- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Explant Culture of the Embryonic Mouse Spinal Cord and Gene Transfer by ex vivo Electroporation


Published: Vol 9, Iss 18, Sep 20, 2019 DOI: 10.21769/BioProtoc.3373 Views: 7237
Reviewed by: Giusy TornilloJackeline Moraes MalheirosAnonymous reviewer(s)
Original research article
The authors used this protocol in:
Feb 2019

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Developing axons change responsiveness to guidance cues during the journey to synapse with target cells. Axon crossing at the ventral midline serves as a model for studying how axons accomplish such a switch in their response. Although primary neuron culture has been a versatile technique for elucidating various developmental mechanisms, many in vivo characteristics of neurons, such as long axon-extending abilities and axonal compartments, are not thoroughly preserved. In explant cultures, such properties of differentiated neurons and tissue architecture are maintained. To examine how the midline repellent Slit regulated the distribution of the Robo receptor in spinal cord commissural axons upon midline crossing and whether Robo trafficking machinery was a determinant of midline crossing, novel explant culture systems were developed. We have combined an “open-book” spinal cord explant method with that devised for flat-mount retinae. Here we present our protocol for explant culture of embryonic mouse spinal cords, which allows flexible manipulation of experimental conditions, immunostaining of extending axons and quantitative analysis of individual axons. In addition, we present a modified method that combines ex vivo electroporation and “closed-book” spinal cord explant culture. These culture systems provide new platforms for detailed analysis of axon guidance, by adapting gene knockdown, knockout and genome editing.
Keywords: Explant cultureBackground
Growing axons sense numerous extracellular cues to reach their final target cells (Stoeckli, 2018). At intermediate targets on the way to the destination, axons timely switch on/off responses to guidance cues. Molecular mechanisms underlying this switch have been a hot topic in neurobiology (Guan and Rao, 2003; Sabatier et al., 2004; Dickson and Zou, 2010; Kolodkin and Tessier-Lavigne, 2011; Stoeckli, 2018; Yang et al., 2018; Ducuing et al., 2019).
The ventral midline of the central nervous system is an important intermediate target for different types of axons. Floor plate (FP) cells at the ventral midline secrete short- and long-range guidance cues, which act as attractants or repellents, and spatiotemporally organize the formation of neural circuits. Commissural axons project across the midline, connect the left and right sides of the nervous system and play roles in information transfer between both sides (Figures 1A and 1B). Commissural axons are initially guided to the midline by attractants derived from the FP and ventricular zones, such as netrin-1 and Sonic hedgehog (Dominici et al., 2017; Varadarajan et al., 2017; Moreno-Bravo et al., 2019; Wu et al., 2019). At the midline, commissural axons lose their responsiveness to the attractants and acquire responsiveness to repellents, such as Slit and semaphorins (Shirasaki et al., 1998; Zou et al., 2000). Thus, the midline is initially an attractive target for commissural axons, but becomes an unfavorable place upon their arrival, so that commissural axons exit the midline and never re-cross it. What mechanism ensures that commissural axons cross the midline only once? One key is the molecular basis by which commissural axons increase Slit sensitivity upon midline crossing.
Here we will summarize how spinal cord explant cultures have contributed to our understanding of axon guidance. The landmark research using explants showed that FP cells produce chemoattractant activity for commissural axons (Tessier-Lavigne et al., 1988). Two-dimensional co-culture assays using explants of commissural neurons and the FP revealed the presence of unknown midline repellent activity, which is unmasked by inhibiting either axonin-1/TAG-1 (a cell adhesion molecule [CAM] expressed in commissural axons) or NrCAM (expressed in FP cells) (Stoeckli et al., 1997). When Slit was identified as a ligand for Robo receptor and a long-awaited midline repellent (Brose et al., 1999; Kidd et al., 1999; Li et al., 1999), its repellent activity for vertebrate commissural axons was not clarified. But soon later, ingenious assay systems using FP-containing and FP-lacking spinal cord explants embedded in collagen gel revealed that Slit inhibits outgrowth of commissural axons that have crossed the midline (post-crossing axons), but not pre-crossing axons (Zou et al., 2000). This was the first demonstration that Slit is an evolutionally conserved midline repellent.
To examine how Slit sensitivity of commissural axons was regulated, we established a primary culture system (Kinoshita-Kawada et al., 2019; Yuasa-Kawada et al., 2009). Commissural neurons dissociated from dorsal spinal cords of mouse embryos at post-crossing stages (embryonic day [E]11.5-12.5) exhibit growth cone collapse and the increase in axonal Robo1 levels in response to Slit. In contrast, commissural neurons harvested from pre-crossing stage embryos show neither collapse responses to Slit nor the Slit-induced increase in axonal Robo1. Therefore, even without FP cells, dissociated commissural neurons recapitulate in vivo behaviors, with respect to the acquisition of Slit responsiveness. We then sought to develop an explant assay to examine Slit-induced Robo redistribution and growth cone responses in individual axons (or small fascicles) in a more physiological context, with subcellular resolution. Unfortunately, it was difficult to use so far developed explant assays for such purposes, because axons tended to form vigorous bundles in collagen gel.
To study retinotectal topography, Drs. Friedrich Bonhoeffer, Uli Schwarz and colleagues developed retinal explant culture systems; a large mass of embryonic retinae were flat-mounted onto nitrocellulose (NC) filters and placed on the tectal cell membrane carpets, allowing robust extension of retinal axons (Halfter et al., 1981 and 1983; Bonhoeffer and Huf, 1982; Walter et al., 1987a and 1987b; Ichijo and Bonhoeffer, 1998; Yuasa-Kawada et al., 2003). By combining these protocols with spinal cord explant techniques (Zou et al., 2000), we prepared FP-lacking dorsal spinal cord [dSC (-FP)] and FP-containing hemisected spinal cord [SC (+FP)] explants (from cervical to lumbar levels) from E11.5 embryos and spread them on NC filters. Such explants were cultured on the Matrigel-coated substratum (Figures 1C-1G). Under these conditions, individual axons robustly extended from explants onto the two-dimensional surface, even without netrin-1, a stimulator of commissural axon growth. In both explant types, the extending axons were negative for TAG-1, a pre-crossing commissural axon marker but positive for L1, a post-crossing commissural axon marker (Dodd et al., 1988), maintaining the characteristics of post-crossing axons (Figure 3). These explant cultures allow simple immunostaining protocols and testing effects of Slit alone on Robo distribution (Kinoshita-Kawada et al., 2019).
Furthermore, to perform RNA interference (RNAi) against transducers of Slit-Robo signals, Arf6 GTPase and cytohesin Arf-guanine nucleotide exchange factors (Arf-GEFs), we modified an RNAi-based assay that combines ex utero electroporation with spinal cord explant culture (Wolf et al., 2008; Parra and Zou, 2010). Because the spinal cord, especially the FP region, becomes too fragile to prepare open-book explants after electroporation, we developed a new culture system of intact, “closed-book” spinal cords embedded in collagen gel (Kinoshita-Kawada et al., 2019) (Figures 2A-2D). The combination of these culture systems with RNAi-mediated knockdown or clustered regularly interspaced short palindromic repeats (CRISPR)-based genome editing, introduced by electroporation, virus or nanocomplex (e.g., Mikuni et al., 2016; Nishiyama et al., 2017; Park et al., 2019), will allow more flexibility to manipulate experimental conditions and to tag endogenous proteins. Such techniques will contribute to the elucidation of molecular mechanisms of axon guidance.
Materials and Reagents
- Animals
Pregnant ICR or C57BL/6J strain mice with embryonic day [E]~11.5 embryos
Note: We usually use 1-2 pregnant mice for each experiment. We assume an average of 6-12 embryos per ICR mouse and 4-8 embryos per C57BL/6J mouse. For timed-pregnancy mating, the day of vaginal plug was dated as E0.5, and embryos were staged according to EMAP (http://www.emouseatlas.org/emap/ema/home.php). In our hands, in terms of axon growth onto the Matrigel-coated substratum and the ease of handling, the best results have been obtained with embryos between E11.0 and E11.5, in order to prepare dSC (-FP) and SC (+FP) explants. At least, it is better to use embryos before E12.0 than later stages. For use of knockout mice, we prepare explants from littermate embryos, in parallel with genotyping. Image acquisition and analysis can be performed in a blinded manner. For ex vivo electroporation and closed-book explant culture, we recommend using ICR mouse embryos just before E11.5, with respect to the efficient introduction of axon-tracer plasmid/shRNA to commissural neurons prior to axon midline crossing and the maintenance of tissue integrity. - Surgical instruments and materials
- Scissors, large
- Scissors, medium
- Ophthalmic scissors (option) (Natsume Seisakusho, catalog number: MB-50-7)
- Forceps (7) (Dumont, No. 5 Inox)
Note: We use relatively blunt forceps (a total of 5; 1 for handling coverslips, 2 for mouse surgery and 2 for spinal cord dissection) and sharpened forceps for fine dissection (2). - Straight metal spatula, with polished edges
- Disposable scalpel (Feather, No. 11)
- Sharpened tungsten needles, bent at a right angle (tungsten wire, diameter: 0.20 mm; Nilaco, catalog number: W-461267) and needle holders (Fine Science Tools, catalog number: 26016-12) (see Figure 1H).
