, which does not contain genomic DNA, can reduce Tn5 efficiency. You may optimize around this issue by reducing contaminating debris in the final sample and/or titrating the amount of Tn5 to compensate for debris.
Recipes
- TTX 0.1 mM (1,000x)
- Dilute TTX in molecular grade H2O
- Aliquot and store at -20 °C for up to six months
- Ketamine 500 mM (1,000x)
- Dilute in molecular grade H2O
- Store at 4 °C for up to six months
- 1% (100x) Digitonin (supplied at 2% in DMSO)
- Dilute 1:1 with molecular grade H2O
- Aliquot and store at -20 °C for up to 6 months or < 5 freeze thaw cycles
- 10% (100x) Tween-20
- Diluted in molecular grade H2O
- Store at 4 °C for up to six months
- 1x Low TE buffer (1x)
10 mM Tris-HCl (pH 8.0)
0.1 mM EDTA - 100x SYBR Green
- Dilute in DMSO
- Aliquot and store at -20 °C for up to six months
- 2x TD buffer
*Adjust the pH to 7.6 with 100% acetic acidFinal [ ] 1 M Tris-HCl pH 7.6 20 mM 1 M MgCl2 10 mM Dimethyl Formamide* 20%
Aliquot and store at -20 °C for up to six months - Lysis buffer
Aliquot and store at 4 °C for up to six monthsFinal [ ] Tris-HCl, pH 7.4 10 mM NaCl 10 mM MgCl2 3 mM IGEPAL CA-630 0.1% - Cutting Solution (CS)
Final [mM] Choline chloride 92 KCl 2.5 NaH2PO4·H2O 1.2 NaHCO3 30 HEPES 20 Na ascorbate 5 Thiourea 2 Na pyruvate 3 N-acetyl-L-cystein 12 MgSO4·7H2O 10 CaCl2·2H2O 0.5 - Modified Artificial Cerebral Spinal Fluid (mACSF)
Final [mM] NaCl 92 KCl 2.5 NaH2PO4·H2O 1.2 NaHCO3 30 HEPES 20 Na ascorbate 5 Thiourea 2 Na pyruvate 3 N-acetyl-L-cystein 12 MgSO4·7H2O 10 CaCl2·2H2O 0.5 - After dissolving all solutes in CS and mACSF, oxygenate with stirring for ~30 min at room temp to stabilize the working pH
- Bring the pH of CS to 7.3-7.4 using NMDG
- CS should have very little sodium to enhance neuroprotection
- Bring the pH of mACSF to 7.3-7.4 using NaOH
- For both CS and mACSF add enough trehalose or glucose (typically 2-6 g for 2 L of solution) to bring the osmolarity to 300-310 mOsm as measured on an osmometer
Note: Trehalose added here is separate and does not need to be considered in step 2 of the procedure. - If preparing more than 24 h in advance of use, filter sterilize and store at 4 °C
Acknowledgments
We thank the National Eye Institute (EY022360), the National Institute of Mental Health (MH105949) and the National Institute of Neurological Disorders and Stroke (NS075007) for funding support.
This protocol was adapted from previous work (Hempel et al., 2007; Buenrostro et al., 2013; Corces et al., 2017).
Competing interests
The authors have no competing interests to report.
Ethics
Animal experimentation: All experiments were conducted in accordance with the requirements of the Institutional Animal Care and Use Committees at Brandeis University (protocol #17001).
References
- Andrews, S. (2010). FastQC: a quality control tool for high throughput sequence data. Available online.
- Broad Institute. (2019). Picard Toolkit. Available on line, GitHub; Broad Institute
- Buenrostro, J. D., Giresi, P. G., Zaba, L. C., Chang, H. Y. and Greenleaf, W. J. (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10(12): 1213-1218.
