- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

A PhoA-STII Based Method for Efficient Extracellular Secretion and Purification of Fab from Escherichia coli
Published: Vol 9, Iss 18, Sep 20, 2019 DOI: 10.21769/BioProtoc.3370 Views: 7349
Reviewed by: Alba BlesaZhenying LiuLi Zhang
Original research article
The authors used this protocol in:

Apr 2019

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
In comparison with full-length IgGs, antigen binding fragments (Fabs) are smaller in size and do not require the complexed post-translational modification. Therefore, Fab can be cost-effectively produced using an Escherichia coli (E. coli) expression system. However, the disulfide-bonds containing exogenous protein, including Fab, tend to form insoluble inclusion bodies in E. coli, which has been the bottleneck for exogenous protein expressions using this system. The secretory expression of proteins in periplasm or extracellular medium are promising strategies to prevent the formation of inclusion bodies to improve the efficiency to produce Fabs from E. coli. The extracellular expression is of particularly interest since it releases the product into the medium, while periplasmic expression yield is limited to the periplasm space. In addition, the extracellular expression allows for the direct harvesting of proteins from the culture supernatant, sparing the procedures of cell lysis and reducing contamination of host cell protein or DNA. Using anti-VEGF Fab as an example, here we provide a protocol based on the alkaline phosphatase (phoA) promoter and the heat-stable enterotoxin II (STII) leader sequence. Using phosphate starvation to induce the secretory expression, the protocol could be generally used for the efficient production of Fabs.
Keywords: FabBackground
Due to its clear genetic background, easy manipulation, and cost-effective production, E. coli is widely employed for exogenous gene expression, especially those of lower molecular weight and simpler conformational structures (Gupta and Shukla, 2017). The expression of foreign proteins in E. coli is mainly divided into three categories, inclusion bodies expression, intracellular soluble expression in periplasmic space, and extracellular secretion into medium (Jalalirad, 2013; Gupta and Shukla, 2017; Zhou et al., 2018). At present, most of mammalian-sourced foreign proteins are expressed in E. coli intracellular region, either in the form of inclusion bodies or soluble. But both forms have their respective disadvantages for subsequent processes. The inclusion bodies need to be tediously denatured and renatured to recover the target proteins with correct refolded structures, and the processes tend to cause reduction in the biological activity and yield (Panda et al., 2003; Nelson and Reichert, 2009). The periplasmic space of E. coli can provide an oxidative environment conducive to the formation of disulfide bonds, but the yield is usually limited by the capacity of the periplasmic space as well as the leading capability of signal peptides (Lobstein et al., 2012; Ellis et al., 2017). Compared with the two processes above, extracellular protein expression is not restricted to the intracellular space, allows convenient enrichment of target proteins with right structures, thus simplifies the downstream purification processes (Zhou et al., 2018).
Fab fragment is of smaller size than full-length antibody, and does not require post-translational modification such as glycosylation modifications, it is particularly suitable to be produced in E. coli (Walsh and Jefferis, 2006; Rezaie et al., 2017). We recently reported a method for efficient extracellular expression and purification of Fabs from E. coli, which has been optimized for many parameters (Luo et al., 2019a). Here, we provide a detailed protocol using anti-VEGF Fab as a model protein, since the anti-VEGF Fab Ranibizumab was the first approved Fab drug on the market (Danyliv et al., 2017). The process consists of three main parts: A) construction of pPhoA-Fab expression vector and transfection of host strain BL21(DE3); B) secretory soluble expression of Fab in E. coli; and C) affinity chromatography purification of Fab. We investigated the combinations with different promoters (phoA and T7) and leader peptides (STII and pelB), among them phoA-STII showed the highest yield in mass and secretion efficiency. The vector was a previously engineered pRSF (Augustine et al., 2016) plasmid containing phoA promoter, hereinafter referred to as the pPhoA plasmid. The secretory expression was induced by phosphate starvation to stimulate the function of phoA promoter (Wang et al., 2005). For affinity purification of Fab, the resin should be selected according to species and light chain types (Kappa or Lambda chain). For anti-VEGF Fab, which has human Kappa light chain, we used a prepacked Capto L column to purify it (Ulmer et al., 2019). After purification, the final product could be further analyzed for purity, yield, and bioactivities.