Note: At the tip, the diameter should be approximately 20 μm. The needles can be sharpened in a 1 N NaOH bath by putting the negative electrode of a power supply (low voltage should be set) into the bath and the positive electrode (alligator clip) onto the end of needle holder. Dip in/out the needle tip to make a pointed, tapered tip. See the Recipe “Sharpened tungsten needles” in Cold Spring Harb Protoc (doi:10.1101/pdb.rec069468). - Disposable transfer pipettes, 3.5 ml (Sarstedt, catalog number: 86.1171.001) or equivalent (approximately 15 cm in length, with an inner diameter of 2 mm)
Note: When handling whole spinal cords, use cut pipettes, which have opening of approximately 4-5 mm. When handling excised, tiny spinal cord explants, we usually use un-cut pipettes. - Acid-washed, sterilized, round coverslips (18 mm in diameter; Matsunami, thickness: No. 1; for preparing the coverslips, see Recipe 1)
- Black nitrocellulose (NC) membrane filter (gridded on one surface, single-packed and sterile) (Sartorius, Cellulose nitrate filter, catalog number: 13006--47----CAN)
- Glass slides (Matsunami, Superfrost, catalog number: S2443)
- 70% Ethanol
- Culture products
- 35-mm sterile tissue culture dishes (Corning, Falcon, catalog number: 353001)
- 60-mm sterile tissue culture dishes (Corning, Falcon, catalog number: 353004)
- 100-mm sterile tissue culture dishes (Corning, Falcon, catalog number: 353003)
- 15-ml conical centrifuge tubes (Thermo Fisher Scientific, catalog number: 14-959-70C)
- 50-ml conical centrifuge tubes (Eppendorf, catalog number: 0030122178)
- 1.5-ml safe-lock microcentrifuge tubes (Eppendorf, catalog number: 0030120086)
- Metal weights (stainless steel; made by a local machine shop)
Note: Before use, wash the weights at least three times with distilled water (washing with sonication for 5-10 min is preferable) and with 100% ethanol overnight, air-dry and sterilize them by baking at 180 °C for 3 h or by UV irradiation (when hurrying). It is convenient to have many metal weights of various sizes, although we usually use the 3 x 3 x 14 mm-size weights (see Figure 1M). After use, wash them three times with distilled water (and perform sonication) and with 100% ethanol overnight. Follow the above sterilization procedure for the next round of use. Avoid using detergent for washing and do not autoclave them. - Matrigel matrix (Corning, catalog number: 354234)
Note: Thaw a bottle of Matrigel overnight on ice. Make aliquots (0.2 ml per 1.5 ml tube) and store at -80 °C. - Concanavalin A (ConA; Type IV, lyophilized powder; Sigma-Aldrich, catalog number: C2010-25MG)
- ConA stock solution (see Recipe 2)
- Culture media
- Hanks’ balanced salt solution, no calcium, no magnesium (HCMF; Thermo Fisher Scientific, catalog number: 14170112)
- Dulbecco’s modified Eagle’s medium (DMEM), high glucose (Thermo Fisher Scientific, catalog number: 11965092)
- L-Glutamine, 200 mM solution (Thermo Fisher Scientific, catalog number: 25030081)
- Fetal bovine serum (FBS) (Thermo Fisher Scientific, Gibco, catalog number: 10270106 or Hyclone, catalog number: SH30910.03)
Note: Heat-inactivation of FBS at 56 °C for 30 min is not essential in this culture protocol. - Neurobasal medium (Thermo Fisher Scientific, catalog number: 21103049)
- B-27 supplement (50x), serum-free (Thermo Fisher Scientific, catalog number: 17504044)
- Opti-MEM I reduced serum medium (Thermo Fisher Scientific, catalog number: 31985062)
- Penicillin-streptomycin, liquid (designated Pen/Strep; used as 100x solution; 10,000 units/ml each) (Thermo Fisher Scientific, catalog number: 15140122)
- HEPES (1 M) (Thermo Fisher Scientific, catalog number: 15630080)
- Purified human recombinant Slit2 (option: used in our experiment; store aliquots at -80 °C)
Note: Recombinant Slit2 proteins were purified from conditioned media from HEK293 cells stably expressing human Slit2-myc (Li et al., 1999) by ion-exchange chromatography using SP Sepharose Fast Flow (GE Healthcare) (Guan et al., 2007). Control purified preparations, made from parental HEK293 cells by employing the same procedure as for Slit purification, were used for explant assays in parallel. Our detailed protocol for Slit purification is available upon request to the authors. - Medium 1 (see Recipe 3)
- Medium 2 (see Recipe 4)
- Medium 3 (see Recipe 5)
- Materials for ex vivo electroporation and closed-book spinal cord explant culture
- Glass capillaries (Narishige, catalog number: GD-1)
Note: To prepare injection pipettes, pull glass capillaries using a vertical puller and break the capillary tip with fine forceps; if possible, cut it diagonally (tip diameter: approximately 30 μm). - Vertical puller (Narishige, current model: PC-100)
- Mouthpiece and pipette joints (for injection before electroporation, we use an apparatus available from Suisaku, Japan (model: slim tube), to which an appropriate length of silicon tube can be attached)
- DPBS, no calcium, no magnesium (Thermo Fisher Scientific, catalog number: 14190144)
- Leibovitz's L-15 (Thermo Fisher Scientific, catalog number: 11415064)
- Horse serum, heat-inactivated (Thermo Fisher Scientific, catalog number: 26050070)
- Axon tracer (EGFP/vYFP plasmid)
Note: We have compared among pCAG-EGFP (available from Nepa Gene), pCAG-vYFP (Venus), pCAG-GAP43-EGFP and pCAG-GAP43-vYFP for axon labeling. In our study, pCAG-vYFP showed the best performance in mouse spinal cords. We usually obtain midi-prep grade DNAs by using NucleoBond Xtra Midi EF kit (Macherey-Nagel). We then clean up the DNAs by conventional phenol-chloroform extraction and ethanol precipitation, and obtain a high-purity DNA stock by dissolving in sterile, endotoxin-free TE buffer at a concentration of 8-10 μg/μl. Store at -30 °C (avoid thawing/refreezing cycles of the stock too many times). - Appropriate short-hairpin RNAs (shRNAs; in this paper, we used shRNA against Arf6 [shArf6] cloned in pRS shRNA vector; Origene) and control shRNA (shControl) for RNAi-mediated knockdown (the DNAs are purified as described above and dissolved at an appropriate concentration; see Kinoshita-Kawada et al., 2019)
- 0.5-1% Fast Green (10x) (Sigma-Aldrich, catalog number: F7252)
Note: After dissolving in DPBS, sterilize by filtration (0.22 μm). Make aliquots and store them at -30 °C. - Native collagen acidic solution I-AC (3 mg/ml, pH3.0) (Koken, catalog number: IAC-30)
- DMEM/F-12, powder (Thermo Fisher Scientific, catalog number: 12500062)
- 5x concentrated DMEM/F-12 medium (Recipe 6)
- Reconstitution buffer (used for gelatinization of collagen; see Recipe 7)
- Medium 4 (see Recipe 8)
- Glass capillaries (Narishige, catalog number: GD-1)
- Fixation
- PBS (Phosphate-buffered saline) tablets (Takara, catalog number: T900)
Note: Sterility is not required, although the above DPBS can also be used. - PBS + 0.025% NaN3
- Paraformaldehyde (PFA) (Fuji film/Wako, catalog number: 162-16065)
- 4% PFA in PBS supplemented with 10% sucrose (~0.3 M sucrose; Walter et al., 1987a; for fixing cultured explants without electroporation)
Note: Prepare a stock in a concentration of 4% PFA in PBS, make aliquots (approximately 30 ml per 50 ml tube) and store at -30 °C. For use in fixation, thaw 4% PFA in PBS and add sucrose to approximately 10 %. - 4% PFA in PBS (for fixing cultured explants of electroporated spinal cords)
- PBS (Phosphate-buffered saline) tablets (Takara, catalog number: T900)
- Immunostaining and observation
- 24 x 60 mm No. 1 Micro cover glass (Matsunami, catalog number: C024601)
Note: This coverslip is used for mounting spinal cord explants that are electroporated and cultured. - Sylgard-coated dissection dishes (for preparation, see page 275 of Banker and Goslin, 1998)
- 0.2% Triton X-100 in PBS
