- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Conjugation of Fab’ Fragments with Fluorescent Dyes for Single-molecule Tracking on Live Cells

Published: Vol 9, Iss 18, Sep 20, 2019 DOI: 10.21769/BioProtoc.3375 Views: 8213
Reviewed by: Imre GáspárShalini Low-NamTrinadh Venkata Satish TammanaWoojong Lee
Original research article
The authors used this protocol in:

Sep 2016
Advertisement





Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics








Abstract
Our understanding of the regulation and functions of cell-surface proteins has progressed rapidly with the advent of advanced optical imaging techniques. In particular, single-molecule tracking (SMT) using bright fluorophores conjugated to antibodies and wide-field microscopy methods such as total internal reflection fluorescence microscopy have become valuable tools to discern how endogenous proteins control cell biology. Yet, some technical challenges remain; in SMT, these revolve around the characteristics of the labeling reagent. A good reagent should have neutrality (in terms of not affecting the target protein’s functions), tagging specificity, and a bright fluorescence signal. In addition, a long shelf-life is desirable due to the time and monetary costs associated with reagent preparation. Semiconductor-based quantum dots (Qdots) or Janelia Fluor (JF) dyes are bright and photostable, and are thus excellent candidates for SMT tagging. Neutral, high-affinity antibodies can selectively bind to target proteins. However, the bivalency of antibodies can cause simultaneous binding to two proteins, and this bridging effect can alter protein functions and behaviors. Bivalency can be avoided using monovalent Fab fragments generated by enzymatic digestion of neutral antibodies. However, conjugation of a Fab with a dye using the chemical cross-linking agent SMCC (succinimidyl 4-(N-maleimidomethyl)cyclohexane-1-carboxylate) requires reduction of the interchain disulfide bond within the Fab fragment, which can decrease the structural stability of the Fab and weaken its antigen-binding capability. To overcome this problem, we perform limited reduction of F(ab’)2 to generate Fab’ fragments using a weak reducer, cysteamine, which yields free sulfhydryl groups in the hinge region, while the interchain disulfide bond in Fab’ is intact. Here, we describe a method that generates Fab’ with high yield from two isoforms of IgG and conjugates the Fab’ fragments with Qdots. This conjugation scheme can be applied easily to other types of dyes with similar chemical characteristics.
Keywords: Single-molecule trackingBackground
Cell-surface protein functions are tightly regulated in their native environment. Gaining a comprehensive understanding of their functions necessitates monitoring their interactions with various cell membrane components, such as other proteins and lipids, and cytoskeletal machinery and cellular organelles below the membranes. Optical tools enabling live-cell based imaging at a molecular level (Joo et al., 2008; Lord et al., 2010; Chung, 2017) include conventional methods such as confocal microscopy and total internal reflection fluorescence microscopy (TIRFM) combined with specific modalities such as Förster resonance energy transfer (FRET) (Sekar and Periasamy, 2003), single-molecule tracking (SMT) (Moerner, 2012), and fluorescence correlation spectroscopy (FCS) (Kim et al., 2007), and cutting-edge super-resolution microscopy, such as photoactivated localization microscopy (PALM) (Betzig et al., 2006), stochastic optical reconstruction microscopy (STORM) (Rust et al., 2006), stimulated emission depletion (STED) microscopy (Hein et al., 2008), structured illumination microscopy (SIM) (Gustafsson, 2000 and Gustafsson et al., 2009), and lattice light sheet (LLS) microscopy (Chen et al., 2014). Using these tools for live-cell imaging to monitor endogenous proteins requires bright fluorophore-coupled reagents that specifically bind to target proteins. To this end, bright dyes such as semiconductor quantum dots (Qdots) (Dahan et al., 2003; Chung and Bawendi, 2004; Lidke et al., 2004; Chung et al., 2010; Bien-Ly et al., 2014; Chung and Mellman, 2015; Chung et al., 2016) and Janelia Fluor (JF) dyes (Grimm et al., 2015 and 2017) conjugated to high-affinity and non-perturbing antibody-based reagents are widely used. However, an antibody can bind to two target proteins simultaneously. This problem is typically circumvented by digesting antibodies to Fab fragments using proteolytic enzymes such as papain, which cleaves at the hinge region of immunoglobulins (Chung et al., 2010; Bien-Ly et al., 2014; Chung and Mellman, 2015; Chung et al., 2016). Conjugation of Fab with fluorescent dyes relies on a thiol-maleimide reaction. This reaction, however, can destabilize Fab when the interchain disulfide bond within a Fab is reduced, which elicits loss of binding capability to target proteins within a relatively short period of time (a few weeks at best). Consequently, laboratories must frequently regenerate the conjugates, imposing higher cost and hours lost. Thus, we use Fab’ fragments containing free sulfhydryl groups in the hinge region (Selis et al., 2016), which can be used for a succinimidyl 4-(N-maleimidomethyl) cyclohexane-1-carboxylate (SMCC)-based conjugation reaction without reducing the interchain disulfide bond within Fab’. To this end, we perform two-step reactions, in which IgG is digested into F(ab’)2 by pepsin and Fab’ is generated by limited reduction of F(ab’)2 using cysteamine. In this protocol, we showcase a method that generates Fab’ fragments from two different types of antibodies and subsequently conjugates one type of Fab’ with quantum dots (Qdots) to monitor EGFR on live cells using SMT. We believe this conjugation scheme will most likely improve the overall yield and stability of the tagging reagents for various types of live-cell imaging of endogenous proteins.
