- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Isolation and Transcriptomic Profiling of Single Myofibers from Mice

(*contributed equally to this work) Published: Vol 9, Iss 19, Oct 5, 2019 DOI: 10.21769/BioProtoc.3378 Views: 6850
Reviewed by: David PaulClara Lubeseder-MartellatoAmriti Rajender Lulla
Original research article
The authors used this protocol in:

Mar 2019
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Skeletal muscle is composed of different cells and myofiber types, with distinct metabolic and structural features. Generally, transcriptomic analysis of skeletal muscle is performed using whole muscle, resulting in average information as all cells composing the organ contribute to the expression value detected for each gene with the loss of information about the distinctive features of each specific myofiber type. Since myofibers are the smallest complete contractile system of skeletal muscle influencing its contraction velocity and metabolism, it would be beneficial to have fiber-specific information about gene expression. Here, we describe a protocol for the isolation and the transcriptomic analysis of single individual myofibers. The protocol was set up using single myofibers isolated from soleus and Extensor Digitorum Longus (EDL) muscles, but it can be applied to all skeletal muscles. Briefly, muscles are enzymatically dissociated and individually collected. Long RNAs (> 200 nt) and short RNAs (< 200 nt) are separately purified from each myofiber and used to produce libraries for microarray or sequencing analysis. Through this approach, myofiber-specific transcriptional profiles can be produced, free from transcripts from other non-contractile cell types, in order to identify mRNA-miRNA-lncRNA regulatory networks specific for each myofiber type.
Keywords: Single myofiberBackground
Skeletal muscle is a complex organ and heterogenous tissue. It is composed by different cell types such as those from blood, vessels, nerves, connective tissue, and myofibers, that are the smallest complete contractile system of skeletal muscle influencing both its mechanical performance and metabolism. In addition, myofibers are classified into different types. In mouse, type I myofibers display slow-twitch contraction and oxidative metabolism, type IIA and IIX myofibers fast-twitch contraction and oxidative metabolism, and type IIB myofibers fast-twitch contraction and glycolytic metabolism (Schiaffino and Reggiani, 2011). The preferential metabolism of each muscle is defined by the different proportion of myofiber types. Soleus muscle is mainly composed by type I, type IIA, and type 2X oxidative myofibers, and preferentially consumes lipids, whereas EDL muscle by type IIB glycolytic myofibers, and primarily uses carbohydrates as energy substrate (Augusto et al., 2004). To investigate differences in muscle metabolism and contraction at transcriptomic level, previous expression profiles were obtained using soleus and EDL whole muscles (Campbell et al., 2001; Wu et al., 2003). Results described in these works are influenced by the different contributions of diverse cells composing whole muscle (Figure 1A) and lose the transcriptional specificity of myofiber types (Figure 1B). We demonstrated the feasibility of scaling down the transcriptomic analysis of skeletal muscle at single isolated myofiber level for mRNA, long non-coding RNA (lncRNA) and microRNA (miRNA) populations (Figure 1C). This approach allowed the identification of expression profiles, free from non-myofiber cells (Chemello et al., 2011), and allowed the generation of mRNA-miRNA-lncRNA regulatory networks specific for each myofiber type (Alessio et al., 2019; Chemello et al., 2019). Moreover, it is easier to characterize perturbations that exclusively affect myofibers (Chemello et al., 2015; Mammucari et al., 2015), and to improve the description and comprehension of muscle atrophy (Alessio et al., 2019). In fact, muscle atrophy induces significant systemic metabolic modifications (Celegato et al., 2006) also in a myofiber-specific manner (Wang and Pessin, 2013).
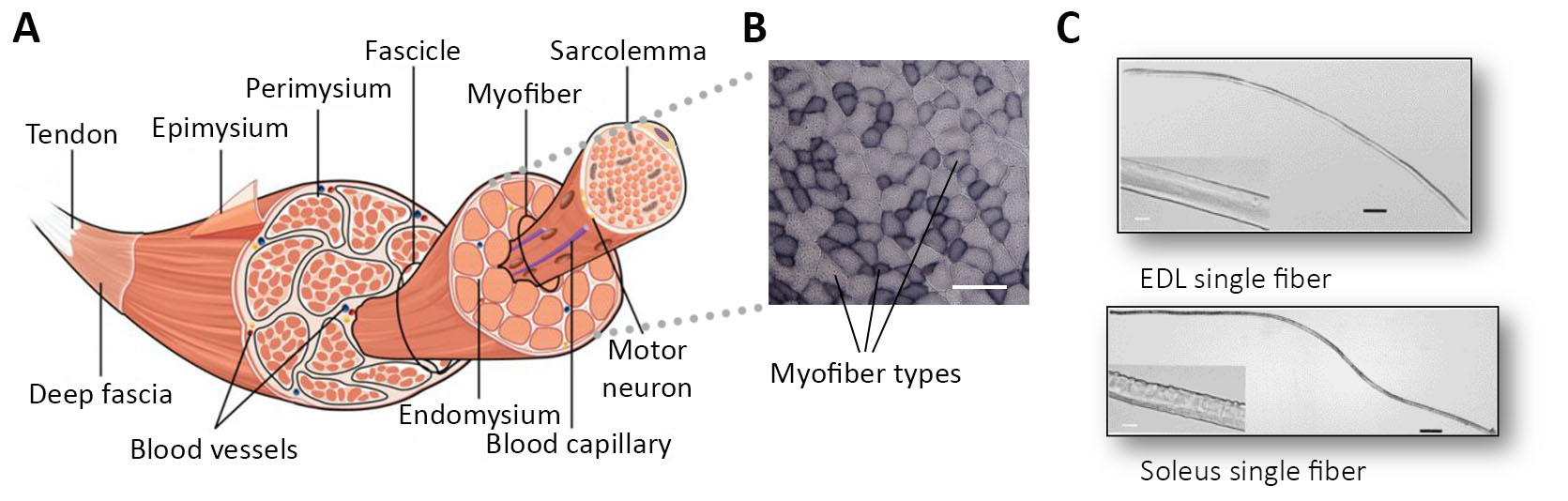
Figure 1. Complexity of skeletal muscle. A. Structure of the skeletal muscle with the different tissues and cells that make up its structure. Image modified from (Biga et al., 2019). B. Succinate Dehydrogenase Staining (SDH) shows different myofiber types in a transversal muscle cross-section. Scale bar is for 100 µm. C. Images of single isolated myofibers from EDL and soleus muscles. Scale bar is for 250 µm. From each single myofibers is possible to profile the expression of long and short RNAs.