Note: We freshly dilute Triton X-100 (Sigma-Aldrich, catalog number: T8787) on the day of immunostaining. - Bovine serum albumin (BSA) (Sigma-Aldrich, catalog number: A2153)
- Goat serum (Sigma-Aldrich, catalog number: G6767)
- Skim milk (BD/Difco, catalog number: 232100)
- Rabbit primary antibodies of interest (in our study, we use anti-Robo antibodies)
- Anti-L1CAM (mouse monoclonal IgG1; clone 2C2; Abcam, catalog number: ab24345)
- Anti-TAG-1 (mouse monoclonal IgM; clone 4D7; Developmental Studies Hybridoma Bank)
- Alexa Fluor 647 mouse anti-β-tubulin, class III (clone TuJ1; BD, catalog number: 560394)
- Goat anti-mouse IgG (H+L) highly cross-adsorbed secondary antibody, Alexa Fluor 555 (Thermo Fisher Scientific, catalog number: A32727)
- Goat anti-mouse IgM (heavy chain) cross-adsorbed secondary antibody, Alexa Fluor 555 (Thermo Fisher Scientific, catalog number: A21426)
- Goat anti-rabbit IgG (H+L) highly cross-adsorbed secondary antibody, Alexa Fluor 488 (Thermo Fisher Scientific, catalog number: A11034)
- PermaFluor aqueous mounting medium (Thermo Fisher Scientific, catalog number: TA-030-FM)
Equipment
- CO2 tank
- CO2 incubator (ASTEC, water-jacket type)
- CO2 single-stage flowmeter regulator
- Dissecting microscope (Carl Zeiss, model: Stemi 2000-C; current models: Stemi 305/508)
- Cold light source (FOSTEC, model: DCRII)
- Phase contrast microscope (Olympus, model: CKX31; current model: CKX53)
- Millipore Milli-Q ultrapure water system
- Laminar flow hood
- 4 °C refrigerator
- -30 °C freezer
- -80 °C freezer
- Electroporator (Nepa Gene, model: NEPA21)
Note: NEPA21 is a versatile electroporator, which is highly programmable and reproducible with many variables controlled and measurable. - Forceps-type electrodes (to electroporate mouse spinal cords, we preferably use CUY665P9-6-2-5, with the tip diameter of 2.5 mm, Nepa Gene)
Note: The shape of electrode tips remarkably affects the efficiency and pattern of gene transfer into the spinal cord. - Confocal laser-scanning microscope (Carl Zeiss, model: LSM780 or LSM880) or all-in-one fluorescence microscope (Keyence, model: BZ-X700)
Note: If possible, use an appropriate confocal microscope with image-tiling functions (see Figure 3A). Although laborious, montage images can also be assembled manually. In our initial studies, we used a wide-field BX61I microscope (Olympus) equipped with a CoolSNAP HQ or ES CCD camera (Roper Industries). We manually assembled images that were processed with AutoQuant X3 deconvolution software (Media Cybermetrics; see Figure 3C).
Software
- MetaMorph (version 7.7 and 7.10; Molecular Devices)
- Photoshop CS5 Extended (Adobe)
- Illustrator CS5 (Adobe)
- Excel (Microsoft)
- Prism 6 (GraphPad)
Procedure
- Coating of coverslips and black NC filters
Notes:- Prepare in advance of experiment.
- The following steps should be performed inside the laminar flow hood.
- Dilute 200 μl of Matrigel (before use, slowly thaw a tube containing aliquoted Matrigel on ice) with 2 ml of HCMF (finally, 1:10 dilution). In our experience, the diluted solution can be stored at 4 °C for a week, but we usually use a freshly diluted stock. It is important to avoid the formation of Matrigel aggregates in the diluted solution. First, pour 2 ml of HCMF into a 15 ml tube placed on ice and wait for a while. Second, transfer approximately 900 μl of the HCMF into the 200 μl Matrigel-containing tube, immediately mix by pipetting and return the mixture to the initial 15 ml tube placed on ice. Mix well again.
- Coat sterile, round 18 mm coverslips using the above Matrigel solution. First, transfer 120 μl of the Matrigel solution onto a 35 mm dish and then cover the solutions with a coverslip (Figure 1I). We handle the coverslips with forceps (their tips should be immersed into 70% ethanol and air-dried before use). Incubate the coverslips at 37 °C in the CO2 incubator for 3-5 h (or more).
- Coat black NC filter(s) with ConA. Dilute 0.5 ml of ConA (before use, slowly thaw a tube containing aliquoted ConA on ice; 2 mg/ml) into 8 ml of HCMF in a 60-mm dish (the resultant working concentration is 0.125 mg/ml). Slowly place one side of the NC filter onto 5 ml of HCMF (without ConA) in another dish. When the whole surface of the filter becomes wet, immerse it completely and transfer the filter into the ConA-containing HCMF (Figure 1I). Incubate at 37 °C in the CO2 incubator for 3-5 h (or more).
- After coating of the coverslips, rinse them with 2 ml of plain DMEM or Medium 3 once (residual Matrigel does not affect axon outgrowth). Place the coverslips, with the coated side up, in new dishes containing 2 ml of Medium 3. Keep them in the CO2 incubator until use. Do not let them dry anytime.
- After coating of the NC filter(s), wash it five times each with 8 ml of HCMF (note again that ConA is toxic). Sterilize both sides of the NC filter, in the immersed condition in HCMF, with UV irradiation in the laminar flow hood (5-10 min/side, twice) (use of a UV-sterilizing box is preferable). Keep them in the HCMF at 37 °C in the CO2 incubator until use. Do not let it dry anytime.
Note: Too long exposure with UV can destroy ConA on the NC filter. - For explant-spreading, cut the coated black NC filer with a disposable scalpel to obtain narrow strips (the width of approximately 0.5-1 scale on the filter, 4-5 scales in length) (Figure 1J).
- Dissecting out spinal cords from mouse embryos and mounting the spinal cord explants onto the NC filter
- Preparation for dissection
Cool the dissecting medium HCMF on ice. Sterilize dissection instruments with 70% ethanol. The following procedure can be performed outside the laminar flow hood. However, to prevent contamination of explant cultures with fungi, bacteria or yeasts, use sterile solutions, sterile dissecting tools and aseptic techniques. Sterilize the dissection instruments frequently with 70% ethanol during manipulation. A caution should also be taken to exclude the inclusion of fur into the dishes, which may be a major source of contamination in the cultures.
Figure 1. Procedure for spinal cord explant culture. A. The typical axon trajectory of dorsal commissural neurons in the spinal cord. RP: roof plate; FP: floor plate; r: rostral; c: caudal. B. Dorsal view of the commissural axon trajectory. C-G. Schematic illustration of the procedure: C. Diagram of an E11.5 embryo. The spinal cord (SC) from cervical to lumbar levels is used for explant culture. D. Cutting the spinal cord at the RP. E. Cutting lines to prepare FP-lacking dorsal spinal cord [dSC (-FP)] and FP-containing spinal cord [SC (+FP)] explants. F. Spreading and nailing of spinal cord explants onto the black NC filter. d: dorsal; v: ventral. G. Mounting and culturing of spinal cord explants on the Matrigel-coated coverslip. H. Sharpened tungsten needle used in cutting the explants. I. Matrigel (MG) coating of a coverslip (left) and ConA coating of a NC filter (right). J. Strips of the ConA-coated NC filter. K. An example of peeling off the meninges from the spinal cord. i: Cutting the spinal cord at the RP to prepare an open-book preparation. ii: Peeling off the meninges from the open-book spinal cord in the rostral-to-caudal direction. iii: The meninges are now completely removed. iv: The spinal cord is ready for cutting out dSC (-FP) and SC (+FP) explants. L. An example of spreading and nailing of an explant with forceps onto the NC filter (i). The spinal cord explant fixed on the filter is ready for mounting (ii). M. Setting up and mounting two explants on the coverslip with two metal weights. - Removal of uterus
- Euthanize the pregnant mouse by CO2 inhalation, followed by cervical dislocation. Lay the mouse on its back onto a paper towel, and wipe the abdominal area thoroughly with 70% ethanol.