Materials and Reagents
- Pipette tips (Olympus, catalog numbers: 24-120RL, 24-150RL, 24-165RL)
- Sterile pipette tips (Olympus, catalog numbers: 24-401, 24-404, 24-412, 24-430)
- Sterile serological pipets (Olympus, catalog numbers: 12-102, 12-104)
- 15 ml centrifuge tubes (Olympus, catalog number: 28-101)
- 50 ml centrifuge tubes (Fisher Scientific, catalog number: 14-955-239)
- 1.5 ml microcentrifuge tubes (Olympus, catalog number: 24-281)
- 1.5 ml Protein LoBind Tubes (Eppendorf, catalog number: 022431081)
- 15 ml Protein LoBind Conical Tubes (Eppendorf, catalog number: 0030122216)
- Adjustable-volume pipettes (Eppendorf, catalog number: 2231300008)
- Pierce disposable columns (Thermo Scientific, catalog number: 29920)
- NAP-5 desalting columns (GE Healthcare, catalog number: 17-0853-01)
- µ-Dish 35 mm, high glass bottom (Ibidi, catalog number: 81158)
- Treated cell culture flasks (Thermo Scientific, catalog number: 12-556-010)
- Pierce Protein Concentrators PES, 30K MWCO, 0.5 ml (Thermo Scientific, catalog number: 88502)
- Pepsin (Sigma Aldrich, catalog number: P6887-250MG)
- Mouse IgG1 Isotype Control (Invitrogen, catalog number: 02-6100); αEGFR (rat IgG2a) antibody (Abcam, catalog number: ab231)
- MDA-MB-468 breast cancer cell line (ATCC, catalog number: HTB-132)
- BCA protein assay kit (Thermo Scientific, catalog number: 23225)
- Superdex G200 (GE Healthcare, catalog number: 17-1043-01)
- Sulfo-SMCC [sulfosuccinimidyl 4-(N-maleimidomethyl) cyclohexane-1-carboxylate], No-Weigh Format (Thermo Scientific, catalog number: A39268)
- DMSO (Dimethylsulfoxide) (Thermo Scientific, catalog number: 20684)
- Qdot 605 ITK Amino (PEG) Quantum Dots (amino-PEG-Qdot605, Invitrogen, catalog number: Q21501MP) or Qdot 565 ITK Amino (PEG) Quantum Dots (amino-PEG-QDot565, Invitrogen, catalog number: Q21531MP)
- HEPES, 1 M solution, pH 7.3, molecular biology grade, ultrapure (Thermo Scientific, catalog number: J16924AE)
- Sodium chloride, 5 M (Lonza, catalog number: 51202)
- Cysteamine (Sigma-Aldrich, catalog number: M9768-5G)
- Dye labeled marker, CAL Fluor Red 610 T10 (LGC Biosearch Technologies, catalog number: RD-5082-5)
- Glycerol (Thermo Scientific, catalog number: J16374AP)
- Dulbecco's PBS (GenClone, catalog number: 25-508)
- Trypsin EDTA (Corning, catalog number: 25-052-CV)
- Pierce F(ab’)2 Preparation Kit (Thermo Scientific, catalog number: 44988, see Note 1), include:
- Zeba Spin Desalting Columns (Thermo Scientific, catalog number: 89889)
- NAb Protein A Plus Spin Columns (Thermo Scientific, catalog number: 89956)
- PBS (Thermo Scientific, catalog number: 1890535)
- Acetate buffer, pH 4.0 (Fisher Scientific, catalog number: 50-255-309)
- EDTA (Fisher Scientific, catalog number: 03-500-506)
- Cell growth medium (RPMI 1640 with 10% fetal bovine serum and 1% penicillin-streptomycin)
- RPMI 1640, with L-Glutamine, 2000 mg/L D-Glucose (GenClone, catalog number: 25-506)
- Fetal bovine serum (FBS), heat-inactivated, U.S. Origin (GenClone, catalog number: 25-514H)
- Penicillin-streptomycin mixture (Lonza, catalog number: 17-602E)
- Acetate digestion buffer, pH 4.0 (see Recipes)
- Exchange buffer, pH 7.2 (see Recipes)
Equipment
- Eppendorf easypet 3 (Eppendorf, catalog number: 4430000018)
- pH meter (Fisherbrand, Accumet, model: 15)
- Vortex mixer (VWR, model: Analog Vortex Mixer)
- Centrifuge (Eppendorf, model: 5810R)
- Thermo mixer (Thermo Scientific, model: 13687720)
- End-over-end mixer (Argos Technologies, RotoFlex, model: R2000)
- CO2 incubator air jacket TC (VWR, catalog number: 10810-902)
- Biosafety cabinet (LabConco, model: A2)
- Nikon Eclipse TE2000 inverted microscope with TIRF illuminator and a 100x/1.49NA Plan Apo objective (Nikon, model: Eclipse TE2000-E)
- iXon back-illuminated EMCCD camera (Andor Technology, catalog number: DU-888E-C00-#BV-500)
- 488 nm line of solid-state lasers (Andor Technology)
Software
- ImageJ 1.52i with Java 1.8.0_172
Procedure
This procedure describes generation of Fab’ fragments from IgG and their subsequent conjugation to fluorescent dyes (here, we use Qdot), as summarized in Figure 1. We optimized the digestion and reduction schemes using a mouse IgG1, and then applied these procedures (see Note 8) to further optimize the conditions for conjugating Fab’ fragments of an αEGFR antibody (rat IgG2a) with Qdots to perform SMT.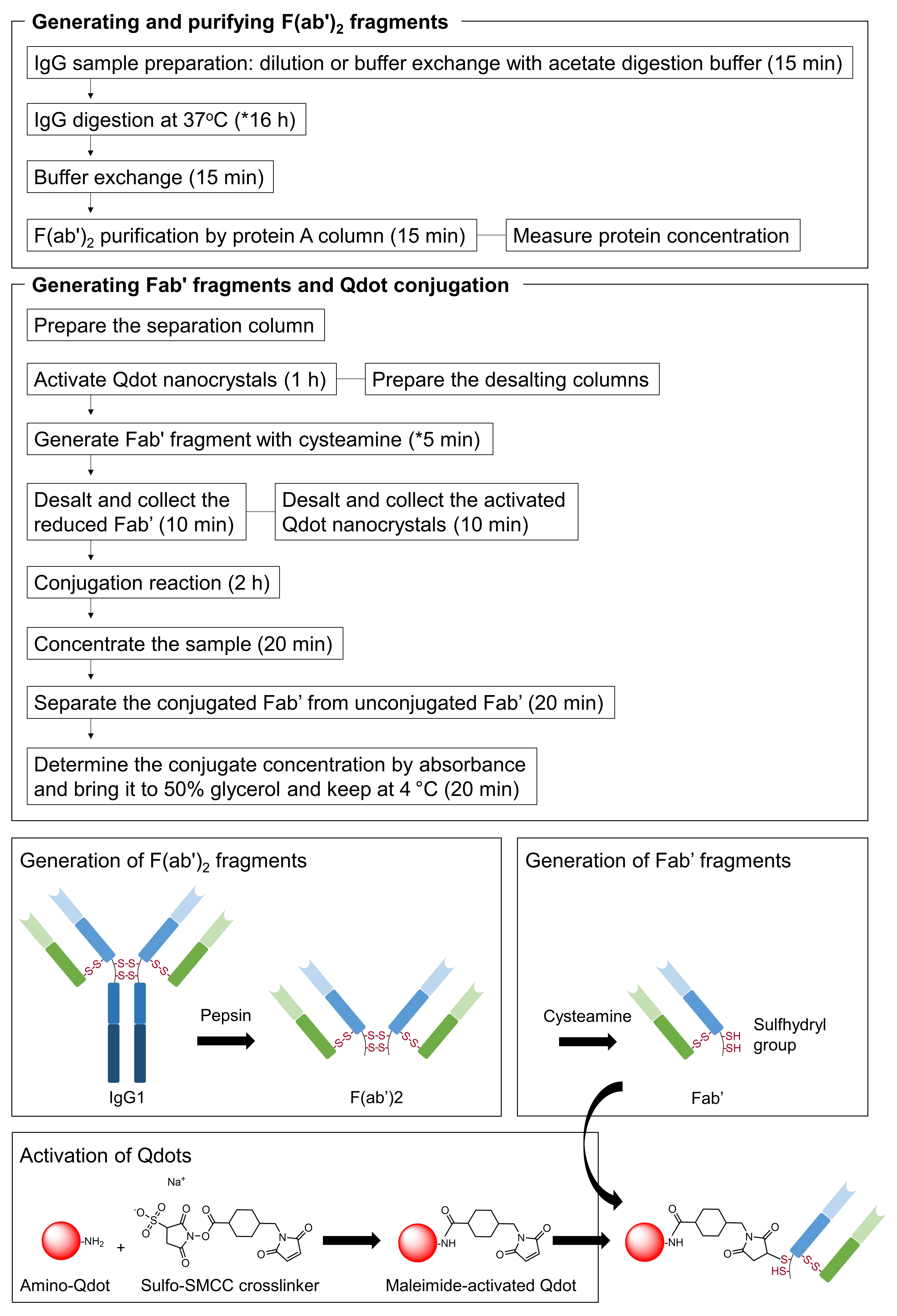
Figure 1. Overview of the procedure with cartoon schematic. The reaction conditions may vary between different antibodies (see Notes 5 and 11).