Materials and Reagents
- 200 µl tips
- DNA LoBind tubes (Eppendorf, catalog number: 0030108051 or Serstedt, catalog number: 72.706.700)
- Spin-X centrifuge tubes (Corning-Costar, catalog number: CLS8160-96EA)
- RNase/DNase free Eppendorf microcentrifuge tube
- 0.2 ml PCR tube
- Hybridization gasket slides, 8 microarrays/slide (Agilent, catalog number: G2534-60014)
- Cell culture plates: 24-well and 6-well plates
- Plastic Pasteur pipettes
- Slide-staining dishes
- Adult wild-type mice, weight: 33-35 g
Note: We used CD1 mouse strain. - High-glucose Dulbecco's modified Eagle medium (DMEM) (Millipore Sigma, catalog number: D5796)
- 2,3-Butanedione monoxime (BDM) (Sigma-Aldrich, catalog number: B0753)
- Agencourt AMPure XP beads (Beckman Coulter, catalog number: A63881)
- Collagenase from Clostridium histolyticum, type I (Millipore Sigma, catalog number: C0130)
- Fetal bovine serum (FBS) (Millipore Sigma, catalog number: F2442)
- Phosphate-buffered saline (PBS) (Thermo Fisher Scientific, catalog number: 20012019)
- TRIzol Reagent (Thermo Fisher Scientific, catalog number: 15596026)
- Nuclease-free water (Thermo Fisher Scientific, catalog number: 10977035)
- Nuclease-free water (Ambion, catalog number: AM9937)
- Chloroform (PanReac AppliChem, catalog number: 121252)
- 70% v/v ethanol (dilute ethanol in nuclease-free water)
- 75% v/v ethanol (dilute ethanol in nuclease-free water)
- 80% v/v ethanol (dilute ethanol in nuclease-free water)
- Absolute ethanol
- miRNeasy kit (Qiagen, catalog number: 217004)
- RNeasy micro kit (Qiagen, catalog number: 74004)
- Complete Whole Transcriptome Amplification Kit (Millipore Sigma, catalog number: WTA2)
- GenElute PCR Clean-Up Kit (Millipore Sigma, catalog number: NA1020)
- SureTag DNA Labeling Kit (Agilent, catalog number: 5190-3400)
- Gene Expression Hybridization Kit (Agilent, catalog number: 5190-0404)
- Gene Expression Wash Buffer Kit (Agilent, catalog number: 5188-5327)
- SurePrint G3 Mouse GE 8x60K Microarray Kit (Agilent, catalog number: G4852A)
- Poly(A) Tailing Kit (Thermo Fisher Scientific, catalog number: AM1350)
- Sodium Acetate 3 M (pH 5.5) (Thermo Fisher Scientific, catalog number: AM9740)
- SuperScript II Reverse Transcriptase (Thermo Fisher Scientific, catalog number: 18064014)
- Oligo-dT-Ion P1 adapter primer (5′-CCTCTCTATGGGCAGTCGGTGATCCTCAGC[dT]20VN-3′)
- SMART primer (5′-CACACACAATTAACCCTCACTAAAggg-3′)
- Ion Xpress Plus gDNA Library (Thermo Fisher Scientific, catalog number: 4471269)
- PGM HI-Q OT2 kit (Thermo Fisher Scientific, catalog number: A27739)
- Platinum Taq DNA Polymerase High Fidelity (Thermo Fisher Scientific, catalog number: 11304011)
- E-Gel SizeSelect Gels (Thermo Fisher Scientific, catalog number: G661012)
- DNA ladder 50 bp (Thermo Fisher Scientific, catalog number: 10416014)
- High Sensitivity DNA kit (Agilent, catalog number: 5067-4626)
- Ion OneTouch Template Kit (Thermo Fisher Scientific, catalog number: A29900)
- 1x Low TE buffer (see Recipes)
Equipment
- Micropipette (P10, P20)
- -80 °C freezer
- -20 °C freezer
- Forceps and scissors for microdissection
- 37 °C incubator
- Stereo-microscope
- Refrigerated microcentrifuge
- SpeedVac concentrator
- Thermocycler
- Nanodrop spectrophotometer
- Heat block
- Centrifuge
- Agilent SureHyb chamber (Agilent, catalog number: G2534A)
- Hybridization oven (Agilent, model: G2545A)
- Hybridization oven rotator (Agilent, model: G2530-60029)
- Agilent scanner system (Agilent, model: G2505C)
- DynaMag (Thermo Fisher Scientific, catalog number: 12321D)
- Ion Torrent, or Ion S5 System (Thermo Fisher Scientific)
- E-GelTM Electrophoresis Device (Thermo Fisher Scientific)
- Agilent 2100 Bioanalyzer (Agilent)
- Ion OneTouch Instrument (Thermo Fisher Scientific)
- Ion PGM sequencing instrument (Thermo Fisher Scientific)
Software
- Bioanalyzer software (Agilent, https://www.agilent.com/en/product/automated-electrophoresis/bioanalyzer-systems/bioanalyzer-instrument/2100-bioanalyzer-laptop-228251 Company or Developer/Provider/Supplier, web address)
- Feature Extraction Software (Agilent, https://www.agilent.com/en/product/mirna-microarray-platform/mirna-microarray-software/feature-extraction-software-228496)
- R statistical software (https://www.r-project.org/)
- MultiExperiment Viewer (https://sourceforge.net/projects/mev-tm4/)
- miRDeep software (https://sourceforge.net/projects/mirdeepstar/)
Procedure
See Figure 2 for a summary of principal steps for the analysis of transcriptional profiling of single myofibers.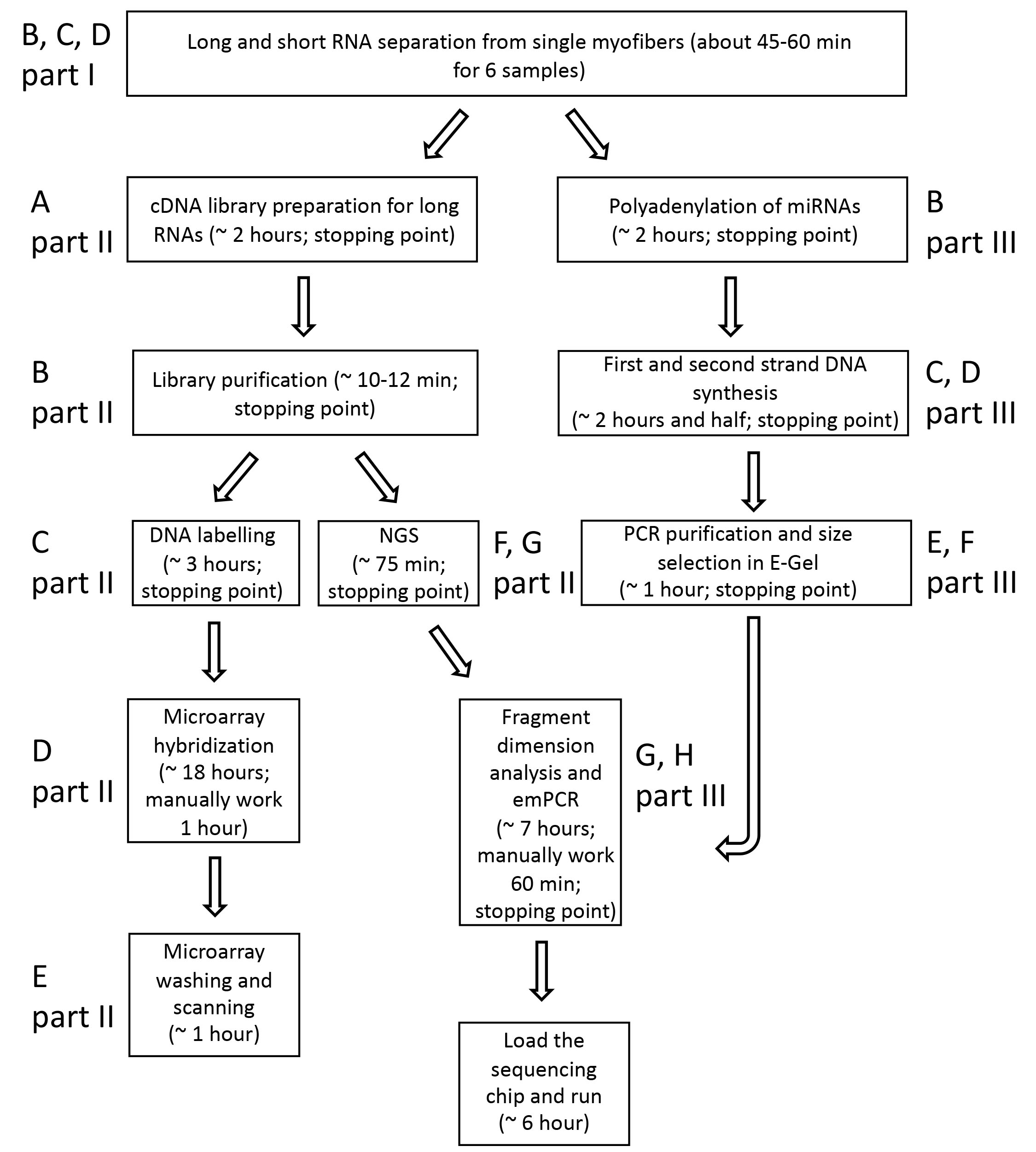
Figure 2. Workflow of transcriptomic analysis of single myofibers. The scheme summarizes principal steps for transcriptomic analysis of single myofibers. On the side of each box, it is reported the corresponding part of the protocol.
Part I: Purification of long RNAs (mRNAs and lncRNAs) and short RNAs (miRNAs) from single isolated myofibers
- Myofiber isolation
Note: Here we will describe myofiber isolation from soleus and Extensor Digitorum Longus (EDL) hindlimb muscles of 3 months old mice, but the same procedure can be used for other types of skeletal muscles optimizing the timing of collagenase dissociation. In principle, younger mice have smaller muscles, we suggest decreasing the time of collagenase incubation and the repetitions of triturating process with muscles of young mice, to avoid damage of myofibers. On the contrary, muscles of older mice have more connective tissue, it could be necessary increase the time of collagenase incubation and the repetitions of triturating process to obtain a sufficient number of single isolated myofibers for the following analyses. In any case, it is important to limit this step to avoid the alteration of gene expression and consequently cell pathways. Sacrifice mice according to the local animal welfare act.- Collect soleus or EDL muscles from hindlimb mouse, taking care to handle them only by their tendons to minimize mechanical damage of myofibers (Figure 3). We used 3 months old CD1 mice. Store them in 1 ml of cold DMEM 3 mM BDM. Muscles collection should take no more than half an hour. Below, step by step procedure to collect soleus and EDL muscles.
- Peel off the leg skin to expose the hindlimb muscles.
- Gently remove the fascia of the hindlimb muscles.
- Position the mouse supine and expose the tendons of tibialis anterior (TA) and EDL.
- Cut the distal TA tendon and use it to remove the TA muscle by cutting it at its proximal attachment.
- Cut the distal EDL tendon and remove it through cutting the proximal EDL tendon.
- Place the muscle in a tube containing 1 ml of DMEM 3 mM BDM.
- Repeat Steps A5-A7 to collect the EDL of the other leg.
- Place the mouse in prone position.
- Cut the tendon of gastrocnemius muscle and use it to peel off the muscle until the exposure of the soleus tendon.
- Cut the soleus tendon and use it to remove the soleus cutting the other end.
- Place the muscle in a tube containing 1 ml of DMEM 3 mM BDM.
- Repeat to collect the soleus of the other leg.