- Using blunt forceps, grasp the skin above the genitalia, and cut the skin and then muscles with large scissors to open the entire peritoneal cavity.
- Remove the uteri, using forceps and medium-size scissors, and transfer them into a 100 mm dish containing ice-cold HCMF. Care should be taken so that the uteri do not touch the body surface of the mouse.
- To reduce the possibility of contamination, wash uteri at least once, twice if possible, by transferring them to HCMF-containing 100 mm dish(es).
- Dissecting out embryos from the uterus
Note: The following procedure can be performed outside the laminar flow hood.- Place a dish containing the uteri under a dissecting microscope (We performed all subsequent procedures under a dissecting microscope). Using two pairs of blunt No. 5 Inox forceps, tear the uterine horn of each uterus carefully and isolate the embryos from their placentas and amniotic membranes. To avoid damages of embryos, we do not separate the uterus during isolation (keep the state of “peas in a pod”). By using a spatula, transfer the embryos into new HCMF-containing dishes. Take care not to damage embryonic bodies, especially the spinal cord. Frequently wipe forceps and other tools with 70% ethanol. These procedures, from isolating embryos to mounting explants before cultivation, can be performed outside the laminar flow hood, but keep the environment and your tools clean. For isolation of E11.5 mouse embryos from the uterus, see also Elhendawi and Davies, 2018.
- Check the pooled embryos, and discard those that appear damaged, malformed, abnormal, or too small and too large. By staging the embryos with the EMAP system (mentioned above), select appropriate embryos at E11.5 or earlier stages.
- Isolation of spinal cords
- Dissection should be performed as quickly as possible, and the dishes containing the remaining embryos should be kept on ice. Transfer 3-4 embryos per 60 mm HCMF-containing dish. First, remove almost all of the ventral organs from the embryo body by using two blunt No. 5 Inox forceps. At this step, we do not use scissors. We obtain the brain and spinal cords together with the surrounding tissues. Next, make an incision at the hindbrain-rostral spinal cord region by using blunt forceps. Dissect out the hindbrain and spinal cord regions from the vertebra, in the rostral-to-caudal direction. Do not peel off the meninges yet; the dorsal root ganglion (DRG) may also be attached. Using a cut pipette (cut the tip of the transfer pipettes with opening of approximately 4-5 mm), transfer the spinal cords (with the meninges) into new HCMF-containing dishes. It is better to quickly proceed to the step of mounting the spinal cords onto ConA-coated black NC filters. In our experience, the shorter period between dissecting out spinal cords and mounting them on NC filters results in better axon growth.
- By using two pairs of sharpened No. 5 Inox forceps (alternatively, ophthalmic scissors), tear (or cut) the dorsal midline (roof plate [RP]) of the spinal cord that is still covered with the meninges (Figure 1D), and open up the entire length of spinal cord like a book (we usually tear the RP by grabbing the adjacent meninges, but not spinal cord proper, with forceps) (see Figure 1K). Next, peel off the meninges from the spinal cord in the rostral-to-caudal direction, by sweeping between the outer surface of spinal cords and meninges, with the forceps. As shown in Figure 1C, at this point, we obtain the spinal cords from the cervical to lumbar levels (by cutting the open-book spinal cord with forceps).
- Preparation of spinal cord explant culture
- For dSC (-FP) explants: immediately dissect out the dorsal half of the spinal cords (see the cutting lines shown in Figure 1E) with a tungsten needle (Figure 1H).
- For SC (+FP) explants: immediately cut the spinal cord at the opposite edge of the FP (Figure 1E) with a tungsten needle.
Note: Do not pool open-book spinal cords or the dSC (-FP)/SC (+FP) explants. If pooled, they will become curled and very difficult to mount onto the NC filter. When one explant is ready, start the Step B5c. - We usually mark the dorso-caudal corner of the explants by cutting diagonally (Figure 1F). Using a wet pipette (it should be noted that explants can easily adhere to dry surfaces and be damaged), immediately transfer the dSC (-FP) or SC (+FP) explant into a coverslip- and Medium 3-containing dish. Spread the explant on a strip of the ConA-coated black NC filter (Figures 1F, 1J and 1L). To fix the position of the explant, nail the explant on the NC filter by using blunt forceps, without using real “nails”, and with the pial side downward (Figure 1L (i)). For dSC (-FP) explants, an extra care not to include any piece of FP tissues into the dish should be taken. We strongly recommend mounting the spinal cord one by one, and immediately after the meninges are peeled off.
Notes:- Use un-cut pipettes for transferring the excised spinal cord explant ready for mounting. The un-cut pipettes, rather than cut pipettes, are suitable for handling small pieces of nerual tissues.
- For preparing retinal explants, researchers usually “blot” retinal tissues that are spread on the NC filter, further onto a dry filter paper, so that the retina becomes firmly attached to the NC filter (e.g., see Bonhoeffer and Huf, 1982). However, we found that this procedure does not improve axon growth in spinal cord explants and may even compromise tissue integrity. Thus, we do not perform the blotting procedure to prepare spinal cord explants.
- Have two spinal cords mounted on the NC filters. Using forceps, place them in parallel on the Matrigel-coated coverslips in the Medium 3-containing 35 mm dish. Reverse the spinal cord explant-mounted filter strips, and hold them with a pair of stainless metal weights (from right above, gently place the weights to stabilize the two NC strips) (Figures 1G and 1M). Now the ventricular side of the spinal cord is contacting the Matrigel-coated surface of the coverslip. Incubate the dish at 5% CO2 at 37 °C for 48 h. In our experiments, the cultures were treated with Slit ligand (see Materials and Reagents D.10.). Usually, we do not replace media during 48 h cultivation, before performing ligand stimulation and fixation.
Notes:- During the flipping step, do not put out the explant on the medium.
- Although netrin-1 has been used to promote axon growth in dSC (-FP) explants (e.g., Zou et al., 2000), we do not use netrin-1, irrespective of dSC (-FP) and SC (+FP) explant cultures. Thus, we have examined effects of Slit alone on Robo1 receptor expression (Kinoshita-Kawada et al., 2019).
- Examine axon outgrowth in the explant culture under a phase-contrast microscope. Much more axons should extend from the ventral side of the explants. Under the conditions used, the axonal front should reach a distance of 500 μm or more from the explant after 48 h in culture.
- For ligand stimulation, we usually remove the half volume (1 ml) of the culture medium and add 1 ml of the ligand-containing fresh medium. After ligand stimulation, perform fixation of the explant cultures at room temperature for 1 h, by adding 4% PFA plus 10% sucrose in PBS. Immediately before fixation, remove the metal weights carefully. Wash the fixed explants three times with PBS each for 30-60 min, and finally add PBS + 0.025% NaN3 for storage. The explants can be subjected to immunostaining. Handle with extra care not to detach the explants from the coverslip during fixation, washing and immunostaining steps.
- Immunostaining
We usually perform immunostaining of the cultured explants 1 day or later after fixation, which gives better results of immunostaining for Robo receptors and L1.- Permeabilize the cultured explants with 0.2% Triton X-100 in PBS for 2 min at room temperature.
- Wash three times with PBS for 5 min each.
- Block the samples with blocking buffer 1 containing 3% BSA, 0.1% skim milk and 0.05% Triton X-100 in PBS for 30 min at room temperature.
- Incubate the explants with the primary antibodies that are diluted in blocking buffer 1 at 4 °C overnight (for approximately 18-20 h). In our experiments, we usually use anti-Robo rabbit polyclonal antibody and anti-L1 (2C2) or anti-TAG-1 (4D7).
- Wash three times with PBS (30-60 min each).
- Block the samples with blocking buffer 2 containing 3% BSA, 3% goat serum, 0.1% skim milk and 0.05% Triton X-100 in PBS for 10 min at room temperature.
- Incubate the explants with the secondary antibodies that are diluted in blocking buffer 2 for 2 h at room temperature. In our experiments, we used Alexa488-conjugated anti-rabbit secondary antibodies and Alexa555-conjugated anti-mouse IgG (or IgM) secondary antibodies.