Generally, secretory expression in E. coli is the most suitable process for producing disulfide bond-containing antibody fragments such as Fab, Fab’, and (Fab’)2 (Ellis et al., 2017). In comparison with previous studies about extracellular expression of Fabs, our work is superior for a considerably reduced time and cost and simplified purification process, due to the use of different expression cassette designs (two separate expression cassettes, phoA promoter, STII leader sequence), host strains (BL21[DE3]) and fermentation conditions (phosphate starvation, low temperature). We have applied the protocol to prepare five Fab fragments which have been marketed successfully (anti IGF1R, anti-Her2, anti-VEGF, anti-RANKL and anti-PD-1) with different types of IgG1/IgG2 or human/humanized structures to cover as wide as possible range of Fab fragments. The results demonstrated that they were all expressed in soluble expression, and the fractions in culture medium were more than the intracellular soluble fractions or inclusion bodies content. By one-step affinity chromatography, the purity of the Fabs reached above 94%, and all products were of correct molecular weight as well as full bioactivity against their antigens. To the best of our knowledge, the current protocol is a universal technique for efficient extracellular expression, secretion and purification of Fabs in E. coli.
Materials and Reagents
Notes:
- All the reagents could be of other brands.
- Resins could be used for both AKTA equipment and manual purification, which is depend on the sample volume to handle. For example, when the sample is more than 100 ml, it saves hand-harbor using AKTA than manual purification.
- Inoculating loop
- 2 L Erlenmeyer flask
- 250 ml centrifuge bottles
- Pipettes
- 200 μl PCR tubes
- 1.5 ml Eppendorf tubes
- 0.22 μm filter
- Pipette tips
- E. coli strains DH5α (Shanghai Weidi Biotechnology, catalog number: DL1001)
- E. coli strain BL21(DE3) (Shanghai Weidi Biotechnology, catalog number: EC1002)
- Bgl II (concentration: 10 units/μl) (NEB, catalog number: R0144S), store at -20 °C
- Nde I (concentration: 20 units/μl ) (NEB, catalog number: R0111S), store at -20 °C
- T4 DNA Ligase (concentration: 350 units/μl) (Takara, catalog number:2011A), store at -20 °C
- NaCl (Sinopharm Chemical Reagent, catalog number: 10019318)
- Tryptone (Oxoid, catalog number: 2336957)
- Yeast (Oxoid, catalog number: 210408)
- Agar (Oxoid, catalog number: LP0011B)
- Kanamycin (Sinopharm Chemical Reagent, catalog number: xw253899403)
- PIPES (Sigma-Aldrich, catalog number: P1851-100G)
- (NH4)2SO4 (Sinopharm Chemical Reagent, catalog number: 10002917)
- MgSO4 (Sinopharm Chemical Reagent, catalog number: 20025117)
- D(+)-Glucose (Sinopharm Chemical Reagent, catalog number: 63005518)
- Tris (hydroxymethyl) aminomethane (Amresco, catalog number: 252859-100G)
- Methanol (Sinopharm Chemical Reagent, catalog number:1001418)
- Isopropanol (Sinopharm Chemical Reagent, catalog number: 80109218)
- Absolute alcohol (Sinopharm Chemical Reagent, catalog number: 10009218)
- Enhanced BCA Protein Assay Kit (Beyotime, catalog number: P0009)
- NaH2PO4 (Sinopharm Chemical Reagent, catalog number: 20040818)
- Na2HPO4 (Sinopharm Chemical Reagent, catalog number: 20040617)
- K2HPO4 (Sinopharm Chemical Reagent, catalog number: 20032117)
- KH2PO4 (Sinopharm Chemical Reagent, catalog number: 10017618)
- NaOH (Sinopharm Chemical Reagent, catalog number: 10019762)
- Glycerol (Sinopharm Chemical Reagent, catalog number: 10010618)
- Citric acid (BBI, catalog number: A610055-0500)
- B-PER (Thermo Fisher, catalog number: 78243)
- Millipore ECL (Millipore, catalog number: WBKLS0010), store at 2-8 °C
- Capto L resins (GE Healthcare)
- KappaSelect prepacked column 1 ml (GE, catalog number: 17545811)
- Prestained 2x FastTaq PCR SuperMix (Miozyme, catalog number: AX111), store at -20 °C