Figure 3. Muscles collection. A. EDL muscle isolated from hindlimb mouse muscles. B. Collection of one muscle in 1 ml of DMEM 3 mM BDM. C. Soleus muscle isolated from hindlimb mouse muscles. - Digest the muscles for 45 min at 37 °C in 1 ml high-glucose DMEM 3 mM BDM containing 10 mg type I collagenase in one well of a 24-well plate.
- Sequentially rinse the muscles for 2 min in 3 ml of DMEM and 3 mM BDM, 3 ml DMEM and 3 mM BDM supplemented with 10% FBS and 3 ml of DMEM and 3 mM BDM in different wells of a 6-well plate (Figures 4A, 4B and 4C). Handle muscle with care and control under the microscope to avoid losing myofibers during these washing steps.
- Transfer the muscles into a well of 6-well plate with 3 ml of DMEM with 10% FBS at about 25 °C to inactivate collagenase (Figure 4D).
- Liberate myofibers by gentle physical trituration (pipetting up and down) using a wide-mouth plastic Pasteur pipette (about 4 mm diameter), rinsed in FBS to prevent sticking.
- Repeat the triturating process several times, eventually transferring non-dissociated muscle in new wells, until about 100 intact fibers are obtained. After each physical trituration, transfer the muscles into a new well, to get rid of collagen wisps and hyper-contracted fibers (Figures 4E, 4F, and 4G).
- Using a stereo-microscope and a micropipette (200 µl tips), pick one by one intact and well-isolated fibers and wash first in a well with DMEM 3 mM BDM 10% FBS and then in a well with PBS (Figure 4H).
- If the researcher chose to avoid the myofiber characterization via MyHC protein expression (possible on a piece of myofiber [Chemello et al., 2011]), after washing, transfer the single myofiber into an RNase/DNase free Eppendorf microcentrifuge tube. The expression of specific genes [Myosin Heavy Chain 4 (Myh4), Myosin Light Chain 3 (Myl3), Myosin Heavy Chain 7 (Myh7), Tropomyosin 3 (Tpm3), actinin alpha 3 (Actn3), myosin heavy chain 2 (Myh2)] will be useful for the myofiber characterization (Chemello et al., 2019). See Figure 5 for the flowchart and timing specifications for each step.

Figure 4. Steps for single myofiber dissociation and isolation. A. After 45 min of incubation with collagenase the muscle is transferred from a 24-well plate (on the left of the picture) into a well of a 6-well plate containing 3 ml of DMEM 3 mM BDM. After 2 min of incubation, the whole muscle is transferred into the second well and then in the third well, waiting 2 min for the incubation in each well. B. Representation of the third transfer of the muscle. C. Scheme of the plate to use for the muscle rinsing after collagenase digestion (L: muscle form the left hindlimb; R: muscle from the right hindlimb; 1, 2, 3: first, second, and third wash). D. Scheme of the plate for collagenase inactivation and muscle dissociation (1, 2, 3: steps for releasing single myofibers from muscle). E. Using a 6-well plate filled as described in D dissociate muscle during collagenase inactivation. F. Transfer the muscle not dissociated in the second well to leave free myofibers in the first one. G. Transfer the muscle not dissociated in the third well to leave free myofibers in the second well and liberate eventually other myofibers. Recover free myofibers from all the wells and transfer them into a new 24-well plate filled as described in H. H. Scheme of the plate used to wash single myofibers (I: first wash; II: second wash; 1, 2, 3: several wells to wash different myofibers. It is not necessary to use a well per myofiber). After the second wash, single myofibers can be collected independently in different Eppendorf tubes or can be cut into two pieces; one used for their characterization using MyHC proteins and the other for RNA extraction and transcriptional analyses.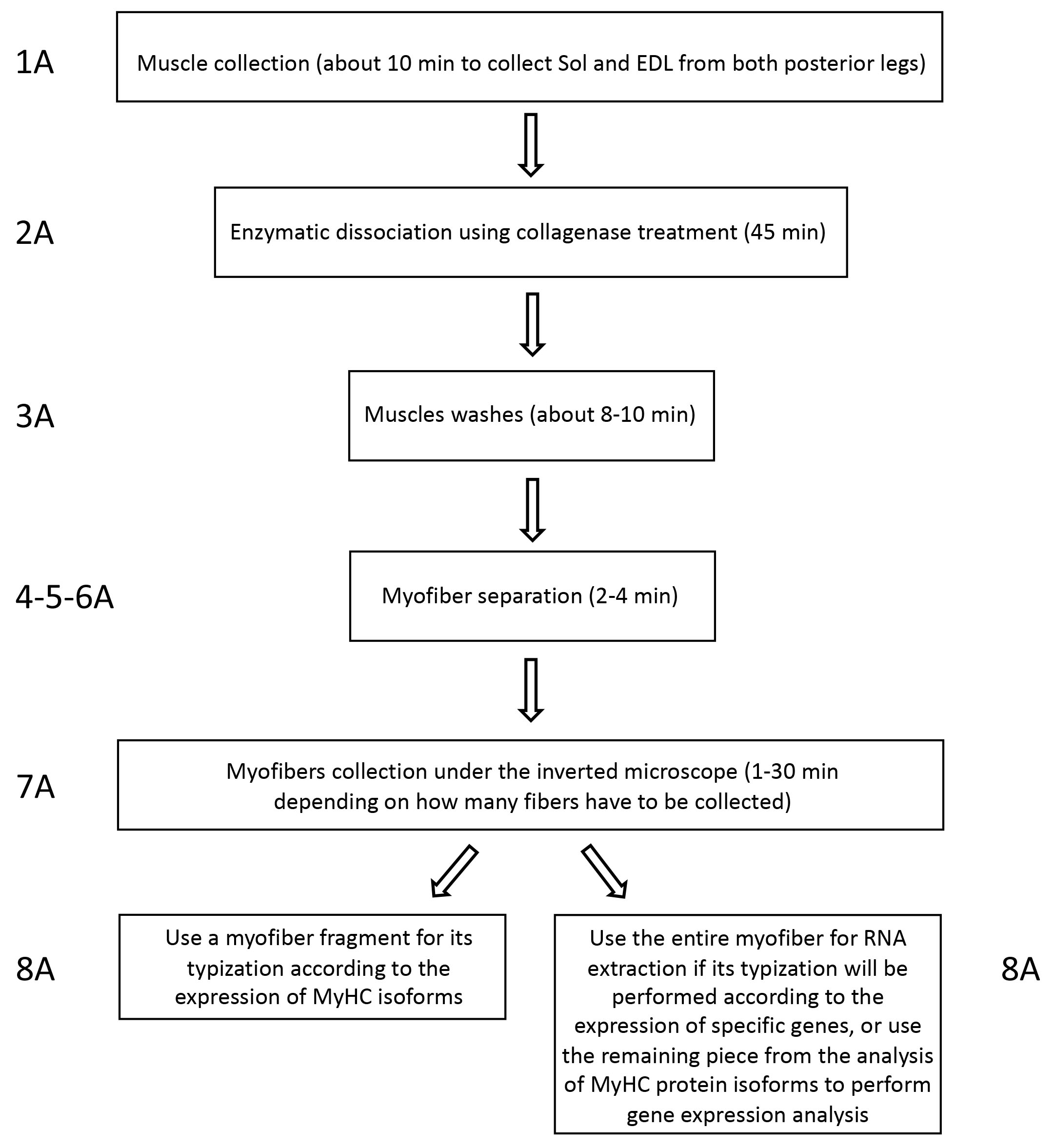
Figure 5. Workflow for the isolation of single myofibers from hindlimb mouse muscles. Each box describes work steps and timing and is associated with the protocol points.
Note: During the isolation of single myofibers it is mandatory to follow exactly the same procedure and time points when processing the same muscle types derived from different mice to avoid introducing errors due to tissue processing. Moreover, it is important to avoid recovering contracted fibers to avoid having a different gene expression due to their peculiar condition. There are no stopping points during the myofiber collection procedure. Only when myofibers are lysed in TRIzol for RNA analysis, in Laemmli buffer (Tris pH 6.8 62.5 mM, glycerol 10%, SDS 2%, β-mercaptoethanol 5%) for MyHC analysis (Chemello et al., 2011), flash freeze in nitrogen and then the tube can be stored at -80 °C. In Figure 1C, it is represented how isolated myofibers appear under microscope.
- Long RNA and short RNA separation using RNeasy kits (Qiagen)
To proceed with RNA purification, it is better to avoid working with too many myofibers. We suggest working with 4-6 myofiber samples.- Lyse each myofiber in 250 µl of TRIzol Reagent (Thermo Fisher Scientific) pipetting up and down the solution.
- Add 50 µl of chloroform, mix well by inverting the tube, and incubate on ice for 15 min.
- Centrifuge the sample for 15 min at 12,000 x g at 4 °C.
- Collect and transfer the aqueous upper phase (about 130 µl) into a new tube.
- Add 1 volume of 70% ethanol and mix.
- Transfer the solution onto a miRNeasy column (Qiagen).
- Spin for 15 s at 12,000 x g.
- Long RNAs are retained by the column, whereas short RNAs are collected in the eluate. Retain the eluate and process as described in Section D in Part I if you are interested in miRNA components.