- Wash three times with PBS (30-60 min each).
- Incubate the explants with Alexa647-conjugated anti-β-tubulin, class III antibody (TuJ1) that is diluted in blocking buffer 2 for 2 h at room temperature.
- Wash three times with PBS (30-60 min each).
- Mount the samples onto glass slides with PermaFluor, usually on the day of observation. The mounted samples are not well maintained for a long time. They should be photographed soon.
- Preparation for dissection
- Ex vivo electroporation (EP) and spinal cord explant culture
The following procedure can be performed outside the laminar flow hood. Spinal cords are dissected from mouse embryos after DNA injection and EP, and are cultured as “closed-book” explants in collagen matrix at 37 °C at 5% CO2 for 4 days (Figures 2A-2H). During this culture period, vYFP-expressing commissural axons reach, cross the ventral midline and turn longitudinally in control spinal cord preparations (Figure 2I). The effects of gene knockdown and knockout can be evaluated in terms of midline axon guidance.- Dissect out E11.5 mouse embryos into HCMF, as described above. Select the embryos appropriate for EP by performing accurate staging of mouse embryos.
Note: It is important that the curving morphology of the neural tube of the embryos matches the shape of the electrode. In our hands, the stages just before E11.5 are appropriate for introduction of plasmids/shRNAs to pre-crossing neurons by EP and axon midline crossing in situ. E10.5-11.0 spinal cords are difficult to electroporate efficiently and to prepare the explants in subsequent steps, resulting in insufficient labeling of midline-crossing axons. Video 1. Injection of an axon-tracer plasmid into the neural tube of a mouse embryo. This video shows an experiment of DNA injection into E11.5 mouse spinal cord before EP under a dissecting microscope. (This video was made at Fukuoka Univ., according to guidelines from the Fukuoka Univ. on Animal Care and approved by the Animal Research Ethics Board of Fukuoka University under protocol # 1815122.)
Video 1. Injection of an axon-tracer plasmid into the neural tube of a mouse embryo. This video shows an experiment of DNA injection into E11.5 mouse spinal cord before EP under a dissecting microscope. (This video was made at Fukuoka Univ., according to guidelines from the Fukuoka Univ. on Animal Care and approved by the Animal Research Ethics Board of Fukuoka University under protocol # 1815122.) - To perform ex utero EP, place an embryo in a new 100 mm dish containing an enough volume of cold sterile DPBS under a dissecting microscope. Manually inject spinal cords of E11.5 embryos with a solution containing shRNA constructs (2 μg/μl) and pCAG-vYFP (0.4 μg/μl) supplemented with 0.05-0.1% Fast Green and diluted in DPBS (Video 1). Immediately thereafter, electroporate embryos unilaterally with three 50-ms pulses of 20 V at 50-ms intervals (for E11.5 ICR embryos) by using the NEPA21 electroporator (Nepa Gene) and forceps-type electrodes (CUY665P9-6-2-5, Nepa Gene) (Figures 2E and 2F). Just before performing EP, sandwich the injected embryo gently between two electrodes (we set the cathode left-side and the anode right-side) and press the foot switch of the NEPA21 machine. Wait until EP is finished. Do not sandwich the embryo with the electrodes too tightly during EP. If too tightly sandwiched, the injected DNA solution will leak out, and the embryo may be damaged during EP.
Notes:- If different types of PBS are used, the actual electric conditions for EP of embryos may become dramatically different. We recommend testing several kinds of PBS for EP, in terms of gene transfer efficiency and tissue integrity. In addition, if we continue to use the same DPBS for EP of many embryos, the current flowed and electric energy tend to increase remarkably, which also affects tissue integrity. We use fresh DPBS in a new dish for every 3 embryos.
- Inject a sufficient volume of DNA solution into the cavity of the neural tube (two approaches are presented in Figure 2A), but too much volume does not increase the efficiency of gene transfer into the spinal cord, because the DNA tends to leak from the embryo after a blowout of the neural tube. EP pulses make the embryonic tissues, especially FP regions fragile. For getting the best results in EP, in terms of the location and efficiency of gene transfer, sufficient practice will be required. If the EP efficiency is too low even after practice, consider whether the shape of your electrodes matches the curving morphology of spinal cords of the embryos, and the electric condition of EP may have to be modified (efficient EP depends on the types of electrodes, pulse voltage, width, numbers and intervals as well as damping factors).
This video shows an experiment of DNA injection into E11.5 mouse spinal cord before EP under a dissecting microscope. (This video was made at Fukuoka Univ., according to guidelines from the Fukuoka Univ. on Animal Care and approved by the Animal Research Ethics Board of Fukuoka University under protocol # 1815122.)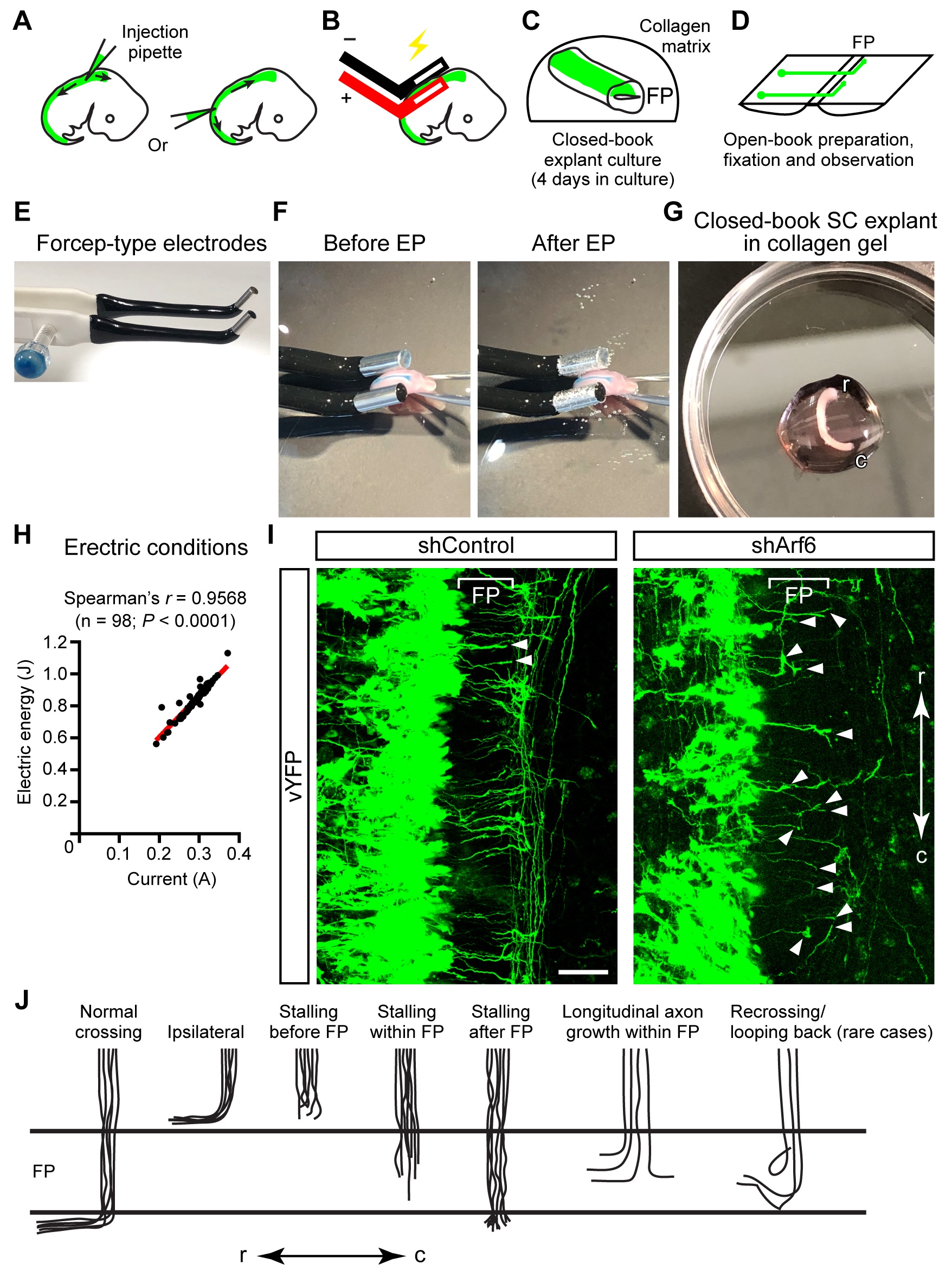
Figure 2. Ex vivo electroporation and closed-book spinal cord explant culture. A-D. Schematic illustration of ex vivo electroporation (EP) and explant culture: A. Two approaches for DNA injection. B. EP of the spinal cord region. C. Explant embedding into collagen matrix, followed by 4-days culture. D. Open-book preparation and observation of axon trajectories. E. CUY665P9-6-2-5 electrodes (Nepa Gene) used for EP of spinal cords. F. An example of the EP experiment. G. An example of a spinal cord explant embedded in collagen gel, before adding the medium. r: rostral; c: caudal. H. The significant Spearman’s correlation between the current and electric energy measured during EP (in case of three 50 ms pulses of 20 V at 50 ms intervals). I. Midline axon trajectories of vYFP/shRNA-coelectroporated spinal cord neurons in open-book preparations. Axons stalling within the FP (marked by brackets) or at the contralateral edge of the FP are indicated by arrowheads. shRNA-mediated Arf6 knockdown in commissural neurons causes axon stalling at the midline. Scale bar: 50 μm. J. Summary of representative axon trajectories at the midline in shControl- and shRNA-electroporated spinal cords.