- PrimeSTAR Max DNA Polymerase (Takara, catalog number: R045A), store at -20 °C
- AxyPrepTM PCR Cleanup Kit (Axygen, catalog number: AP-PCR-250G)
- AxyPrepTM Plasmid Miniprep Kit (Axygen, catalog number: AP-MN-P-250G)
- AxyPrepTM DNA Gel Extraction Kit (Axygen, catalog number: AP-GX-250G)
- PLM medium (see Recipes)
- Buffer A (see Recipes)
- Buffer B (see Recipes)
- TAE buffer (see Recipes)
Equipment
- BS 210S electronic balance (Sartorius)
- Electronic analytical balance (Mettler Toledo)
- Vertical protein electrophoresis tank (Bio-Rad)
- Tanon GIS Gel Imaging Analysis System (Tanon)
- BECKMAN COULTER Avanti J-E Centrifuge (BECKMAN COULTER)
- Centrifuge (Eppendorf, Model: Centrifuge 5418R)
- LRH-70F biochemical incubator (TENSUC)
- TS-200B desktop constant temperature oscillation culture shaker (TENSUC)
- HH-4 constant temperature water bath (Youlian)
- SW-CJ-IFD type ultra-clean workbench (Sujing)
- HYCD-205 refrigerator freezer (Haier)
- -80°C cryopreservation box (Haier)
- Benchtop pH meter FE20K (Mettler Toledo)
- SS325 autoclave (Tomy)
- TS-2 type bleaching shaker (Qilinbeier)
- Milli-Q Advantage A10 UltraPure Water Meter (Millipore)
- Electric blast drying oven (Yiheng)
- Thermal Cyclers for PCR (Bio-Rad)
- Multi-function microplate reader Infinite M200 PRO (Tecan)
- AKTA Avant 150 (GE)
Software
- Primer 5.0
Procedure
- Construct of anti-VEGF Fab expressing plasmids and transformation of E. coli host strain
Notes:- According to the Tm of the primers and the polymerases, set appropriate PCR reaction conditions, and select proper restriction enzymes depending on the restriction sites of the plasmids.
- The DNA sequence can be amplified from a template or synthesized by companies such as General Biosystems (Chuzhou, Anhui, China) we used.
- The backbone plasmid pPhoA can be derived from commercial plasmids such as pRSF-Duet by inserting dual phoA promoters followed by STII signal peptides.
- The transformation approach adapts to all the transformation mentioned in this protocol.
- According to the process shown in Figure 1, synthesize the entire DNA fragment encoding VL-CL, phoA promoter, STII peptide and VH-CH1 of anti-VEGF Fab expression cassette according to the published sequences.
- Analyze the sequences of backbone vector and Fab fragments to determine the sites for double restriction enzymes digestion and ligation.
Note: If there are no appreciated sites, recombinase mediated homology infusion method can be used. - Design the primers with the selected restriction enzymes using Primer 5.0 software. To simplify the insertion of Fab fragments into the vector, it’s optimal to synthesize STII-VL-CL-phoA-STII-VH-CH1 by a company (for example General Biosystems, Chuzhou, Anhui, China).
Note: To insert the target fragment into the backbone vector, we firstly determined the restriction enzymes to produce pairing sticky ends. We chose Nde I which holds the translational starting codon ATG as the upstream site. For the downstream site, it’s fine to choose any one listed in the multiple clone sites of the vector. In our study, we chose Bgl II, and the vector could be digested by Nde I and Bgl II to get the linear backbone. However, the target fragment is rather long (about 2 kb), and there are another two Bgl II sites, making it impossible to digest the target fragment with Bgl II. Therefore, we used Bsa I enzyme and designed its sticky ends the same with that of Bgl II. Then we designed the forward primer with an Nde I site and the reverse primer with a Bsa I site to amplify the fragment from the synthesized template. Finally the Nde I and Bsa I enzymes digested fragment was ligated with the Nde I and Bgl II enzymes digested backbone to construct the recombinant plasmid expressing anti-VEGF Fab. - Extract backbone vector.
- Prepare target fragments of STII-VL-CL-phoA-STII-VH-CH1.
- Amplify STII-VL-CL-phoA-STII-VH-CH1 fragments with above-designed primers (Table 1) by PCR.