- Long RNA purification
- Add 700 µl of RWT Buffer (Qiagen) to the column containing long RNAs (Section B in Part I).
- Spin for 15 s at ≥ 8,000 x g and discard the eluate.
- Add 500 µl of RPE Buffer (Qiagen) to the column.
- Spin for 15 s at ≥ 8,000 x g and discard the eluate.
- Add again 500 µl of RPE Buffer (Qiagen) to the column.
- Spin for 15 s at ≥ 8,000 x g and discard the eluate.
- Place the column in a new collection tube.
- Add 50 µl of nuclease-free water to the center of the column and incubate at room temperature for 2 min.
- Spin for 1 min at ≥ 8,000 x g to eluate long RNAs. Eventually, store the eluate at -80 °C in the eluted volume.
- Use SpeedVac concentrator to reduce the volume to 14 µl. It is better to control the volume during its reduction to avoid drying the sample.
- Store at -80 °C until use. To avoid RNA binding to the plastic tube it is strongly suggested to use LoBind tubes.
- miRNA purification using miRNeasy kit (Qiagen)
- Add 0.65 volumes of absolute ethanol (about 165 µl) to the eluate produced after loading the aqueous phase of the TRIzol extraction into the miRNeasy columns (Section B in Part I) and mix well.
- Transfer the solution onto an RNeasy micro column (Qiagen).
- Spin for 15 s at ≥ 8,000 x g and discard the eluate.
- Add 500 µl of RPE Buffer (Qiagen) to the column.
- Spin for 15 s at ≥ 8,000 x g and discard the eluate.
- Add 500 µl of 80% ethanol to the column.
- Spin for 2 min at ≥ 8,000 x g and discard the eluate.
- Spin again for 5 min at ≥ 8,000 x g with the cap opened.
- Transfer the column to a new collection tube.
- Add 14 µl of nuclease-free water to the center of the column and incubate at room temperature for 2 min.
- Spin for 1 min at ≥ 8,000 x g to eluate miRNAs. Eventually, store the eluate at -80 °C in the eluted volume.
- Use SpeedVac concentrator to reduce the volume to 6.5 µl. If you dry the solution, resuspend miRNAs in nuclease-free water and preferably use LoBind tubes for the SpeedVac step.
- Store at -80 °C until use.
- Preparation of the cDNA library of long RNAs for microarray hybridization using Complete Whole Transcriptome Amplification Kit (Sigma)
- Add 2.5 µl of Library Synthesis Solution to the 14 µl of long RNA solution prepared in Section C in Part I.
- Incubate at 70 °C for 5 min and cool down the temperature by putting the Eppendorf on ice (~4 °C) for 5 min.
- Add the mix indicated below to the solution previously prepared (Steps A1 and A2 in Part II) (Table 1).
Table 1. Library synthesis enzyme mix
- Incubate in the thermocycler using the parameters shown in (Table 2).
Table 2. Parameters for library synthesis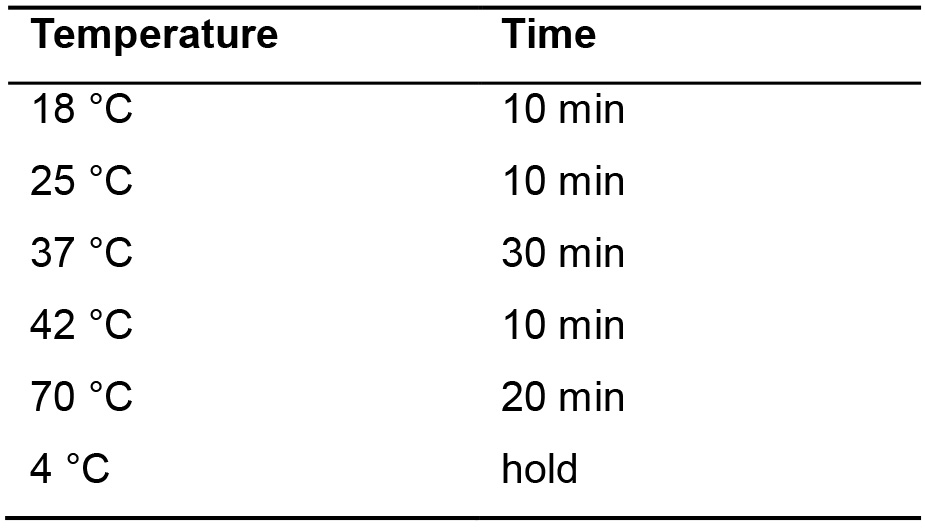
- Remove tubes from the thermocycler and spin briefly.
- Add the mix indicated into the solution (Table 3).
Table 3. Library amplification solution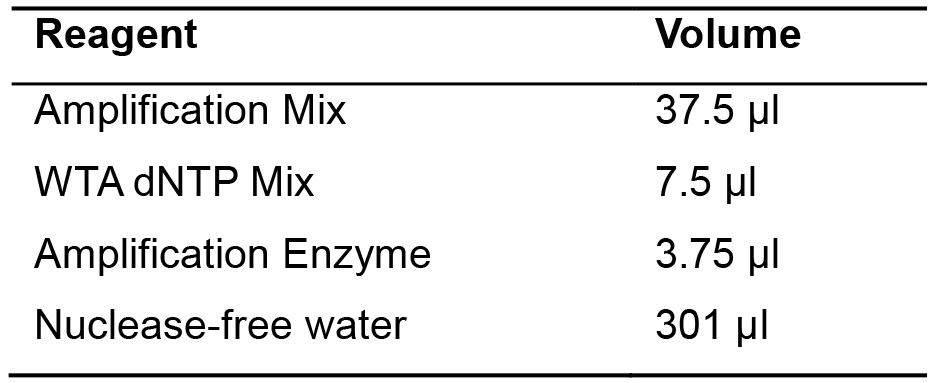
- Divide previously prepared mix into five Eppendorf each containing 75 µl of solution (for the last aliquot the reaction volume is < 75 µl).
- Incubate in the thermocycler using the following parameters (Table 4).
Table 4. Parameters for Library amplification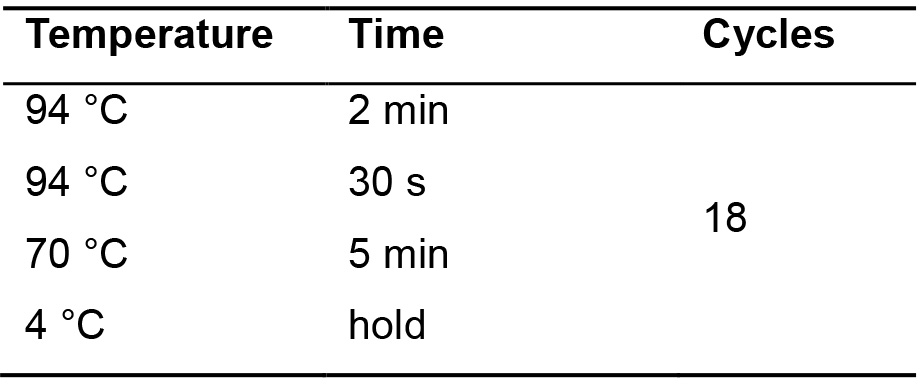
- Store the WTA DNA at -20 °C until use.
- WTA DNA purification using GenElute PCR Clean-Up Kit (Sigma)
- Purify separately each mix (each Eppendorf contains 75 µl of solution prepared as described in Section A of Part II).
- Add 375 µl of Binding Solution and transfer the solution into the binding column.
- Spin for 1 min at 12,000 x g and discard the eluate.
- Add 500 µl of Wash Solution to the column.
- Spin for 1 min at 12,000 x g and discard the eluate.
- Spin for 2 min at maximum speed without any additional wash.
- Transfer the column to a new collection tube.
- Add 50 µl of nuclease-free water to the center of the column and incubate at room temperature for 2 min
- Spin for 1 min at 12,000 x g.
- Quantify DNA using Nanodrop spectrophotometer.
- Store the purified WTA DNA at -20 °C until use.
- WTA DNA can be labeled for microarray hybridization (Section C in Part II) or can be used for NGS library preparation (Section F in Part II).
- Labeling WTA DNA for microarray hybridization using SureTag DNA Labeling Kit (Agilent)
- Prepare 2 µg of WTA DNA in 13 µl of nuclease-free water.
- Add 2.5 µl of Random Primers.
- Incubate at 98 °C for 5 min.
- Move onto ice and incubate for 5 min.
- Spin for 1 min at 12,000 x g.
- Add the mix indicated to the solution (Table 5).
Table 5. DNA labeling solution
- Incubate in the thermocycler using the parameters shown in Table 6.
Table 6. Parameters for DNA labeling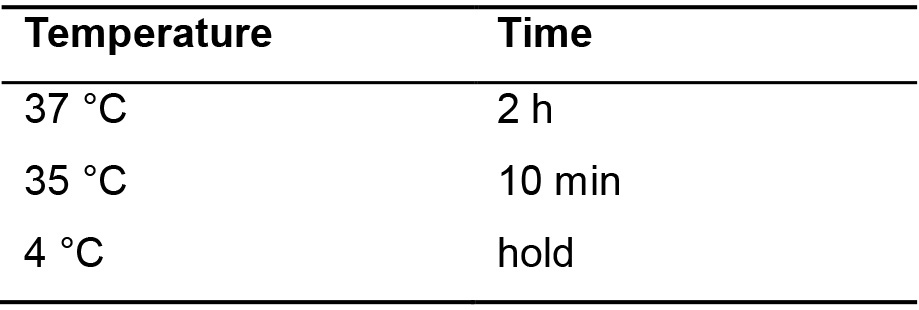
- Spin for 1 min at 10,000 x g.