- Immediately after EP, transfer the embryo into numbered 35 mm dishes containing L-15 media. Keep them on ice for recovery. After performing EP of each embryo, gently wipe the electrodes with 70% ethanol.
Note: We found that the tissue integrity of eletroporated spinal cords is better when keeping in L-15 than in HCMF. After finishing all the EP steps, clean up the electrodes, brush them softly using a toothbrush (or a cotton swab) and toothpaste. Wash them with distilled water and finally with 70% ethanol, and let them air-dry. - After EP of all the embryos are finished, transfer the embryos to fresh HCMF-containing dishes and dissect out spinal cords carefully, one-by-one. When isolating the electroporated spinal cord from the embryo, we usually start dissection from the regions near the roof plate, to avoid damaging the FP. Do not remove the meninges from the spinal cord. Prepare the closed-book explant, with the meninges attached.
Note: We recover the electroporated spinal cords from the cervical to lumbar levels for explant culture. In our hands, successful vYFP expression in the spinal cord, robust axon extension toward the ventral region and axon midline crossing were observed usually from the upper-limbs to lower-limbs levels. - Embed each isolated spinal cord in collagen gel. Before starting embedding, have acid collagen solution, 5x concentrated medium (DMEM/F12) and reconstruction buffer, and keep them on ice. Mix 7 volumes of acid collagen solution (for example, 350 μl/tube, in our experiments) and 2 volumes of 5x concentrated medium (DMEM/F12) (for example, 100 μl/tube) and keep on ice until use. Just before use, add approximately 0.9-1 volume of reconstruction buffer (for example, 45 μl/tube) to the above mixture, mix well by pipetting and if needed, adjust pH to ~7.4 by referring to the color of phenol red included in the mixture. When kept on ice or at 4 °C, the collagen mixture can be maintained in the sol state for a while. When kept at 37 °C in a CO2 incubator, the mixture will gelatinize.
- Place one spinal cord per 35 mm dish (we usually keep the FP the right side [Figure 2G], so that the electroplated side of the spinal cord is located upward). Cover a “closed-book” preparation of the explant with 120 μl of the sol-state collagen mixture and incubate in a CO2 incubator to gelatinize collagen for 15-30 min (not too long). When the collagen has gelatinized, add 2-2.5 ml of Medium 4 to the dish slowly.
- Culture at 37 °C for 4 days (exchange daily the half volume, ~1 ml, of the medium).
- After 4 days of culture, dissect out spinal cords from collagen gel. Transfer them into PBS or HCMF. Remove the meninges, cut the roof plate and prepare open-book preparations. Fix the spinal cord with 4% PFA in PBS on Sylgard-coated dissection dishes for 15 min at room temperature (for this fixation step, Sylgard-coated dishes appear to be much better than usual culture dishes). Continue fixation as immersion fixation of the spinal cord at 4 °C overnight. Wash the spinal cords twice with PBS and finally replace with PBS + 0.025% NaN3 for storage.
Notes:- When recovering the spinal cords from the gel, their appearance should be similar to those before cultivation, without remarkable swelling. If so different, consider the milder EP condition (e.g., try fewer pulses, lower voltages). Use 4% PFA in PBS without sucrose for fixation. In the presence of sucrose in the fixative, spinal cords will be floating and damaged due to surface tension.
- We do not use collagenase to recover the spinal cords from the gel.
- Mount the electroprated spinal cords for observation. Transfer the open-book spinal cord explant, together with PBS, onto a 24 x 60 mm rectangular coverslip, remove most of PBS, add PermaFluor and cover the explant with another coverslip (same size) to make a sandwich. Visualize them under a confocal microscope. We acquire confocal stacks of green-channel images for visualizing vYFP signals and DIC images to show the accurate FP position. We usually observe the spinal cord explant first from the ventricular side and then from the pial side, when necessary.
- Dissect out E11.5 mouse embryos into HCMF, as described above. Select the embryos appropriate for EP by performing accurate staging of mouse embryos.
Data analysis
- Quantification of neuronal guidance receptor expression in commissural axons extending from spinal cord explants (in our cases, we analyze the distribution of Robo receptors; see Figure 3).
- Take images of immunostained explants by using appropriate tiling functions on a laser-scanning microscope or Keyence all-in-one fluorescence microscope. Alternatively, take images of the explant by manual-hand acquisition using a conventional confocal microscope. Obtain the merged images by assembling maximal-intensity projection images.
- Measure signal intensity in the individual axons (or small axon fascicles) by using MetaMorph (or ImageJ) software. We usually quantify the immunosignal intensity in the most distal part and shaft (30 μm length) of L1-positive commissural axons, which is normalized to TuJ1 immunoreactivity (these signals reflect axon density). We usually prepare 3-8 spinal cord explants for each experimental group, thus from the same numbers of mouse embryos (one explant per embryo), and analyze more than 100 imaging fields for axons per experimental group (30 images per explant). For statistical analysis, we perform the nonparametric Mann-Whitney test (see Kinoshita-Kawada et al., 2019; Figures 1 and 4).
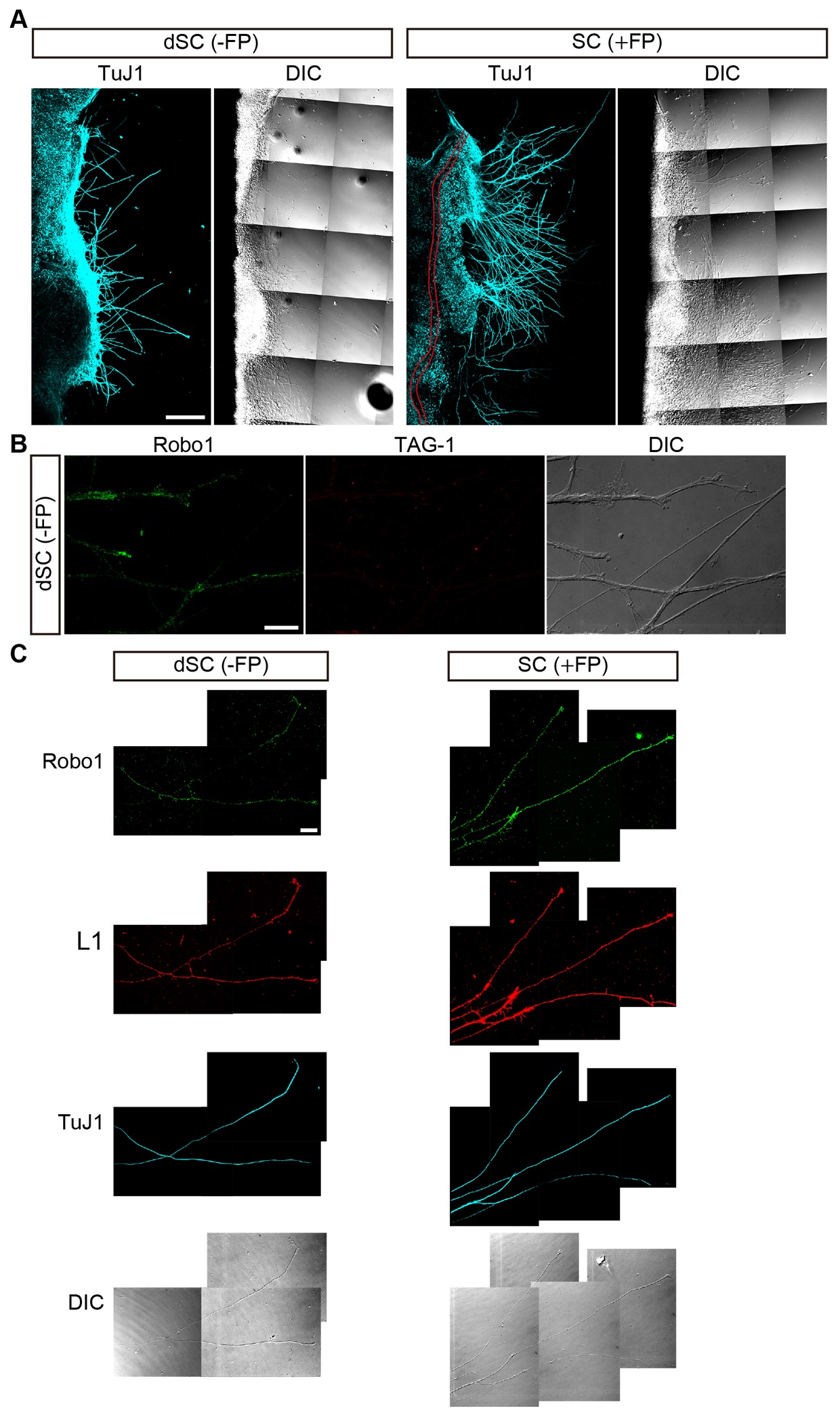
Figure 3. Immunocytochemical characterizaion of commissural axons growing from spinal cord explants. A. axon growth from dSC (-FP) and SC (+FP) explants prepared from E11.5 embryos and cultured on Matrigel-coated coverslips. The explants were immunostained for TuJ1. Differential interference contact (DIC) images are also shown. The FP is marked with red lines. B. Axons extending from dSC are positive for Robo1 but negative for TAG-1, indicating that they do not have characteristics of pre-crossing commissural axons. C. Triple immunostaining of dSC (-FP) and SC (+FP) explants for Robo1, L1 and TuJ1. Axons extending from dSC (-FP) and SC (+FP) explants are double positive for Robo1 and for L1, a post-crossing commissural axon marker. Scale bars: 250 μm in A; 20 μm in B and C.