- Preparation of acetate digestion buffer and pepsin solution
- Prepare acetate digestion buffer (Recipe 1).
- Prepare a 10 mg/ml pepsin solution in acetate digestion buffer (see Notes 2 and 3).
- IgG sample preparation (see Note 4)
- Twist off the bottom closure of a Zeba Spin Desalting Column and loosen cap. Place the column in a 15 ml centrifuge tube.
- Centrifuge the column at 1,000 x g for 2 min to remove storage solution. Discard the flow-through.
- Add 1 ml of acetate digestion buffer to the column. Centrifuge at 1,000 x g for 2 min and remove the flow-through. Repeat this step three times.
- Place the equilibrated column in a 15 ml Protein LoBind Conical Tube. Remove the cap and slowly add 0.5 ml of antibody sample to the center of the resin bed in the column. Be careful not to disturb the resin bed.
- Replace the cap and centrifuge at 1,000 x g for 2 min. Collect the IgG sample. Figure 2 shows that the total IgG amount was slightly decreased (Lane 2) relative to the original amount (Lane 1) from this buffer exchange step.
- Pepsin digestion of IgG into F(ab’)2
- Transfer 0.5 ml of the prepared IgG sample to a 1.5 ml Protein LoBind Conical Tube.
- For 1 mg/ml IgG, add 2.5 μl of 10 mg/ml pepsin solution to the tube. Antibody/pepsin w/w ratio is 20:1.
- Let the digestion reaction proceed for 16 h on a thermo mixer at 37 °C with agitation at 1,000 rpm. This condition can vary for different IgG molecules (see Note 5). The band of ~100 kDa in lane 3 in Figure 2 shows F(ab’)2.
- Termination of the digestion reaction (see Note 6)
- Twist off the bottom closure of a Zeba Spin Desalting Column and loosen the cap. Place the column in a 15 ml centrifuge tube.
- Centrifuge the column at 1,000 x g for 2 min to remove storage solution.
- Add 1 ml of PBS (pH 7.4) to the column. Centrifuge at 1,000 x g for 2 min and discard the flow-through. Repeat this step three times.
- Place the equilibrated column in a 15 ml Protein LoBind Conical Tube. Remove the cap and slowly apply 0.5 ml of the digested IgG sample to the center of the resin bed. Be careful not to disturb the resin bed. Pepsin digestion of IgG will be terminated at this step.
- Replace the cap and centrifuge at 1,000 x g for 2 min to collect the flow-through. See Lane 4 in Figure 2.
- Purification of F(ab’)2 sample
- Allow the NAb Protein A Plus Spin Column and PBS to come to room temperature. Set the centrifuge speed to 1,000 x g.
- Loosen the top cap on the NAb Protein A Plus Spin Column and snap off the bottom closure. Place the column in a 15 ml centrifuge tube and centrifuge for 1 min to remove storage solution.
- Disperse the resin by adding 2 ml of PBS. For a 0.5 ml sample, use half of the resin.
- Equilibrate the resin in the column with 2 ml of PBS. Centrifuge for 1 min and discard the flow-through. Repeat this step once.
- Cap the bottom of the column with the included rubber cap. Apply the digestion mixture to the column and cap the top tightly. Resuspend the resin and sample by inversion. Incubate at room temperature on an end-over-end mixer for 15 min.
- Loosen the top cap and remove the bottom cap. Place the column in a 15 ml Protein LoBind Conical Tube and centrifuge for 1 min. Save the flow-through (this fraction contains F(ab’)2). See Lane 5 in Figure 2.
- For optimal recovery, wash the column with 0.5 ml PBS. Centrifuge for 1 min and collect the flow-through. Repeat and collect two wash fractions containing additional F(ab’)2 (see Lanes 6 and 7 in Figure 2 and Notes 7 and 8).
- Measure protein concentration using BCA protein assay.
- Prepare the BCA reagents and a serial dilution of the protein standard following the manufacturer’s instruction. Mix 200 μl of the BCA reagents with 10 μl of either the standards or the samples collected above.
- After incubation at 37 °C for 30 min, the color of the mixture will turn from pale green to purple in response to the protein concentration. See Figure 3.
- Measure the absorbance at 562 nm with a spectrophotometer or a plate reader to estimate the protein concentration by comparison to the standard curve.