- Add the reaction reagents in 200 μl PCR tubes as shown in Table 2.
Table 1. Sequences of primers used in PCR or sequencing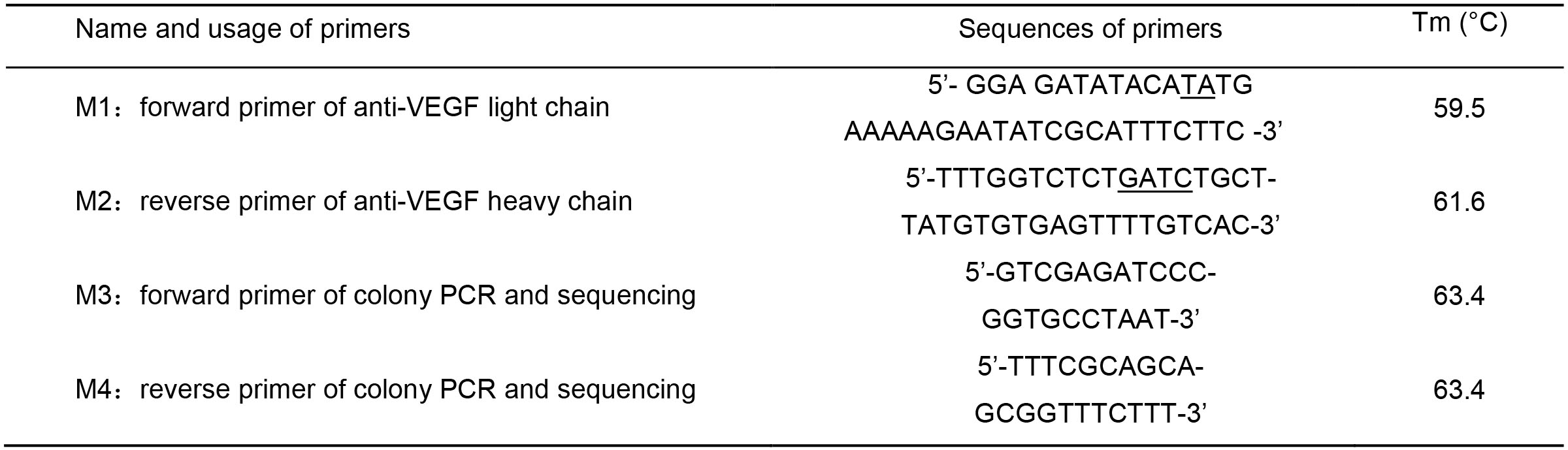
*Primers M1 and M2 hold Nde I and Bsa I digestion sites, respectively.
Table 2. PCR reaction recipes for amplifying STII-VL-CL-phoA-STII-VH-CH1 fragments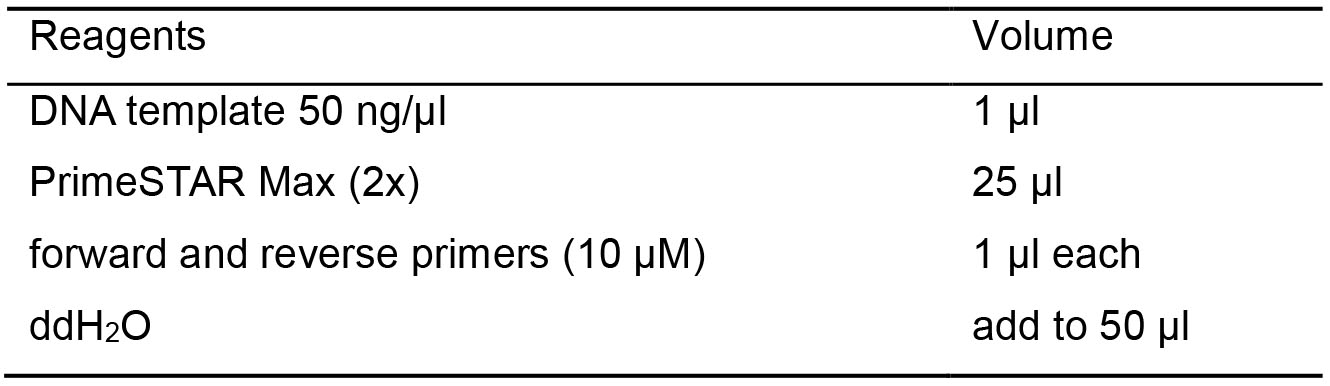
- Set the PCR program to be: denaturation at 98 °C for 10 s, annealing at 55 °C for 5 s (according to the manual of PrimeSTAR Max, the annealing temperature could be set at 55 °C when the calculated Tm values are around 60 °C) and extension at 72 °C for 30 s (the extension time depends on the polymerase as well as the length of the target fragment, here the target fragment is about 2 kb), recycling for 35 cycles.