- Quantification of midline-crossing by commissural axons in electroporated and cultured spinal cord explants.
- Mount spinal cord explants between two coverslips on the day of observation.
- Take images by an LSM confocal microscope (at least green and DIC channels of images should be obtained).
- For quantification, count the number of vYFP-positive axons that exhibited turning contralaterally (crossing) or ipsilaterally, stalling (before reaching the FP, within the FP [we include longitudinal axon growth within the FP] or after crossing the FP) or re-crossing/looping back in each imaging field, and present the data as the percentage of the total number of vYFP-positive axons (for categorization of the axon trajectories, see Figure 2J). We usually analyze the axonal trajectories in at least 4 embryos for each experimental group (tracing approximately 200-3000 labeled axons) and perform Kruskal-Wallis test with Dunn’s post-hoc test for statistical analysis. In these analyses, no data for vYFP-positive axons were excluded, with exceptions of over-shooting axons (see below; see also Kinoshita-Kawada et al., 2019; Figure 7). We found that axonal phenotypes in commissural neurons vary markedly along the rostrocaudal axis, even within the same embryo. Therefore, we have been analyzing values quantified in electroporated spinal cords at different rostrocaudal levels of individual embryos as distinct data points and as biological replicates.
Note: In our study, midline-overshooting phenotypes of the vYFP-labeled axons were excluded from analysis, because we detected such overshooting phenotypes in the tissue areas damaged during EP and/or manipulation, even in control-electroporated mouse embryos.
Notes
- All experimental procedures involving animals should be approved by your institutional animal care committee.
- When working with Matrigel or collagen, keep the original bottle and solution on ice, as much as possible, to prevent solidifying or forming aggregates, which may affect axon growth from explants.
Recipes
- Acid washing of coverslips
- Wash coverslips with enough amounts of distilled water, with sonication (if available), three times for 5 min
- Wash the coverslips with 70% ethanol for 10 min and wash with distilled water for 10 min, while shaking
- Wash the coverslips with 1 N HCl, while occasionally shaking for more than 1 h
- Wash the coverslips with distilled water, while shaking, five times for 10 min, and then three times for 30 min
- Air-dry the coverslips on KimWipe papers
Note: Place one by one. Separate them with each other at this step. After baking, it is almost impossible to separate them. - Transfer them to a 100 ml beaker and sterilize by baking at 180 °C for 3 h
- 2 mg/ml ConA stock solution
- Dissolve 25 mg of ConA in 12.5 ml of HCMF
- Dispense 0.5 ml/1.5 ml tube and store at -80 °C
- Medium 1
DMEM
10% FBS
2 mM L-Glutamine
100 units/ml Pen/100 units/ml Strep
Store at 4 °C - Medium 2
Neurobasal + B-27
10 mM HEPES
2 mM L-Glutamine
100 units/ml Pen/100 units/ml Strep
Store at 4 °C - Medium 3
1:1 mixture of Medium 1 and Medium 2 (store at 4 °C) - 5x concentrated DMEM/F-12 medium
- Prepare 5x concentrated medium and add Pen/Strep, but not NaHCO3
- Sterilize by filtration (0.22 μm) and store at 4 °C. This medium contains phenol red
- Reconstitution buffer (used for gelatinization of collagen)
- Dissolve 0.05 N NaOH (0.2 g), 0.26 M NaHCO3 (2.2 g) and 0.18 M HEPES (4.8 g) per 100 ml of distilled water
- Sterilize by filtration (0.22 μm) and store at 4 °C
- Medium 4 (Sabatier et al., 2004)
45% Opti-MEM I
50% F-12
5% Heat-inactivated horse serum
2 mM Glutamine
40 mM Glucose
100 units/ml Pen/100 units/ml Strep
Store at 4 °C
Acknowledgments
This work was supported by grants from the National Institutes of Health and the Japan Society for the Promotion of Science. We thank all of present and previous members of the Wu and Rao labs for advice, Drs. Fujio Murakami and Atsushi Tamada for generously providing anti-Robo antibodies, Dr. Chen-bing Guan for generously providing his Slit2 purification protocol, Animal/Biology Resources Sections at OIST, Yasutomo Kubota (Molecular Devices) and Nepa Gene for their valuable support. We are grateful to Dr. Hiroyuki Ichijo for introducing us retinal explant culture techniques that were originally developed by Dr. Friedrich Bonhoeffer and colleagues. Our dSC (-FP)/SC (+FP) explant culture protocols were adapted from the previously published protocols (Bonhoeffer and Huf, 1982; Halfter et al., 1981 and 1983; Ichijo and Bonhoeffer, 1998; Walter et al., 1987a and 1987b; Zou et al., 2000). Our ex vivo RNAi assay system was adapted based on the previously published protocols (Parra and Zou, 2010; Wolf et al., 2008). Finally, we would like to thank Dr. Zhao Chen for her kind invitation to publish our protocols here.
Competing interests
The authors have no conflicts of interest to disclose.
Ethics
All animal procedures were approved by the Institutional Animal Care and Use Committee at Northwestern University, OIST, Kyushu University and Fukuoka University.
References
- Banker, G. and Goslin, K. (Eds.) (1998). Culturing nerve cells. 2nd edition. The MIT Press.
- Bonhoeffer, F. and Huf, J. (1982). In vitro experiments on axon guidance demonstrating an anterior-posterior gradient on the tectum. EMBO J 1(4): 427-431.
- Brose, K., Bland, K. S., Wang, K. H., Arnott, D., Henzel, W., Goodman, C. S., Tessier-Lavigne, M. and Kidd, T. (1999). Slit proteins bind Robo receptors and have an evolutionarily conserved role in repulsive axon guidance. Cell 96(6): 795-806.
- Dickson, B. J. and Zou, Y. (2010). Navigating intermediate targets: the nervous system midline. Cold Spring Harb Perspect Biol 2(8): a002055.
- Dodd, J., Morton, S. B., Karagogeos, D., Yamamoto, M. and Jessell, T. M. (1988). Spatial regulation of axonal glycoprotein expression on subsets of embryonic spinal neurons. Neuron 1(2): 105-116.