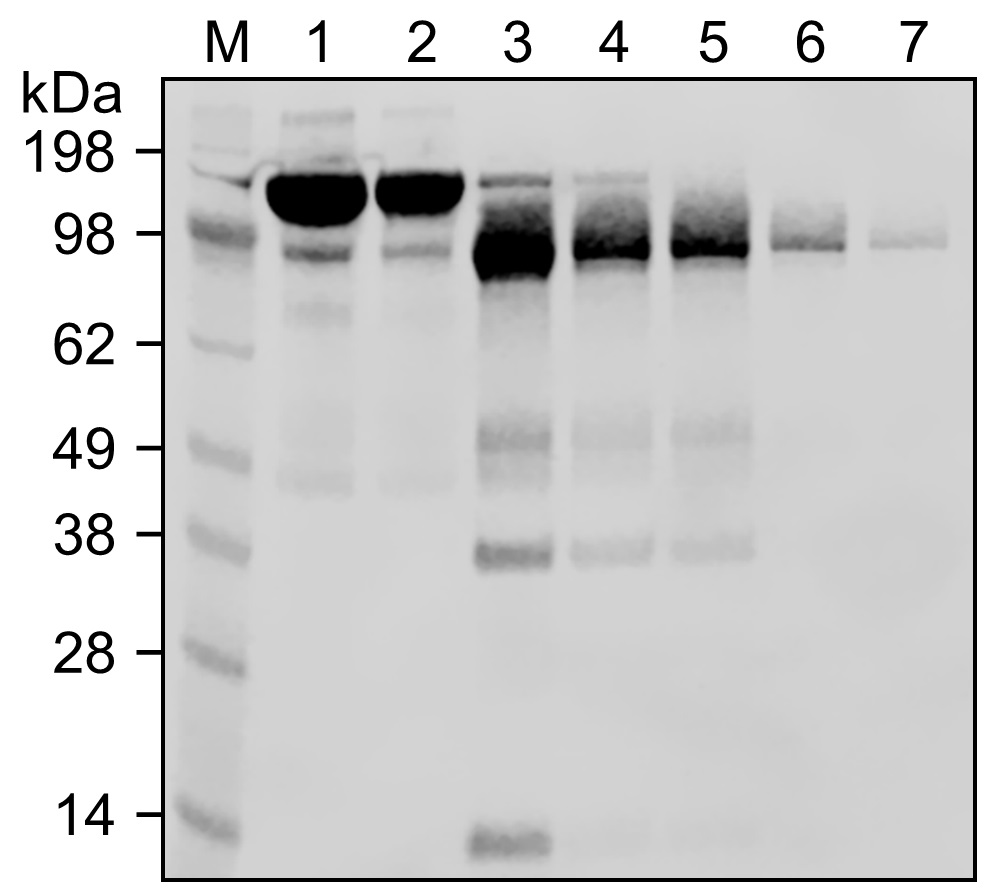
Figure 2. SDS-PAGE analysis of samples from each step before and after pepsin digestion of mouse IgG1. Lane 1: The whole IgG1 sample that was originally stored in PBS (pH 7.4). Lane 2: The IgG1 sample after a buffer exchange to bring it to the acetate digestion buffer (pH 4.0). Lane 3: The sample resulting from a 16 h digestion on a thermo shaker at 37 °C (a strong band at ~100 kDa position indicates F(ab’)2). Lane 4: The sample after a buffer exchange to bring it to PBS (pH 7.4) to terminate the reaction. Lane 5: The flow-through from a Protein A column after a 15-min incubation at room temperature. Lanes 6 and 7: Two consecutive portions collected from washing the protein A column to maximize the output.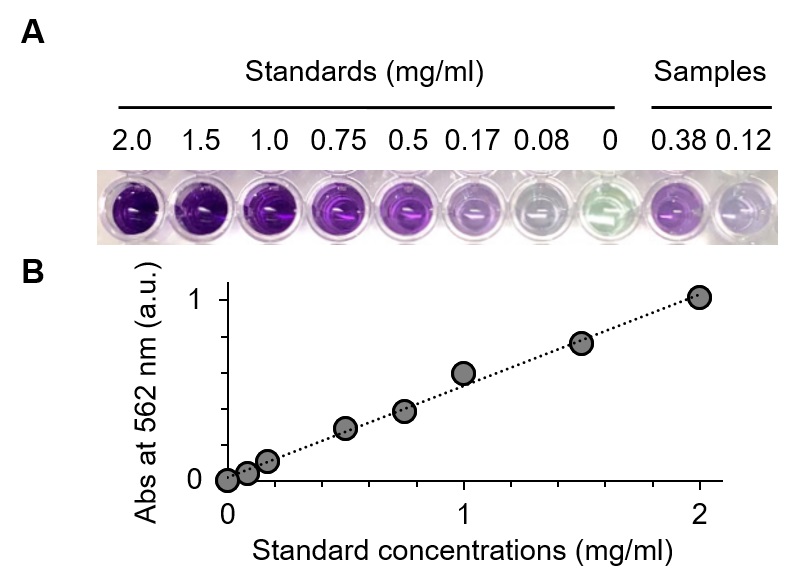
Figure 3. BCA assay for determining the protein concentrations after purification. A. The degree of green-to-purple color conversion of BCA solutions is proportional to the protein concentration. B. A standard curve of absorbance (at 562 nm) versus known protein concentrations (y = 0.507x + 0.0188) is used to determine the concentrations of F(ab')2 samples collected from the Protein A column flow-through.
- Preparation of the separation column for Fab’-Qdot conjugate
Note: This segment is a modification of the Qdot® Antibody Conjugation protocol by Invitrogen.- Suspend the separation medium (Superdex G200) in the bottle by gentle shaking or vortexing. Ensure the medium is fully suspended before starting column preparation with Pierce disposable columns.
- Mark the column with two lines, one at 45 mm above the top of the frit, and a second at 55 mm above the frit (Figure 4A).
- Wet the frit with pure water before loading the media. After ensuring that the separation medium is a uniform suspension, load media into the column with a 1 ml pipette to the second line at the 55 mm mark (Figure 4B) and let it settle into a packed gel bed that is ~45 mm high (Figure 4C).
- Gently add 0.5 ml distilled water to the top of the gel while maintaining a level bed surface.
- As the solvent level drops to near the top of the settled gel bed, fill the column with PBS, and allow the PBS level to drip down to just above the top of the gel bed. Repeat this two more times.
- Replace the bottom and the top caps when the PBS level from the last fill drops to 2 to 3 mm above the top of the settled gel bed.

Figure 4. Preparation of the separation column. A. The image shows two marks (45 and 55 mm) above the frit. B. The gel suspension fills the column to the upper mark (55 mm). C. The gel settles to the lower mark (45 mm).
- Activation of Qdot nanocrystals (see Note 9)
- Prepare a freshly dissolved 20 mM solution of sulfo-SMCC in DMSO. To do this, dissolve 2 mg Pierce No-Weigh Sulfo-SMCC in 229 μl of DMSO.
- Add 1.75 μl of 20 mM solution of sulfo-SMCC in DMSO to 62.5 μl of an 8 μM stock solution of amino-PEG-QDot605. Vortex briefly.
- Incubate for 1 h at room temperature with agitation at 500 rpm to activate the Qdots. Avoid light.
- Prepare desalting columns while the activation step is proceeding.
- Equilibration of desalting columns
- Prepare exchange buffer (see Recipe 2).
- Label the NAP-5 desalting columns. Mark one with “reduced Fab’” and the other with “activated Qdot”.
- Remove the top and bottom caps from both columns and allow the storage liquid in the columns to drain. Just as the liquid in each column is approaching the top of the column gel bed, begin adding exchange buffer.
- Equilibrate each column with 10 ml of exchange buffer.
- While there is still exchange buffer visible above the gel bed in each column, cap the bottom of each column and set that aside until the F(ab’)2 reduction and activation of Qdot are completed.
- Generation of Fab’ fragments
- Prepare 100 μg of F(ab’)2 in 300 μl of PBS by dilution or concentration, as necessary.
- When the Qdot activation is almost finished, incubate the F(ab’)2 sample with cysteamine (5 mM) and EDTA (2 mM) at 10 °C for 5 min, mixing them at 500 rpm (see Note 8 and 10). The bands in Lane 2 of Figure 5 represent Fab’ (50 kDa) and further reduced fragments (25 kDa); the latter fragments are unlikely to retain antigen binding capacity. To maximize Fab’ yield, we recommend testing various reduction conditions (see Notes 11 and 12) for different F(ab’)2 samples using small aliquots (~5-10 μl each lane) for gel analysis.