- Add the reaction reagents in 200 μl PCR tubes as shown in Table 2.
- Digest the STII-VL-CL-phoA-STII-VH-CH1 PCR products with Nde I and Bsa I, and digest the backbone vectors with double enzymes Bgl II and Nde I to get desired sticky ends.
- Add the reaction reagents in a 1.5 ml Eppendorf tubes as Table 3.
Table 3. Double enzyme digestion reaction recipes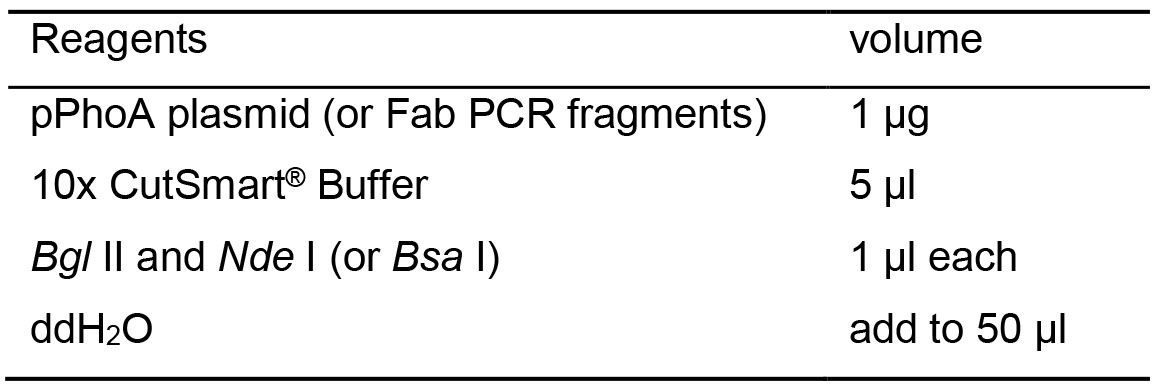
- Incubate the mixture in a 37 °C water bath for 1 h.
- Add the reaction reagents in a 1.5 ml Eppendorf tubes as Table 3.
- Separate the digested PCR fragments or backbone vectors by a 1% DNA agarose gel electrophoresis.
- Retrieve the target bands from the gel with the AxyPrepTM DNA Gel Extraction Kit.
- Amplify STII-VL-CL-phoA-STII-VH-CH1 fragments with above-designed primers (Table 1) by PCR.
- Ligate the fragments onto the backbone vector and then transform them into E. coli.
- Add the reaction reagents in 200 μl PCR tubes as listed in Table 4.
Table 4. Ligation reaction recipes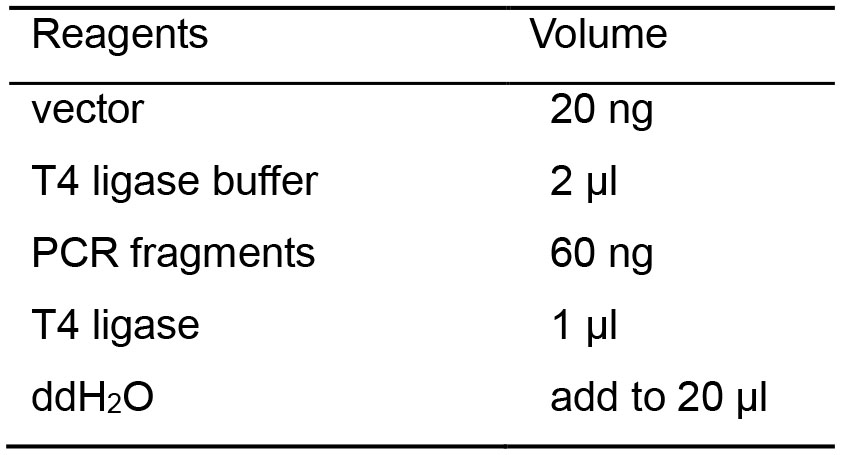
- Incubate the mixture in the thermal cycler at 16 °C for 30 min.