- Dominici, C., Moreno-Bravo, J. A., Puiggros, S. R., Rappeneau, Q., Rama, N., Vieugue, P., Bernet, A., Mehlen, P. and Chedotal, A. (2017). Floor-plate-derived netrin-1 is dispensable for commissural axon guidance. Nature 545(7654): 350-354.
- Ducuing, H., Gardette, T., Pignata, A., Tauszig-Delamasure, S. and Castellani, V. (2019). Commissural axon navigation in the spinal cord: A repertoire of repulsive forces is in command. Semin Cell Dev Biol 85: 3-12.
- Elhendawi, M. and Davies, J. A. (2018). Sebinger Culture: A System Optimized for Morphological Maturation and Imaging of Cultured Mouse Metanephric Primordia. Bio-protocol 8(4): e2730.
- Guan, C. B., Xu, H. T., Jin, M., Yuan, X. B. and Poo, M. M. (2007). Long-range Ca2+ signaling from growth cone to soma mediates reversal of neuronal migration induced by slit-2. Cell 129(2): 385-395.
- Guan, K. L. and Rao, Y. (2003). Signalling mechanisms mediating neuronal responses to guidance cues. Nat Rev Neurosci 4(12): 941-956.
- Halfter, W., Claviez, M. and Schwarz, U. (1981). Preferential adhesion of tectal membranes to anterior embryonic chick retina neurites. Nature 292(5818): 67-70.
- Halfter, W., Newgreen, D. F., Sauter, J. and Schwarz, U. (1983). Oriented axon outgrowth from avian embryonic retinae in culture. Dev Biol 95(1): 56-64.
- Ichijo, H. and Bonhoeffer, F. (1998). Differential withdrawal of retinal axons induced by a secreted factor. J Neurosci 18(13): 5008-5018.
- Kidd, T., Bland, K. S. and Goodman, C. S. (1999). Slit is the midline repellent for the Robo receptor in Drosophila. Cell 96(6): 785-794.
- Kinoshita-Kawada, M., Hasegawa, H., Hongu, T., Yanagi, S., Kanaho, Y., Masai, I., Mishima, T., Chen, X., Tsuboi, Y., Rao, Y., Yuasa-Kawada, J. and Wu, J. Y. (2019). A crucial role for Arf6 in the response of commissural axons to Slit. Development 146(3): pii: dev172106.
- Kolodkin, A. L. and Tessier-Lavigne, M. (2011). Mechanisms and molecules of neuronal wiring: a primer. Cold Spring Harb Perspect Biol 3(6): a001727.
- Li, H. S., Chen, J. H., Wu, W., Fagaly, T., Zhou, L., Yuan, W., Dupuis, S., Jiang, Z. H., Nash, W., Gick, C., Ornitz, D. M., Wu, J. Y. and Rao, Y. (1999). Vertebrate slit, a secreted ligand for the transmembrane protein roundabout, is a repellent for olfactory bulb axons. Cell 96(6): 807-818.
- Mikuni, T., Nishiyama, J., Sun, Y., Kamasawa, N. and Yasuda, R. (2016). High-throughput, high-resolution mapping of protein localization in mammalian brain by in vivo genome editing. Cell 165(7): 1803-1817.
- Moreno-Bravo, J. A., Roig Puiggros, S., Mehlen, P. and Chedotal, A. (2019). Synergistic activity of floor-plate- and ventricular-zone-derived netrin-1 in spinal cord commissural axon guidance. Neuron 101(4): 625-634 e3.
- Nishiyama, J., Mikuni, T. and Yasuda, R. (2017). Virus-mediated genome editing via homology-directed repair in mitotic and postmitotic cells in mammalian brain. Neuron 96(4): 755-768 e5.
- Park, H., Oh, J., Shim, G., Cho, B., Chang, Y., Kim, S., Baek, S., Kim, H., Shin, J., Choi, H., Yoo, J., Kim, J., Jun, W., Lee, M., Lengner, C. J., Oh, Y. K. and Kim, J. (2019). In vivo neuronal gene editing via CRISPR-Cas9 amphiphilic nanocomplexes alleviates deficits in mouse models of Alzheimer's disease. Nat Neurosci 22(4): 524-528.
- Parra, L. M. and Zou, Y. (2010). Sonic hedgehog induces response of commissural axons to Semaphorin repulsion during midline crossing. Nat Neurosci 13(1): 29-35.
- Sabatier, C., Plump, A. S., Le, M., Brose, K., Tamada, A., Murakami, F., Lee, E. Y. and Tessier-Lavigne, M. (2004). The divergent Robo family protein rig-1/Robo3 is a negative regulator of slit responsiveness required for midline crossing by commissural axons. Cell 117(2): 157-169.
- Shirasaki, R., Katsumata, R. and Murakami, F. (1998). Change in chemoattractant responsiveness of developing axons at an intermediate target. Science 279(5347): 105-107.
- Stoeckli, E. T. (2018). Understanding axon guidance: are we nearly there yet? Development 145(10): pii: dev151415.
- Stoeckli, E. T., Sonderegger, P., Pollerberg, G. E. and Landmesser, L. T. (1997). Interference with axonin-1 and NrCAM interactions unmasks a floor-plate activity inhibitory for commissural axons. Neuron 18(2): 209-221.
- Tessier-Lavigne, M., Placzek, M., Lumsden, A. G., Dodd, J. and Jessell, T. M. (1988). Chemotropic guidance of developing axons in the mammalian central nervous system. Nature 336(6201): 775-778.
- Varadarajan, S. G., Kong, J. H., Phan, K. D., Kao, T. J., Panaitof, S. C., Cardin, J., Eltzschig, H., Kania, A., Novitch, B. G. and Butler, S. J. (2017). Netrin1 produced by neural progenitors, not floor plate cells, is required for axon guidance in the spinal cord. Neuron 94(4): 790-799 e3.
- Walter, J., Kern-Veits, B., Huf, J., Stolze, B. and Bonhoeffer, F. (1987a). Recognition of position-specific properties of tectal cell membranes by retinal axons in vitro. Development 101(4): 685-696.
- Walter, J., Henke-Fahle, S. and Bonhoeffer, F. (1987b). Avoidance of posterior tectal membranes by temporal retinal axons. Development 101(4): 909-913.
- Wolf, A. M., Lyuksyutova, A. I., Fenstermaker, A. G., Shafer, B., Lo, C. G. and Zou, Y. (2008). Phosphatidylinositol-3-kinase-atypical protein kinase C signaling is required for Wnt attraction and anterior-posterior axon guidance. J Neurosci 28(13): 3456-3467.
- Wu, Z., Makihara, S., Yam, P. T., Teo, S., Renier, N., Balekoglu, N., Moreno-Bravo, J. A., Olsen, O., Chedotal, A., Charron, F. and Tessier-Lavigne, M. (2019). Long-range guidance of spinal commissural axons by Netrin1 and sonic hedgehog from midline floor plate cells. Neuron 101(4): 635-647 e4.
- Yang, T., Huang, H., Shao, Q., Yee, S., Majumder, T. and Liu, G. (2018). miR-92 suppresses Robo1 translation to modulate Slit sensitivity in commissural axon guidance. Cell Rep 24(10): 2694-2708 e6.
- Yuasa-Kawada, J., Kinoshita-Kawada, M., Wu, G., Rao, Y. and Wu, J. Y. (2009). Midline crossing and Slit responsiveness of commissural axons require USP33. Nat Neurosci 12(9): 1087-1089.
- Yuasa-Kawada, J., Suzuki, R., Kano, F., Ohkawara, T., Murata, M. and Noda, M. (2003). Axonal morphogenesis controlled by antagonistic roles of two CRMP subtypes in microtubule organization. Eur J Neurosci 17(11): 2329-2343.
- Zou, Y., Stoeckli, E., Chen, H. and Tessier-Lavigne, M. (2000). Squeezing axons out of the gray matter: a role for slit and semaphorin proteins from midline and ventral spinal cord. Cell 102(3): 363-375.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Kinoshita-Kawada, M., Hasegawa, H., Hongu, T., Yanagi, S., Kanaho, Y., Masai, I., Mishima, T., Chen, X., Tsuboi, Y., Rao, Y., Yuasa-Kawada, J. and Wu, J. Y. (2019). Explant Culture of the Embryonic Mouse Spinal Cord and Gene Transfer by ex vivo Electroporation. Bio-protocol 9(18): e3373. DOI: 10.21769/BioProtoc.3373.
Category
Neuroscience > Development > Explant culture
Neuroscience > Development > Electroporation
Cell Biology > Cell movement > Cell migration
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.