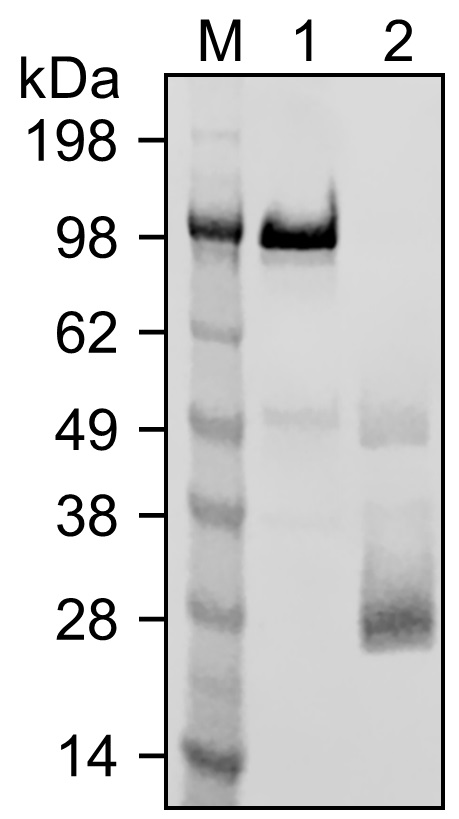
Figure 5. SDS-PAGE analysis of F(ab’)2 reduction to Fab’ with cysteamine. Lane 1: The F(ab’)2 sample from Lane 5 in Figure 2. Lane 2: The sample after a reduction with 5 mM of cysteamine for 5 min at pH 7 and 10 °C (see Note 11), before buffer exchange. The band at ~50 kDa indicates Fab’.
- Desalting and collection of the reduced Fab’ fragment
- Add 40 μl of distilled water to one vial of the dye-labeled marker and mix. This makes enough dye-labeled marker for two conjugation reactions. Store at 2-6 °C when not in use.
- Add 500 μl of water to a 1.5 ml microcentrifuge tube and mark the outside of the tube at the meniscus. Add another 500 μl of water and make a second mark on the outside of the tube corresponding to a 1,000 μl volume. Discard the water. This tube is used to collect the reduced Fab’ in Step J7 and the activated Qdot nanocrystals in Step K4.
- When the F(ab’)2 reduction is completed (Step I2), add 20 μl of dye-labeled marker (prepared in Step J1) to the reduced Fab’.
- Uncap the desalting column labeled “reduced Fab’” and allow the remaining exchange buffer to enter the gel bed. Immediately following this, add the reduced mixture (prepared in Step J3) to the top of the gel bed.
- Allow the reduced Fab’ mixture to completely enter the gel.
- Add 1 ml of exchange buffer to the top of the gel bed to elute the Fab’.
- Begin collecting reduced Fab’ into a centrifugation tube (marked in Step J2) when the first colored drop elutes (see Figure 6); collect no more than 500 μl (to the lower marked line).
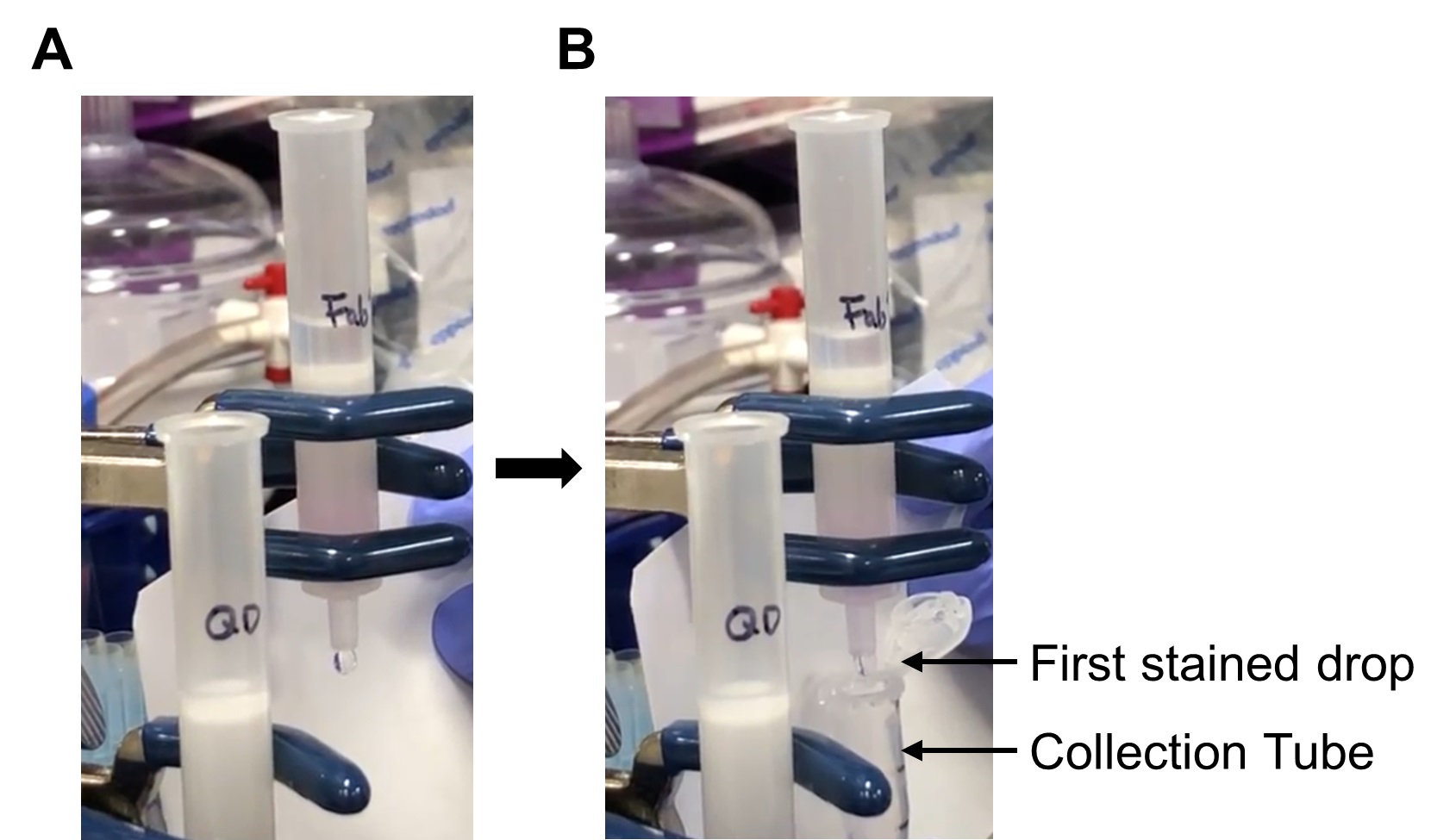
Figure 6. Collection of the reduced Fab’ fragment from the desalting column. A. One milliliter of exchange buffer was added to the desalting column after the mixture of the reduced Fab’ with the dye marker entered the column completely. B. The first stained drop is being collected in the tube marked in Step J2.