- Add 430 µl 1x TE (pH 8.0).
- Transfer the solution onto a purification column provided with the SureTag DNA Labeling Kit (Agilent).
- Spin for 10 min at 14,000 x g and discard the eluate.
- Add 480 µl of 1x TE (pH 8.0) to the column.
- Spin for 10 min at 14,000 x g and discard the eluate.
- Invert the column into a new tube.
- Spin for 1 min at 1,000 x g to collect the purified sample. The volume per sample will be approximatively 20-32 µl.
- Measure DNA quantity and labeling using Nanodrop spectrophotometer. DNA yield will be about 4 µg and the specific activity 30 pmol of Cy3 per µg of cDNA.
- Store the labeled DNA at -20 °C until use.
- Hybridization on Agilent microarrays
Note: If you are interested in measuring the expression of lncRNAs make sure that the microarray platform you are using contains probes for this class of transcripts.- Prepare 800 ng of labeled DNA in 20 µl of nuclease-free water.
- Add 5 µl of 10x Blocking Agent.
- Incubate at 95 °C for 2 min.
- Add 25 µl of 2x GEx Hybridization Buffer HI-RPM.
- Dispense 40 µl of the solution on hybridization gasket slide.
- Put the 8x60K microarray platform slide facing down the hybridization gasket slide.
- Load the microarray sandwich into the Agilent SureHyb chamber.
- Load the assembled chamber into the oven rotator rack.
- Hybridize at 65 °C for 17 h at 10 rpm.
- Scan of the microarray slides
- Disassemble the hybridization chamber.
- Disassemble the microarray sandwich in slide-staining dish filled with Wash Buffer 1.
- Wash the microarray slide in Wash Buffer 1 for 1 min.
- Wash the microarray slide in 37 °C pre-warmed Wash Buffer 2 for 1 min.
- Slowly remove the microarray slide, to minimize droplets on the slide.
- Put the microarray slide in a slide holder.
- Scan the slide using G2505C scanner at 3 µm resolution to obtain single myofiber mRNA and lncRNA expression profile.
- NGS of RNA extracted from a single myofiber
- The bulk of SeqPlex amplification product (WTA DNA) ranges in size from 200-400 base pairs and therefore it is not needed DNA fragmentation for its sequencing. Here we will describe the sequencing protocol to use Ion Torrent or Proton sequencers but changing adapters, amplified RNA (WTA DNA) can also be sequenced with Illumina sequencer.
- The terminus of each amplicon possesses a 5′-phosphate and 2-base 3′-over-hang.
- Sequencing library preparation
- In a 0.2 ml PCR tube, prepare Adapters mix as indicated in the table below (non-barcoded), and mix by pipetting up and down. Reagents are in the Ion Xpress Plus gDNA Library preparation (Thermo Fisher Scientific) (Table 7).
Table 7. Adapters mix
- Prepare the following mix (Table 8).
Table 8. DNA repair and adapters ligation mix
- Incubate in thermal cycler following the program below (Table 9).
Table 9. Parameters for DNA repair and adapters ligation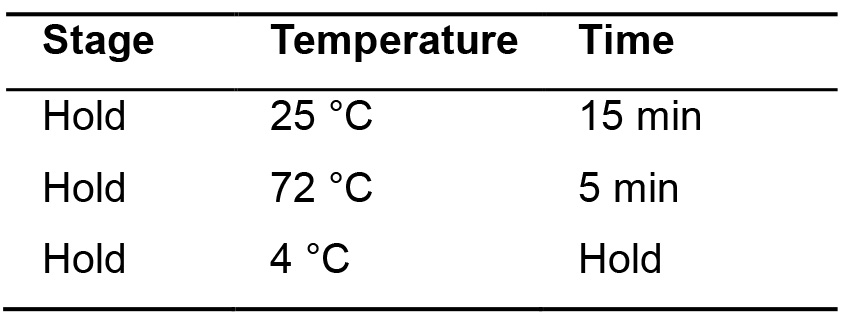
- Purify adapter-ligated DNA using Agencourt AMPure XP beads.
According to the desired reads length it will be used different amounts of Agencourt AMPure XP beads: add 100 µl of Agencourt AMPure XP beads to 100 µl of reaction from Step G3 in Part II for 400-600 bp reads length, 120 µl of Agencourt AMPure XP beads to 100 µl of reaction from Step G3 in Part II for 200-300 bp reads length. - Mix well by pipetting up and down and avoid bubble formation. Then place the tube in a magnetic rack (e.g., DynaMag) for 3 min and wait until the solution becomes clear.
- Discard the supernatant and avoid disturbing the pellet.
- Add 500 µl of freshly prepared 70% ethanol leaving the tube on the magnet.
- Incubate for 30 s rotating the tube around the vertical axis 3 times and maintaining the tube on the magnet to agitate the beads avoiding that they go in the solution. After the solution is clear, remove the supernatant avoiding disturbing the pellet.
- Wash the beads with 70% ethanol for a second time as indicated in the previous step.
- Remove residual of ethanol centrifuging the tube, placing it on the magnet and pipetting out the supernatant using a tip for a P10 micropipette.
- Keep the cap-opened tube on the magnet for 5 min to air dry the beads (avoid over dry).
- Remove the tube from the magnet, add 20 µl of Low TE on the beads and mix by pipetting up and down the solution. Vortex the sample for 10 s.
- Spin the solution for a few seconds and place the tube on the magnet for at least 1 min.
- Transfer the supernatant into a new LoBind Eppendorf without disturbing the pellet.
- Store at -30 °C to -10 °C.
- Before performing emulsion-PCR, amplification library can or cannot be selected in a gel according to DNA size. If the range selected with Agencourt AMPure XP beads it is fine for NGS researcher to go throughout emPCR. For method about size selection see Section F in Part III. For emulsion-PCR, see Section H in Part III.
- In a 0.2 ml PCR tube, prepare Adapters mix as indicated in the table below (non-barcoded), and mix by pipetting up and down. Reagents are in the Ion Xpress Plus gDNA Library preparation (Thermo Fisher Scientific) (Table 7).
- Purification of miRNAs from a single myofiber (see Section D in Part I)
- Polyadenylation of miRNAs using Poly(A) Tailing Kit (Thermo Fisher Scientific)
- Add the mix indicated in Table 10 to the solution of miRNAs purified as in Section D in Part I (6.5 µl of miRNA solution).
Table 10. Poly(A) tailing mix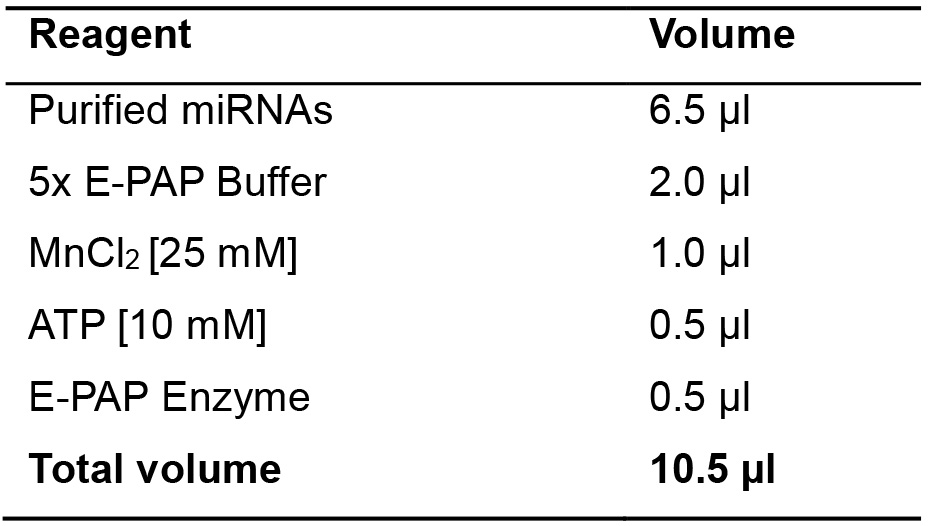
- Incubate at 37 °C for 1 h.
- Add 1 µl NaOAc 3 M (pH 5.5).
- Add 44 µl of absolute ethanol and mix well.
- Precipitate at -20 °C overnight.
- Spin for 20 min at maximum speed and carefully remove the supernatant without disturbing the pellet.
- Wash with 50 µl of 75% ethanol.
- Spin for 10 min at maximum speed and carefully remove the supernatant without disturbing the pellet.
- Resuspend in 3.2 µl of nuclease-free water.
- Add the mix indicated in Table 10 to the solution of miRNAs purified as in Section D in Part I (6.5 µl of miRNA solution).