- Transform the E. coli strain DH5α with the ligated mixture following the procedures as described below.
- Place the E. coli competent cells stored at -80 °C on ice to thaw.
- Add 10 μl of ligated products into the melted E. coli competent cells, incubate on ice for 20 min.
- Heat the mixture in a preheated 42 °C water bath for 80 s, followed by another 2 min incubation on ice.
- Add 600 μl of fresh LB broth to the tube in a clean bench.
- Incubate the bacteria at 37 °C, 220 rpm for 40 min in a constant temperature shaker.
- Centrifuge the tube at 2,150 x g for 5 min, discard most of the supernatant, retain about 200 μl of the liquid, and re-suspend the bacteria in the clean bench.
- Then spread it on a pre-prepared LB plate (include 100 μg/ml kanamycin).
- Incubate the plate upside-down in a 37 °C incubator overnight (Placing the plate upside down can prevent condensation from dripping onto the medium, avoiding contamination, and facilitating the growth, reproducibility and colony counting), then verify the clones with correct insertion.
- Pick a single clone in 5 μl sterilized ddH2O for colony PCR verification using the primers listed in Table 1.
- Add the reaction system in 200 μl PCR tubes as Table 5.
Table 5. Colony PCR reaction recipes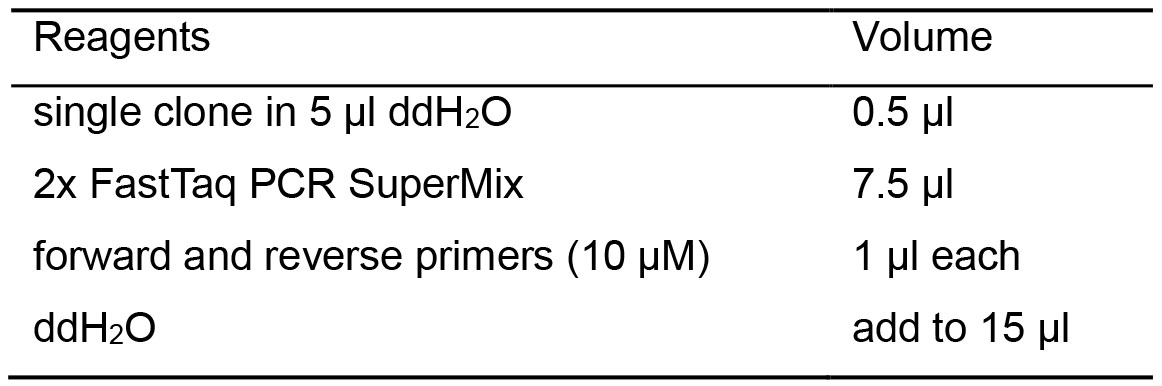
- Set the PCR program to be: denaturation at 95 °C for 10 s, annealing at 54 °C for 5 s and extension at 72 °C for 30 s and together for 35 cycles. The primers were listed in Table 1.
- Use 1% DNA agarose gel electrophoresis using TAE buffer (Recipe 4) to verify the PCR products.
- The remaining of the clones with positive PCR fragments were further cultured in LB broth for sequencing to confirm the correct insertion of desired Fab sequences.
- Confirm the correct insertion of STII-VL-CL-phoA-STII- VH-CH1 fragments by sequencing, and get the recombinant pPhoA-Fab plasmid as shown in Figure 1.