- Desalting and collecting the activated Qdot nanocrystals
- Uncap the desalting column labeled “activated Qdot”. Allow remaining exchange buffer to enter the gel bed. Immediately after this, add the activated Qdot nanocrystals (from Step G3) to the top of the gel bed.
- Allow the activated Qdot nanocrystals mixture to completely enter the gel bed.
- Add 1 ml of exchange buffer to the top of the gel bed to elute the Qdot nanocrystals.
- When the first drop of colored material elutes from the column, begin collecting directly into the centrifugation tube containing the reduced and desalted Fab’. See Figure 7.
- Stop collecting when the final volume reaches 1 ml (up to the top line marked in Step J2; 500 μl of activated Qdot nanocrystals).
- Mix briefly.
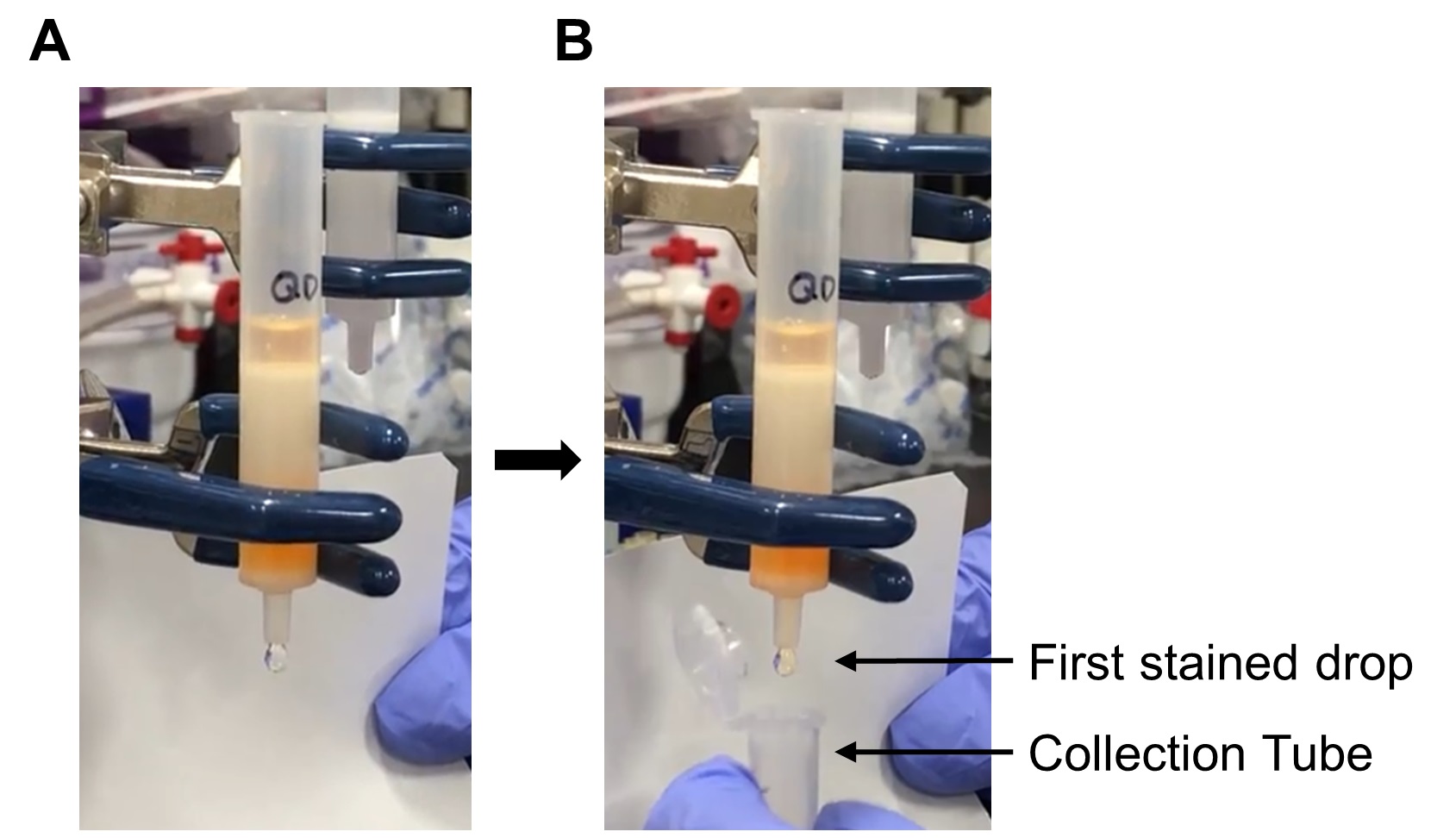
Figure 7. Collection of the activated Qdot from the desalting column. A. One milliliter of exchange buffer was added to the desalting column after the activated Qdots completely entered the gel. B. The first stained drop is being collected in a tube.
- Conjugation reaction
Allow the reduced Fab’ and activated Qdot nanocrystals to react for 2 h at room temperature. Avoid light. - Concentrating the sample
- Split the volume (from Step L1) into two protein concentrators.
- Concentrate each volume to ~20 μl by centrifuging at 4,000 x g for 15 min. If the volume is > 20 μl after the centrifugation, continue centrifuging for another 5 min.
- Separation of the Qdot-conjugated Fab’ from unconjugated Fab’
- Uncap the separation column (from Step F6) and allow the remaining PBS to enter the gel bed by gravity. Immediately following this, add the concentrated conjugate reaction solution combined from the two protein concentrators (~40 μl total volume) to the top of the column. Avoid disturbing the gel bed.
- Allow the conjugate reaction solution to enter the gel and then gently add 50 μl PBS. Let this buffer run into the gel bed.
- Gently fill the reservoir above the column with PBS and allow the sample to elute by gravity. Visually monitor the “dead space” between the frit and the column tip.
- When color appears in the “dead space,” collect only the first 8-10 drops of colored solution in a centrifugation tube. See Figure 8.
- Determine the Qdot concentration by absorbance measurements at the first absorption peak of the Qdot.
- Bring the final conjugate pool to 50% v/v of glycerol and store at 4 °C.

Figure 8. Collection of the Qdot-conjugated Fab’ from the separation column. A. The separation column is filled with PBS after the conjugation mixture completely enters the column. B. The collection of the eluate begins when the color appears in the dead space.
- Single-molecule tracking of EGFR on live cells using αEGFR Fab’-Qdot605 conjugates
- Digest αEGFR antibodies into F(ab’)2 using pepsin [20:1 antibody/pepsin (w/w ratio)] in acetate digestion buffer (pH 4.0) at 37 °C for 16 h with agitation, followed by a Protein A column purification (Figure 9A).
- Reduce the resulting F(ab')2 to Fab' with 5 mM cysteamine at 10 °C for 5 min (Figure 9B).
- Conjugate Fab’ with activated Qdot605 for generating αEGFR Fab’-Qdot605 conjugates.