- Synthesis of the first stranded cDNA from polyadenylated miRNAs (from Section B in Part III)
- Prepare the mix A in a 0.2 ml tube as indicated in Table 11.
Table 11. Retrotranscription mix A
- Prepare the mix B in a 0.2 ml tube as indicated below (Table 12).
Table 12. Retrotranscription mix B
- Incubate the mix B at 65 °C for 5 min in a thermoblock or thermocycler without heated lid.
- Spin the mix B and incubate it on ice for 5 min.
- Mix the mix A and B. You should have a final volume of 10 µl. Mix well the solution by pipetting up and down.
- Incubate at 42 °C for 60 min using a thermocycler without heated lid.
- Inactivate the enzyme at 70 °C for 15 min.
- Prepare the mix A in a 0.2 ml tube as indicated in Table 11.
- Synthesis of the second stranded cDNA
Second strand cDNA is synthesized by a PCR step avoiding reaching the plateau phase. In fact, it is important to maintain the original ratios of miRNAs. This step allows the production of double stranded DNA that, after its purification, can be sequenced because presenting on the 5′ and 3′ end sequencing primers for the Ion Torrent platform. It is important to use a High-Fidelity Taq DNA polymerase to avoid the introduction of errors during PCR amplification. For a scheme of the SMART protocol see Figure 6.- Prepare the following mix to perform two PCR reactions per sample (each of 50 µl) (Table 13).
Table 13. PCR amplification of poly(A) tagged miRNAs
- Incubate in the thermocycler according to the following cycle (Table 14).
Table 14. Parameters for PCR amplification of poly(A) tagged miRNAs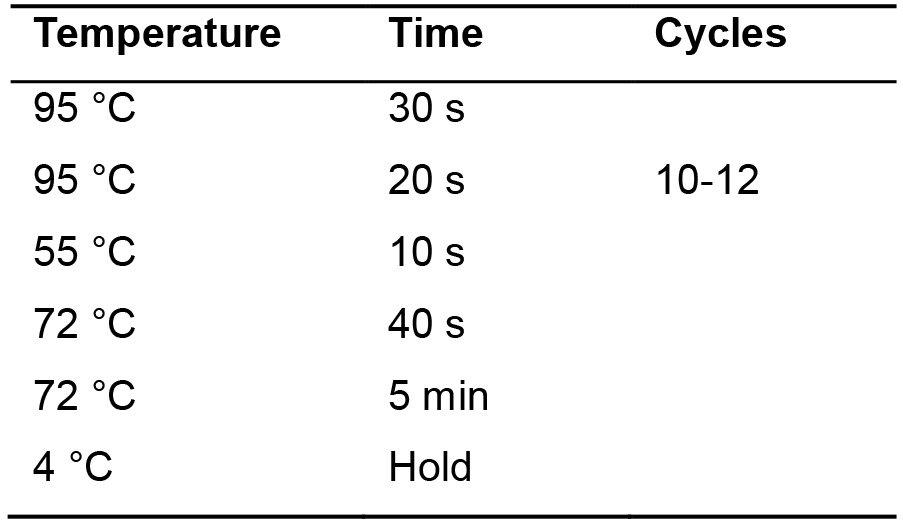

Figure 6. Scheme for miRNA amplification. miRNAs extracted from a single myofiber are polyadenylated using the PAP enzyme (1, and Section B in Part III). After that, using the SMART technique (Wellenreuther et al., 2004) miRNAs are retrotranscribed. The primer used for the retrotranscription is an oligo-d(T) that includes the Ion P1 adapter sequence at the 5′ end (2). When the Superscript II reaches the 5′ end of miRNA it adds a stretch of no template C to the cDNA that serves for the binding of the SMART primer (3). In the SMART primer, the three terminal g are RNA nucleotides. After the binding of the SMART primer to the cDNA it is used from the Superscript II for the synthesis of the complementary sequence (4, and Section C in Part III). Using this method, two anchors at the 3′ and 5′ end of the miRNAs are produced. The two anchors can be used for the miRNA amplification by PCR reaction. During PCR amplification (Section D in Part III) a specific primer it is used to include the A adapter in the 5′ end of miRNAs (5) that will be useful for miRNA sequencing in a strand specific way. After 10-12 cycles of PCR amplification (6, 7, 8), sufficient quantity of double stranded DNA is obtained with P1 and A adaptors on the miRNA 3′ and 5′ end, respectively to allow emulsion-PCR and following sequencing.
- Prepare the following mix to perform two PCR reactions per sample (each of 50 µl) (Table 13).
- DNA purification using GenElute PCR Clean-Up Kit (Sigma)
- Pool the two PCRs (50 + 50 µl). If you have more than one sample remember to maintain them separated.
- Add 500 µl of Binding Solution to the pooled PCRs and transfer the solution into the binding column.
- Spin for 1 min at 12,000 x g and discard the eluate.
- Add 500 µl of Wash Solution to the column.
- Spin for 1 min at 12,000 x g and discard the eluate.
- Spin for 2 min at maximum speed without any additional wash.
- Transfer the column to a new collection tube.
- Add 50 µl of nuclease-free water to the center of the column and incubate at room temperature for 2 min.
- Spin for 1 min at 12,000 x g.
- Quantify DNA using Nanodrop spectrophotometer.
- Store the purified PCR at -20 °C until use.
- Size selection for NGS using the E-Gel SizeSelect Gels (Thermo Fisher Scientific)
For DNA sequencing it is important to select the size range of DNA to be sequenced. In the case of long RNAs dimensional purification it was described in the Step G4 in Part II. Retained reads to identify miRNAs are 18-35 nt long (He et al., 2019) or purified small RNAs for miRNA libraries covers the same dimensional range ( Krishna et al., 2013; Fu et al., 2018) and adapters added during the library construction (Sections D and E in Part III) are ~107 nucleotides (Primer A is 57 nt and Oligo-dT-Ion P1 50 nt ) (Figure 6). We, therefore, selected the smear between 125 and 142 nt. See below for the full procedure.- Use a 2% agarose gel.
- Each well of the gel should be loaded with no more than 20-25 µl and 500 ng of DNA.
- If DNA prepared from Section E in Part III needed to be diluted use Low TE to dilute it.
- Dilute DNA ladder (50 bp) to 25 ng/µl with Low TE and load central well of the E-Gel with 10 µl of diluted ladder.
- Load sample wells avoiding using adjacent wells for different samples (same samples split in different volumes can be loaded in adjacent wells).
- Empty wells have to be filled with 25 µl of nuclease-free water.
- Run E-Gel SizeSelect instrument (Thermo Fisher Scientific) selecting the correct program (SizeSelect 2%).
- Visualize the run using the instrument and stop it when desired DNA dimension filled recovery well.
- Recover the solution from the recovery well using a P20 pipette without touching the gel. Usually, 12-15 µl can be recovered.
- Fill the recovery well with nuclease-free water replacing the withdrawn volume.
- Start again the instrument and repeat Step F9 in Part III until all the desired dimensional range is recovered.
- Dimensional analysis and library quantification (Agilent Bioanalyzer analysis)
Run purified samples according to the High Sensitivity DNA Chip (Agilent).- Thaw High Sensitivity DNA kit at room temperature for at least 30 min.
- Prepare gel dye mix by adding 15 µl of High Sensitivity DNA dye to High Sensitivity DNA gel matrix.
- Mix by vortexing and spin down.
- Put the solution onto Spin-X Centrifuge tubes 0.22 µM pore diameter (Corning Costar) and centrifuge at 2,200 x g for 10 min at room temperature.
- Load the High Sensitivity DNA chip as described in the table below (Table 15).