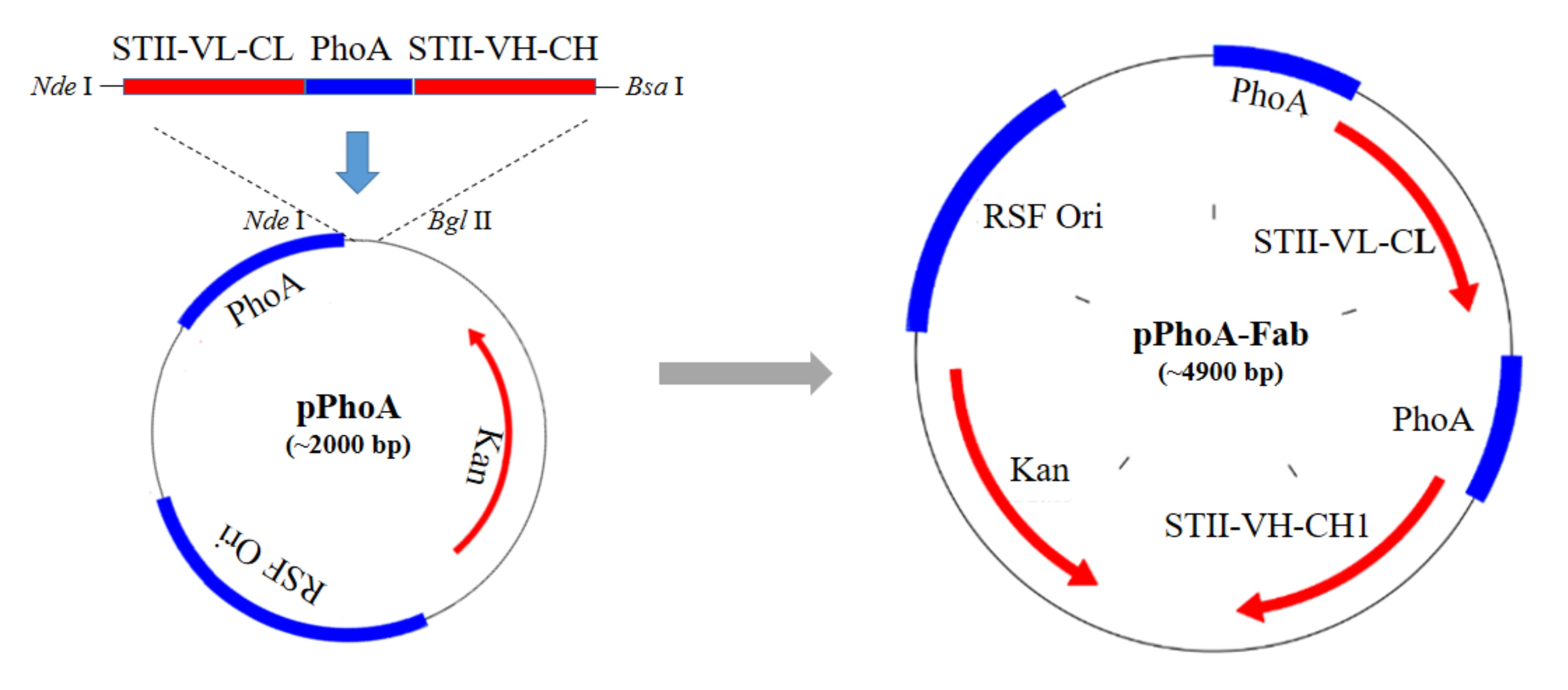
Figure 1. Construction process of pPhoA-Fab plasmid. STII-VL-CL-phoA-STII- VH-CH1 fragment was synthesized, PCR amplified, digested by Nde I and Bsa I, and ligated with backbone vector digested by Nde I and Bgl II, to obtain the recombinant plasmid pPhoA-Fab. - Culture the DH5α clone that sequenced to be with the correct insert overnight to extract recombinant plasmid pPhoA-Fab as shown in Figure 1 by the AxyPrepTM Plasmid Miniprep Kit.
- Using the constructed pPhoA-Fab to further transform BL21 (DE3) using the same heat shock procedures (Steps A6e to A6l), and the successfully transformed clone is used as the expression host strain for anti-VEGF Fab expression.
- Make the transformed bacteria DH5α and BL21 (DE3) aliquots, add glycerol to a final concentration of 20% and store at -80°C for future use.
- Add the reaction reagents in 200 μl PCR tubes as listed in Table 4.
- Extracellular expression of anti-VEGF Fab
- Take a vial of BL21 (DE3) containing pPhoA-Fab from -80 °C, put it on ice for 5 min.
- Transfer the thawed BL21 (DE3) to an LB agar plate (containing 100 μg/ml kanamycin) using inoculating loop, incubate at 37 °C overnight.
- Pick a single clone from the LB plate, and inoculate it into a tube containing 5 ml of LB broth (containing 100 μg/ml kanamycin).