- Allow 2 nM conjugates in the full growth medium to bind to target proteins (EGFR) for 10 min at room temperature on live cells (MDA-MB-468 breast cancer cell line that overexpresses EGFR) plated on glass bottom dishes. Wash three times with full growth medium (See Note 13).
- Perform single-molecule tracking (SMT) (we followed the methods described in Chung and Mellman, 2015; Chung, 2017) with light excitation at 488 nm using the TIRFM on an inverted microscope with a 100x/1.49NA Plan Apo objective. Image acquisition was done at ~11 Hz using an EMCCD camera. One snapshot image of individually labeled Qdots that was rendered using ImageJ is shown in Figure 9C.
- Estimate the binding specificity of the conjugates by determining the density of bound conjugates after saturating the epitope binding with original antibody (~100 times the Kd will suffice), relative to the density without the saturation. The binding specificity of the Fab’-QD conjugates was ~89%.
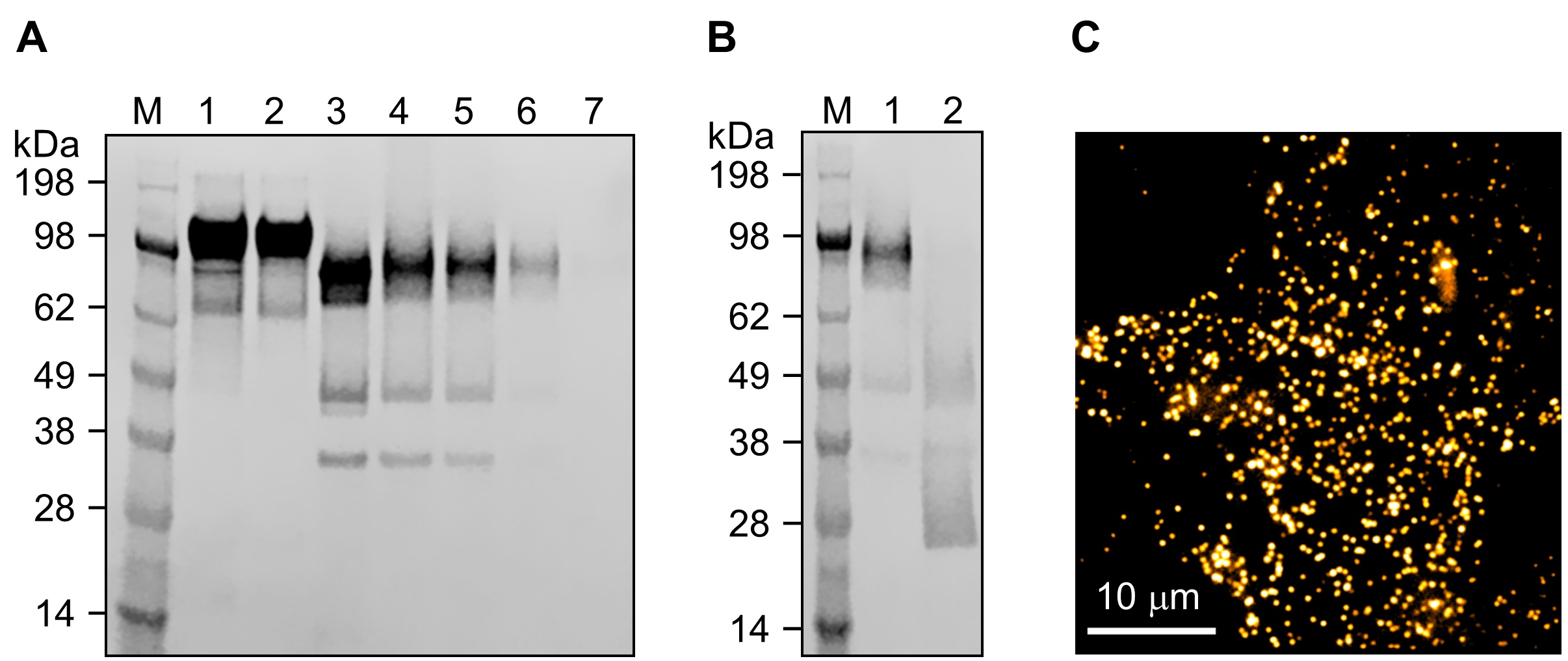
Figure 9. Generation of Fab’-Qdot conjugates for SMT using αEGFR antibodies (IgG2a). A and B. An αEGFR IgG2a was used to demonstrate the applicability of the Fab’-Qdot605 conjugates in SMT on live cells. SDS-PAGE analyses of the pepsin digestion (A) and cysteamine reduction (B) of αEGFR rat IgG2a. (B) The Fab’ throughput of the IgG from the reduction showed similar dependencies to changes of temperature, cysteamine concentration, and pH to those for the mouse IgG1 F(ab’)2 shown in Figure 5 (see Note 11). C. One snapshot (10.72 Hz) SMT image (488 nm illumination, 100x oil objective) by a total internal reflection fluorescence microscope (TIRFM) after labeling EGFR with the αEGFR Fab’-Qdot605 conjugates on ~ 60% confluent MDA-MB-468 cells.
Notes
- Procedures A-E are modified from the protocol for Pierce F(ab’)2 Preparation Kit (Thermo Scientific, catalog number: 44988). Zeba Spin Desalting Columns, PBS, and NAb Protein A Plus Spin Columns used in this protocol can be found in this kit.
- Pepsin solution should be prepared freshly for each reaction.
- Pepsin will be irreversibly denatured in higher pH buffer. Never prepare the pepsin solution in neutral buffer.
- When using concentrated IgG samples, simply dilute the samples to desired concentration in acetate digestion buffer and skip Procedure B.
- Pepsin digestion varies for different IgG molecules. A time course (1-5 and 16 h) test using small aliquots of desired IgG before the preparation of F(ab’)2 is recommended. If the digestion time is set to be 16 h as proposed in Procedure C, start this reaction at the end of a day and stop the reaction the next morning.
- The purpose of changing the acetate buffer to PBS after the pepsin digestion is to terminate the reaction and to prepare the sample for Protein A purification (for removal of undigested IgG) since the Protein A column is ineffective at acidic pH.
- Protein A column can be regenerated by following the manufacturer’s instructions using IgG elution buffer (pH 2.8).
- The digestion/reduction products can be verified using non-reducing SDS polyacrylamide gel electrophoresis as shown in Figures 2, 5, and 9, as necessary.
- This conjugation scheme can be applied directly to a maleimide linked dye, where active maleimide can react with sulfhydryl groups within the Fab’.
- Cysteamine is sensitive to air and moisture. Store the original bottle in a vacuum desiccator. Prepare cysteamine solution right before use.
- The reduction yield by cysteamine may vary depending on different F(ab’)2 samples. A pilot test on a range of pH (5, 6, and 7), temperature (4 °C, 10 °C, 22 °C, and 37 °C), and cysteamine concentration (0.5-20 mM) is highly recommended.
- HPLC or FPLC can be used to further purify the reduced sample for Fab’.