Table 15. Agilent Bioanalyzer chip loading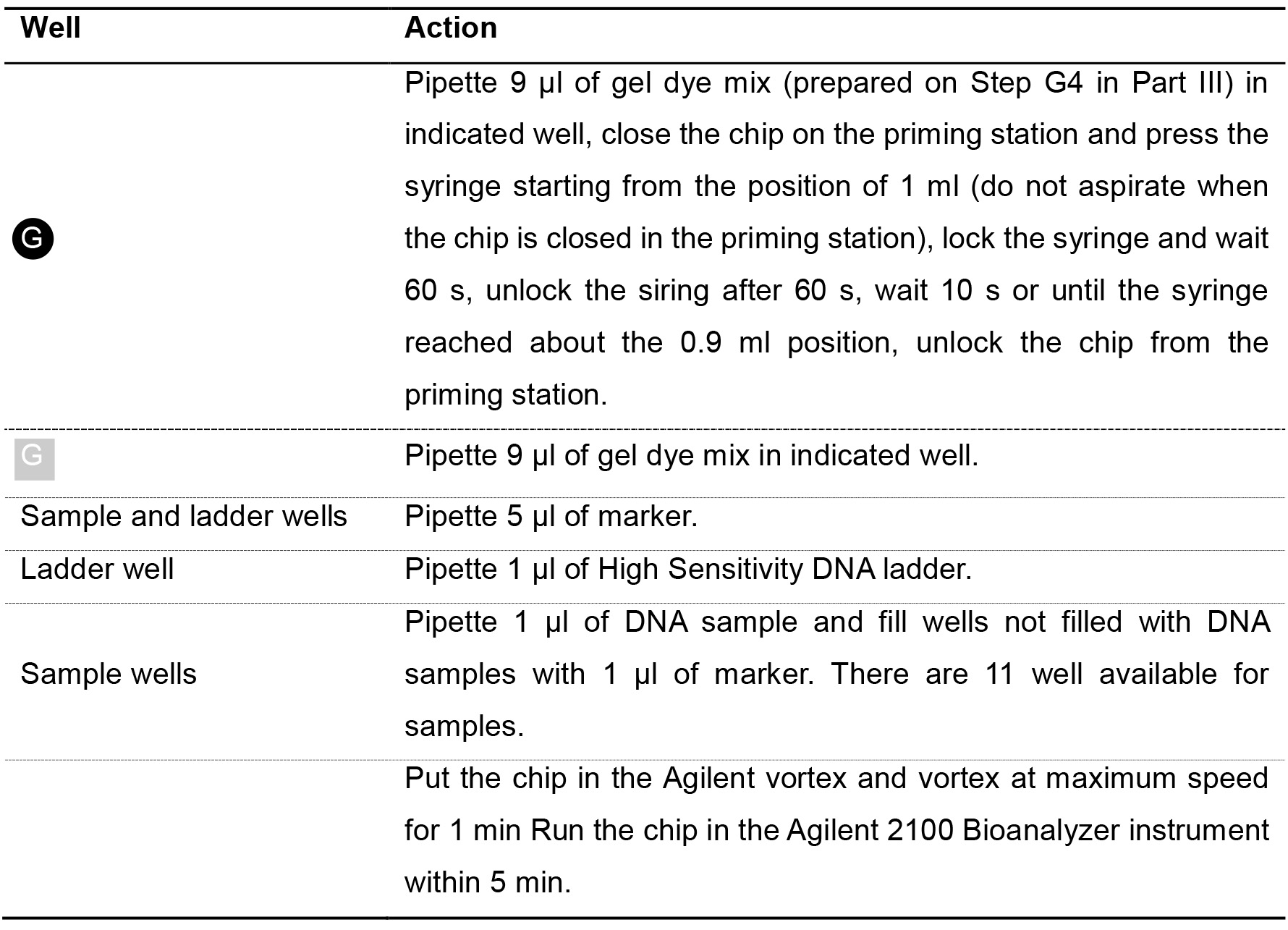
- From the Bioanalyzer analysis used to assess the library size distribution, determine the molar library concentration in pmol/L using the Bioanalyzer software.
- Determine the dilution factor that results in a concentration of the library of ~100 pM.
- This concentration is suitable for template preparation using Ion OneTouch Template Kit (Thermo Fisher Scientific). Use the following formula: Dilution factor = (Library concentration in pM)/100 pM.
- Dilute the library in a new LoBind Eppendorf using a small amount of DNA library and necessary Low TE to yield approximately a concentration of 100 pM. Use this library dilution for template preparation.
- Emulsion-PCR (template preparation for DNA sequencing) using the Ion OneTouch system (Thermo
I.Fisher Scientific)
Note: Described material is included in the Ion PGM Hi‑Q OT2 Kit (Thermo Fisher Scientific).- Set up the Ion OneTouch system.
- Prepare the amplification solution as follow (Table 16).
Table 16. DNA library dilution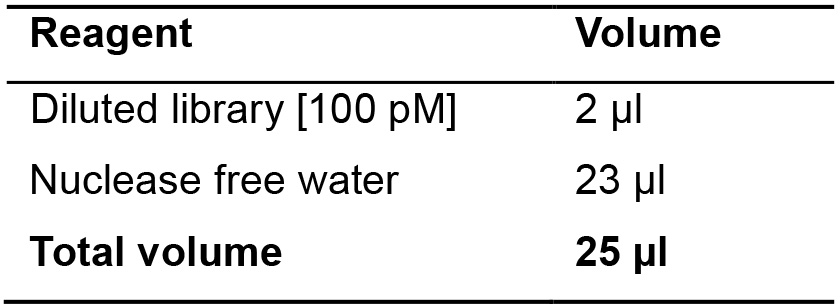
- Vortex for 5 s and spin for 2 s.
- Prepare the Ion PGM Hi-Q ISPs and mix thoroughly by vortexing the tube for 1 min and spinning for 2 s.
- Prepare the following mix in a 2-ml LoBind tube (Table 17).
Table 17. OneTouch reaction mix
- Load the Ion OneTouch Reaction Filter with the solution prepared in Step H5 of Part III, install the filter onto the Ion OneTouch Instrument and start the run.
- At the end of the run, enrich for the template-positive IPSs following the Ion OneTouch instructions and load the sequencing chip.
Data analysis
- Microarray data analysis for the expression analysis of long RNAs
- Data pre-processing
Probes features are extracted using Feature Extraction Software (Agilent Technologies). We published the following description of data treatment in (Alessio et al., 2019; Chemello et al., 2019). Data have to be intra-array and inter-array normalized. For intra-array normalizations, we suggest using the Feature Extraction Software while for the inter-array the quantile normalization (normalizeQuantiles, limma R package). For filtering out bad expression values we used Found and/or Well Above Background flags calculated from the Feature Extraction Software (Agilent Technologies). If the chosen filter was not positive, we assign to the particular expression value the value of NA. This allows maintaining in the expression matrix only transcripts that have a defined expression value in a sufficient number of biological replicates. For example, in (Chemello et al., 2019) we considered only gene/probes with at least 8 available values out of a total of 10 biological replicates. - Statistical analyses of microarray data
Microarray data can be analyzed using the R statistical suite or a more user-friendly platform such as the MultiExperiment Viewer, a tool of TM4 Microarray Software Suite (Saeed et al., 2003). To identify the differentially expressed probes among myofibers, we performed one-way analysis of variance (ANOVA), but different alternatives can be explored, such as the identification of differentially expressed probes using the False Discovery Rate implemented in the Significance Analysis of Microarrays (SAM) algorithm (Tusher et al., 2001). - Gene ontology
Differentially expressed coding genes can be categorized according to gene ontology (GO) using different web tools (e.g., the Functional Annotation Clustering method, implemented in the DAVID database [Huang da et al., 2009] or according to pathway analyses [e.g., (Sales et al., 2013)] that implement different algorithms for the pathway analysis). Regarding lncRNAs, there are no enrichment analyses that can be performed. To infer their function, it can be performed a gene expression correlation with coding genes such as in (Guo et al., 2017; Fang et al., 2018; Cagnin et al., 2019).
- Data pre-processing
- NGS expression analysis
In all sequencing experiments, raw reads have to be trimmed for the presence of adaptors and then filtered out for their length and quality. In Chemello et al. (2019) reads longer than 18 nucleotides were used for the following analyses: The expression of miRNAs was quantized using miRDeep software (Friedlander et al., 2008). The processed reads were mapped to the known mouse miRNA precursors from miRBase database (Ver. 19) using the mapper module of miRDeep with default values. Basically, in this process equal reads are counted and collapsed. Reads that mapped more than 5 different locations are automatically excluded. Quantize module in miRDeep was used to normalize read counts of mature miRNAs. For a discussion about problems, software used in the analysis (normalization, read count, clustering) of single cell data see (Risso et al., 2018a; Risso et al., 2018b; Chen et al., 2019; Cole et al., 2019).
Recipes
- 1x Low TE buffer
TE buffer is a buffer solution containing Tris, a common pH buffer, and EDTA, a molecule that chelates cations like Mg2+. It is used to solubilize DNA or RNA, to protect them from degradation
To make 1x Low TE buffer, see Table 18.
Table 18. Recipe for making 1x Low TE buffer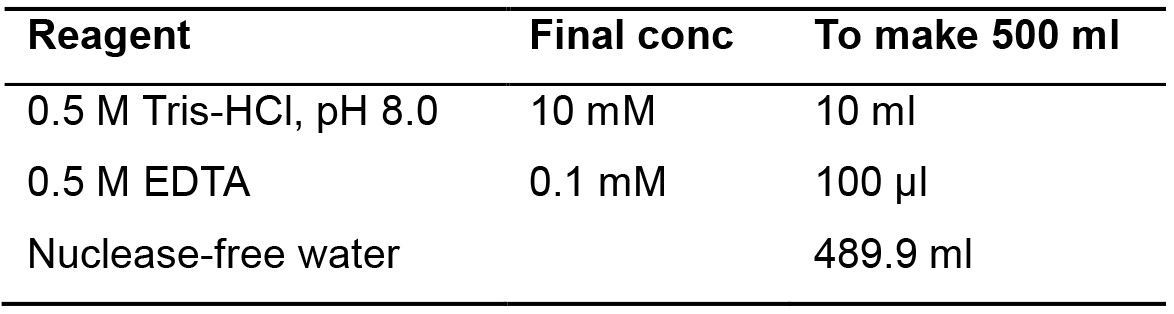
Acknowledgments
Founding sources: CARIPARO Foundation (FIBRE-GEXP) and the University of Padova (CPDA139317) to S.C. and G.L.; the Cariplo Foundation (2016-1006) to S.C. and G.L.; the Italian Ministry of Health (GR-2011-02346845) to S.C.; the AIRC Foundation (Italy project IG2015-ID17773) to G.L.; Agenzia Nazionale di Valutazione del Sistema Universitario e della Ricerca (ANVUR) [FFABR- 2017 to S.C.]; Bando Grandi Attrezzature 2015 University of Padova (Italy) (Prot. 5086).