- Corces, M. R., Trevino, A. E., Hamilton, E. G., Greenside, P. G., Sinnott-Armstrong, N. A., Vesuna, S., Satpathy, A. T., Rubin, A. J., Montine, K. S., Wu, B., Kathiria, A., Cho, S. W., Mumbach, M. R., Carter, A. C., Kasowski, M., Orloff, L. A., Risca, V. I., Kundaje, A., Khavari, P. A., Montine, T. J., Greenleaf, W. J. and Chang, H. Y. (2017). An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat Methods 14(10): 959-962.
- Gage, G. J., Kipke, D. R., Shain, W. (2012). Whole animal perfusion fixation for rodents. J Vis Exp 65: 3564.
- Hempel, C. M., Sugino, K. and Nelson, S. B. (2007). A manual method for the purification of fluorescently labeled neurons from the mammalian brain. Nat Protoc 2(11): 2924-2929.
- Kent, W. J., Zweig, A. S., Barber, G., Hinrichs, A. S. and Karolchik, D. (2010). BigWig and BigBed: enabling browsing of large distributed datasets. Bioinformatics 26(17): 2204-2207.
- Langmead, B. and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9(4): 357-359.
- Lee, H. J., Yoon, Y. S. and Lee, S. J. (2018). Mechanism of neuroprotection by trehalose: controversy surrounding autophagy induction. Cell Death Dis 9(7): 712.
- Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., Marth, G., Abecasis, G., Durbin, R. and Genome Project Data Processing, S. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25(16): 2078-2079.
- Martin M. (2015). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet Journal 17(1): 10-12.
- Picelli, S., Bjorklund, A. K., Reinius, B., Sagasser, S., Winberg, G. and Sandberg, R. (2014). Tn5 transposase and tagmentation procedures for massively scaled sequencing projects. Genome Res 24(12): 2033-2040.
- Papouin, T. and Haydon, P. G. (2018). Obtaining acute brain slices. Bio-protocol 8(2): e2699.
- Quinlan, A. R. and Hall, I. M. (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26(6): 841-842.
- Saxena, A., Wagatsuma, A., Noro, Y., Kuji, T., Asaka-Oba, A., Watahiki, A., Gurnot, C., Fagiolini, M., Hensch, T. K. and Carninci, P. (2012). Trehalose-enhanced isolation of neuronal sub-types from adult mouse brain. Biotechniques 52(6): 381-385.
- Shima, Y., Sugino, K., Hempel, C. M., Shima, M., Taneja, P., Bullis, J. B., Mehta, S., Lois, C. and Nelson, S. B. (2016). A Mammalian enhancer trap resource for discovering and manipulating neuronal cell types. Elife 5: e13503.
- Sugino, K., Clark, E., Schulmann, A., Shima, Y., Wang, L., Hunt, D. L., Hooks, B. M., Trankner, D., Chandrashekar, J., Picard, S., Lemire, A. L., Spruston, N., Hantman, A. W. and Nelson, S. B. (2019). Mapping the transcriptional diversity of genetically and anatomically defined cell populations in the mouse brain. Elife 8: e38619.
- Ting, J. T., Daigle, T. L., Chen, Q. and Feng, G. (2014). Acute brain slice methods for adult and aging animals: application of targeted patch clamp analysis and optogenetics. Methods Mol Biol 1183: 221-242.
Article Information
Copyright
![]() Clark et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Clark et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Clark, E. A., Shima, Y. and Nelson, S. (2019). ATAC-seq on Sorted Adult Mouse Neurons. Bio-protocol 9(19): e3382. DOI: 10.21769/BioProtoc.3382.
- Sugino, K., Clark, E., Schulmann, A., Shima, Y., Wang, L., Hunt, D. L., Hooks, B. M., Trankner, D., Chandrashekar, J., Picard, S., Lemire, A. L., Spruston, N., Hantman, A. W. and Nelson, S. B. (2019). Mapping the transcriptional diversity of genetically and anatomically defined cell populations in the mouse brain. Elife 8: e38619.
Category
Neuroscience > Basic technology > Chromatin biology
Molecular Biology > DNA > Chromatin accessibility
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.