- The incubation time and concentration of the conjugates can vary depending on experiments, affinity of the Fab’, etc., We typically vary these in the ranges of 2-10 min (incubation time) and 0.5 to 10 nM (conjugate concentration).
Recipes
- Acetate digestion buffer, pH 4.0
0.1 M acetate buffer
0.01 M EDTA - Exchange buffer, pH 7.2
50 mM HEPES
150 mM NaCl
Acknowledgments
We thank the members of the Chung Laboratory for useful discussion and Jore Kotryna Vismante for photographing the procedure. This work was supported by the GW Cancer Center and Katzen Research Cancer Research Pilot Award. The procedure introduced here was modified from past single-molecule tracking studies (Chung et al., 2010; Bien-Ly et al., 2014; Chung et al., 2016).
Competing interests
The authors declare no financial or non-financial competing interests related to this work.
References
- Betzig, E., Patterson, G. H., Sougrat, R., Lindwasser, O. W., Olenych, S., Bonifacino, J. S., Davidson, M. W., Lippincott-Schwartz, J. and Hess, H. F. (2006). Imaging intracellular fluorescent proteins at nanometer resolution. Science 313(5793): 1642-1645.
- Bien-Ly, N., Yu, Y. J., Bumbaca, D., Elstrott, J., Boswell, C. A., Zhang, Y., Luk, W., Lu, Y., Dennis, M. S., Weimer, R. M., Chung, I. and Watts, R. J. (2014). Transferrin receptor (TfR) trafficking determines brain uptake of TfR antibody affinity variants. J Exp Med 211(2): 233-244.
- Chen, B. C., Legant, W. R., Wang, K., Shao, L., Milkie, D. E., Davidson, M. W., Janetopoulos, C., Wu, X. S., Hammer, J. A., 3rd, Liu, Z., English, B. P., Mimori-Kiyosue, Y., Romero, D. P., Ritter, A. T., Lippincott-Schwartz, J., Fritz-Laylin, L., Mullins, R. D., Mitchell, D. M., Bembenek, J. N., Reymann, A. C., Bohme, R., Grill, S. W., Wang, J. T., Seydoux, G., Tulu, U. S., Kiehart, D. P. and Betzig, E. (2014). Lattice light-sheet microscopy: imaging molecules to embryos at high spatiotemporal resolution. Science 346(6208): 1257998.
- Chung, I. (2017). Optical measurement of receptor tyrosine kinase oligomerization on live cells. Biochim Biophys Acta Biomembr 1859 (9 Pt A): 1436-1444.
- Chung, I., Akita, R., Vandlen, R., Toomre, D., Schlessinger, J. and Mellman, I. (2010). Spatial control of EGF receptor activation by reversible dimerization on living cells. Nature 464(7289): 783-787.
- Chung, I. and Bawendi, M. G. (2004). Relationship between single quantum-dot intermittency and fluorescence intensity decays from collections of dots. Physical Review B 70(16): 165304.
- Chung, I. and Mellman, I. (2015). Single-molecule optical methods analyzing receptor tyrosine kinase activation in living cells. Methods Mol Biol 1233: 35-44.
- Chung, I., Reichelt, M., Shao, L., Akita, R. W., Koeppen, H., Rangell, L., Schaefer, G., Mellman, I. and Sliwkowski, M. X. (2016). High cell-surface density of HER2 deforms cell membranes. Nat Commun 7: 12742.
- Dahan, M., Levi, S., Luccardini, C., Rostaing, P., Riveau, B. and Triller, A. (2003). Diffusion dynamics of glycine receptors revealed by single-quantum dot tracking. Science 302(5644): 442-445.
- Grimm, J. B., Brown, T. A., English, B. P., Lionnet, T. and Lavis, L. D. (2017). Synthesis of Janelia Fluor HaloTag and SNAP-Tag ligands and their use in cellular imaging experiments. Methods Mol Biol 1663: 179-188.
- Grimm, J. B., English, B. P., Chen, J., Slaughter, J. P., Zhang, Z., Revyakin, A., Patel, R., Macklin, J. J., Normanno, D., Singer, R. H., Lionnet, T. and Lavis, L. D. (2015). A general method to improve fluorophores for live-cell and single-molecule microscopy. Nat Methods 12(3): 244-250, 243 p following 250.
- Gustafsson, M. G. (2000). Surpassing the lateral resolution limit by a factor of two using structured illumination microscopy. J Microsc 198(Pt 2): 82-87.
- Gustafsson, M. G., Kner, P., Chhun, B. B. and Griffis E. R. (2009). Structured-illumination microscopy of living cells. Abstracts of Papers of the American Chemical Society 238.
- Hein, B., Willig, K. I. and Hell, S. W. (2008). Stimulated emission depletion (STED) nanoscopy of a fluorescent protein-labeled organelle inside a living cell. Proc Natl Acad Sci U S A 105(38): 14271-14276.
- Joo, C., Balci, H., Ishitsuka, Y., Buranachai, C. and Ha, T. (2008). Advances in single-molecule fluorescence methods for molecular biology. Annu Rev Biochem 77: 51-76.
- Kim, S. A., Heinze, K. G. and Schwille, P. (2007). Fluorescence correlation spectroscopy in living cells. Nat Methods 4(11): 963-973.
- Lidke, D. S., Nagy, P., Heintzmann, R., Arndt-Jovin, D. J., Post, J. N., Grecco, H. E., Jares-Erijman, E. A. and Jovin, T. M. (2004). Quantum dot ligands provide new insights into erbB/HER receptor-mediated signal transduction. Nat Biotechnol 22(2): 198-203.
- Lord, S. J., Lee, H. L. and Moerner, W. E. (2010). Single-molecule spectroscopy and imaging of biomolecules in living cells. Anal Chem 82(6): 2192-2203.
- Moerner, W. E. (2012). Microscopy beyond the diffraction limit using actively controlled single molecules. J Microsc 246(3): 213-220.
- Rust, M. J., Bates, M. and Zhuang, X. (2006). Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat Methods 3(10): 793-795.
- Sekar, R. B. and Periasamy, A. (2003). Fluorescence resonance energy transfer (FRET) microscopy imaging of live cell protein localizations. J Cell Biol 160(5): 629-633.
- Selis, F., Foca, G., Sandomenico, A., Marra, C., Di Mauro, C., Saccani Jotti, G., Scaramuzza, S., Politano, A., Sanna, R., Ruvo, M. and Tonon, G. (2016). Pegylated trastuzumab fragments acquire an increased in vivo stability but show a largely reduced affinity for the target antigen. Int J Mol Sci 17(4): 491.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Teng, I., Bu, X. and Chung, I. (2019). Conjugation of Fab’ Fragments with Fluorescent Dyes for Single-molecule Tracking on Live Cells. Bio-protocol 9(18): e3375. DOI: 10.21769/BioProtoc.3375.
Category
Cell Biology > Cell imaging > Live-cell imaging
Cancer Biology > Cancer biochemistry > Protein
Biochemistry > Protein > Single-molecule Activity > Imaging
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.