This protocol was modified from our previous works by Alessio et al., 2019 and Chemello et al., 2019. We thank all authors of the previous works.
Competing interests
The authors declare no competing interests.
Ethics
All aspects of animal care and experimentation were performed in accordance with the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (NIH Publication No. 85-23, Revised 1996) and the Italian regulations (DL 116/92) concerning the maintenance and use of laboratory animals. Experimental procedures were approved by the he Italian Ministry of Health (authorization N. 305/2017-PR), and Ethical Committee of the University of Padova. All efforts were made to minimize animal suffering.
References
- Alessio, E., Buson, L., Chemello, F., Peggion, C., Grespi, F., Martini, P., Massimino, M. L., Pacchioni, B., Millino, C., Romualdi, C., Bertoli, A., Scorrano, L., Lanfranchi, G. and Cagnin, S. (2019). Single cell analysis reveals the involvement of the long non-coding RNA Pvt1 in the modulation of muscle atrophy and mitochondrial network. Nucleic Acids Res 47(4): 1653-1670.
- Augusto, V., Padovani, C.R., and Campos, G.E.R. (2004). Skeletal muscle fiber types in C57BL6J mice. Braz J Morphol Sci 21(2): 89-94.
- Biga, L. M., Dawson, S., Harwell, A., Hopkins, R., Kaufmann, J., LeMaster, M., Matern, P., Morrison-Graham, K., Quick, D. and Runyeon, J. (2019). Anatomy & Physiology, Open Oregon State, Oregon State University.gaiw
- Cagnin, S., Chemello, F., Alessio, E. and Lanfranchi, G. (2019). Single-cell transcriptomics and proteomics of skeletal muscle. Technology and Applications, Elsevier.
- Campbell, W. G., Gordon, S. E., Carlson, C. J., Pattison, J. S., Hamilton, M. T. and Booth, F. W. (2001). Differential global gene expression in red and white skeletal muscle. Am J Physiol Cell Physiol 280(4): C763-768.
- Celegato, B., Capitanio, D., Pescatori, M., Romualdi, C., Pacchioni, B., Cagnin, S., Vigano, A., Colantoni, L., Begum, S., Ricci, E., Wait, R., Lanfranchi, G. and Gelfi, C. (2006). Parallel protein and transcript profiles of FSHD patient muscles correlate to the D4Z4 arrangement and reveal a common impairment of slow to fast fibre differentiation and a general deregulation of MyoD-dependent genes. Proteomics 6(19): 5303-5321.
- Chemello, F., Bean, C., Cancellara, P., Laveder, P., Reggiani, C. and Lanfranchi, G. (2011). Microgenomic analysis in skeletal muscle: expression signatures of individual fast and slow myofibers. PLoS One 6(2): e16807.
- Chemello, F., Grespi, F., Zulian, A., Cancellara, P., Hebert-Chatelain, E., Martini, P., Bean, C., Alessio, E., Buson, L., Bazzega, M., Armani, A., Sandri, M., Ferrazza, R., Laveder, P., Guella, G., Reggiani, C., Romualdi, C., Bernardi, P., Scorrano, L., Cagnin, S. and Lanfranchi, G. (2019). Transcriptomic analysis of single isolated myofibers identifies miR-27a-3p and miR-142-3p as regulators of metabolism in skeletal muscle. Cell Rep 26(13): 3784-3797 e3788.
- Chemello, F., Mammucari, C., Gherardi, G., Rizzuto, R., Lanfranchi, G. and Cagnin, S. (2015). Gene expression changes of single skeletal muscle fibers in response to modulation of the mitochondrial calcium uniporter (MCU). Genom Data 5: 64-67.
- Chen, G., Ning, B. and Shi, T. (2019). Single-cell RNA-Seq technologies and related computational data analysis. Front Genet 10: 317.
- Cole, M. B., Risso, D., Wagner, A., DeTomaso, D., Ngai, J., Purdom, E., Dudoit, S. and Yosef, N. (2019). Performance assessment and selection of normalization procedures for Single-Cell RNA-Seq. Cell Syst 8(4): 315-328 e318.
- Fang, L., Wang, H. and Li, P. (2018). Systematic analysis reveals a lncRNA-mRNA co-expression network associated with platinum resistance in high-grade serous ovarian cancer. Invest New Drugs 36(2): 187-194.
- Friedlander, M. R., Chen, W., Adamidi, C., Maaskola, J., Einspanier, R., Knespel, S. and Rajewsky, N. (2008). Discovering microRNAs from deep sequencing data using miRDeep. Nat Biotechnol 26(4): 407-415.
- Fu, Y., Wu, P. H., Beane, T., Zamore, P. D. and Weng, Z. (2018). Elimination of PCR duplicates in RNA-seq and small RNA-seq using unique molecular identifiers. BMC Genomics 19(1): 531.
- Guo, X., Chang, Q., Pei, H., Sun, X., Qian, X., Tian, C. and Lin, H. (2017). Long non-coding RNA-mRNA correlation analysis reveals the potential role of HOTAIR in pathogenesis of sporadic thoracic aortic aneurysm. Eur J Vasc Endovasc Surg 54(3): 303-314.
- He, P., Wei, P., Chen, X., Lin, Y. and Peng, J. (2019). Identification and characterization of microRNAs in the gonad of Trachinotus ovatus using Solexa sequencing. Comp Biochem Physiol Part D Genomics Proteomics 30: 312-320.
- Huang da, W., Sherman, B. T. and Lempicki, R. A. (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4(1): 44-57.
- Krishna, S., Nair, A., Cheedipudi, S., Poduval, D., Dhawan, J., Palakodeti, D. and Ghanekar, Y. (2013). Deep sequencing reveals unique small RNA repertoire that is regulated during head regeneration in Hydra magnipapillata. Nucleic Acids Res 41(1): 599-616.
- Mammucari, C., Gherardi, G., Zamparo, I., Raffaello, A., Boncompagni, S., Chemello, F., Cagnin, S., Braga, A., Zanin, S., Pallafacchina, G., Zentilin, L., Sandri, M., De Stefani, D., Protasi, F., Lanfranchi, G. and Rizzuto, R. (2015). The mitochondrial calcium uniporter controls skeletal muscle trophism in vivo. Cell Rep 10(8): 1269-1279.
- Risso, D., Perraudeau, F., Gribkova, S., Dudoit, S. and Vert, J. P. (2018a). A general and flexible method for signal extraction from single-cell RNA-seq data. Nat Commun 9(1): 284.
- Risso, D., Purvis, L., Fletcher, R. B., Das, D., Ngai, J., Dudoit, S. and Purdom, E. (2018b). clusterExperiment and RSEC: A Bioconductor package and framework for clustering of single-cell and other large gene expression datasets. PLoS Comput Biol 14(9): e1006378.
- Saeed, A. I., Sharov, V., White, J., Li, J., Liang, W., Bhagabati, N., Braisted, J., Klapa, M., Currier, T., Thiagarajan, M., Sturn, A., Snuffin, M., Rezantsev, A., Popov, D., Ryltsov, A., Kostukovich, E., Borisovsky, I., Liu, Z., Vinsavich, A., Trush, V. and Quackenbush, J. (2003). TM4: a free, open-source system for microarray data management and analysis. Biotechniques 34(2): 374-378.
- Sales, G., Calura, E., Martini, P. and Romualdi, C. (2013). Graphite Web: Web tool for gene set analysis exploiting pathway topology. Nucleic Acids Res 41(Web Server issue): W89-97.
- Schiaffino, S. and Reggiani, C. (2011). Fiber types in mammalian skeletal muscles. Physiol Rev 91(4): 1447-1531.
- Tusher, V. G., Tibshirani, R. and Chu, G. (2001). Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A 98(9): 5116-5121.
- Wang, Y., and Pessin, J.E. (2013). Mechanisms for fiber-type specificity of skeletal muscle atrophy. Curr Opin Clin Nutr Metab Care 16, 243-250.
- Wellenreuther, R., Schupp, I., Poustka, A., Wiemann, S., and German c, D.N.A.C. (2004). SMART amplification combined with cDNA size fractionation in order to obtain large full-length clones. BMC Genomics 5, 36.
- Wu, H., Gallardo, T., Olson, E.N., Williams, R.S., and Shohet, R.V. (2003). Transcriptional analysis of mouse skeletal myofiber diversity and adaptation to endurance exercise. J Muscle Res Cell Motil 24, 587-592.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Chemello, F., Alessio, E., Buson, L., Pacchioni, B., Millino, C., Lanfranchi, G. and Cagnin, S. (2019). Isolation and Transcriptomic Profiling of Single Myofibers from Mice. Bio-protocol 9(19): e3378. DOI: 10.21769/BioProtoc.3378.
Category
Molecular Biology > RNA > Transcription
Molecular Biology > RNA > miRNA-mRNA interaction
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.