Notes:- It takes about 12-14 h for preculture, so it’s best to carry out this step in the evening to ensure using fresh E.coli to inoculate.
- The ingredients of LB media of various brands are consistent, so any qualified brand of LB medium can be used.
- Culture the bacteria in the tube in a shaking incubator at 37 °C, 220 rpm for about 12-14 h. No more than 16 h.
- Inoculate 5 ml culture into 800 ml of fresh LB broth containing 100 μg/ml kanamycin.
Note: Use a 2 L Erlenmeyer flask. The LB medium volume should not occupy over 40% of the full capacity of the container. - Culture the bacteria at 37 °C, 220 rpm until OD600 reaches 0.8-1.0.
- Transfer the bacteria to a sterilized 250 ml centrifuge bottles to pellet the bacteria at room temperature by centrifugation at 4,000 x g for 10 min.
Note: This step should be performed in the clean bench. - Discard the supernatant, re-suspend the bacterial pellet with 800 ml sterilized PLM medium.
Note: PLM medium is chemically modified and contains no phosphate. This step should be performed in the clean bench. Here take 1 ml of culture for analysis, marked as ‘Pre’. - Incubate the bacteria in PLM medium overnight at a temperature of 20 °C with a shaking speed of 200 rpm for about 12-14 h. The absence of phosphate in PLM medium promotes protein induction.
- After induction, quickly raise the incubation temperature to 59 °C in the water bath and incubate for 1 h to improve Fab yields.
Note: This step can improve the extracellular yield for some level, it is also OK to skip it. - Then, quickly cool down the culture to room temperature in a water bath and centrifuge at 3,500 x g for 10 min to separate media and bacterial pellet, the expressed anti-VEGF Fab should mostly remain in the medium.
- Collect the culture medium and further centrifuge at 12,000 x g for 10 min to further remove the contaminants.
Note: Immediately before centrifugation, take 2 aliquots of culture for analysis, 1 ml for each, marked as ‘Ind’.
- Purification of anti-VEGF Fab
Notes:- The Capto L binds to kappa light chains of type I, III, and IV of human IgG but not type II.
- KappaSelect resin can be used to purify protein with a type II IgG kappa light chain. The purification process is similar to that of Capto L, but using PBS buffer of pH 7.4 as Buffer A, and 0.1 M glycine buffer of pH 2.7 as Buffer B.
- Filter the expression medium through a 0.22 μm filter for subsequent column chromatography purification steps.
- Prepare AKTA Avant 150 by washing the A, B pumps and tubes with 20% ethanol at 2 ml/min until the UV280 reaches baseline.
- Assemble the prepacked Capto L column with a column volume (CV) of 1 ml at a flow rate of 0.5 ml/min, and continue to wash the system with 20% ethanol at 2 ml/min until the UV 280 nm and Cond baselines are stable.
- Then, repeat the washing with ddH2O with the same procedure until the UV 280 nm and Cond baselines are stable.
- Place the B1 tube head in Buffer B and fill the tube with Buffer B.
- Place the A1 tube head in Buffer A, and equilibrate the system and column for 10-20 CV until the UV 280 nm and Cond reach baseline.
- Load the sample onto the column at a flow rate of 0.5 ml/min while collecting flow through.
Note: Collect 1 ml of flow through for analysis, marked as ‘FL’. - After loading, wash the column with Buffer A until the UV 280 nm reaches the baseline.
- Set the B pump solution ratio to 100%, elute the target protein by Buffer B with a flow rate of 1.0 ml/min, and collect the target protein when there are UV280 peaks.
- Immediately adjust the pH of the collected sample with 1 M Tris buffer (pH 9.0) to prevent protein inactivation.
Note: Take eluted protein samples for analysis, marked as ‘E’. - After the baseline of the elution is stable, flush the tubing with 10 to 20 CV ddH2O until the baselines of UV and Cond are stable.
- Then, rinse the tubing and column with 15 mM NaOH to clean the column. After the UV280 baseline is stabilized, wash the system, column, and tubes with ddH2O and 20% ethanol successively.
- Finally, fill the pipeline with 20% ethanol and disassemble the pre-packed column, which should also be kept in 20% ethanol.
- Test the collected protein by SDS-PAGE and Western blot (WB).
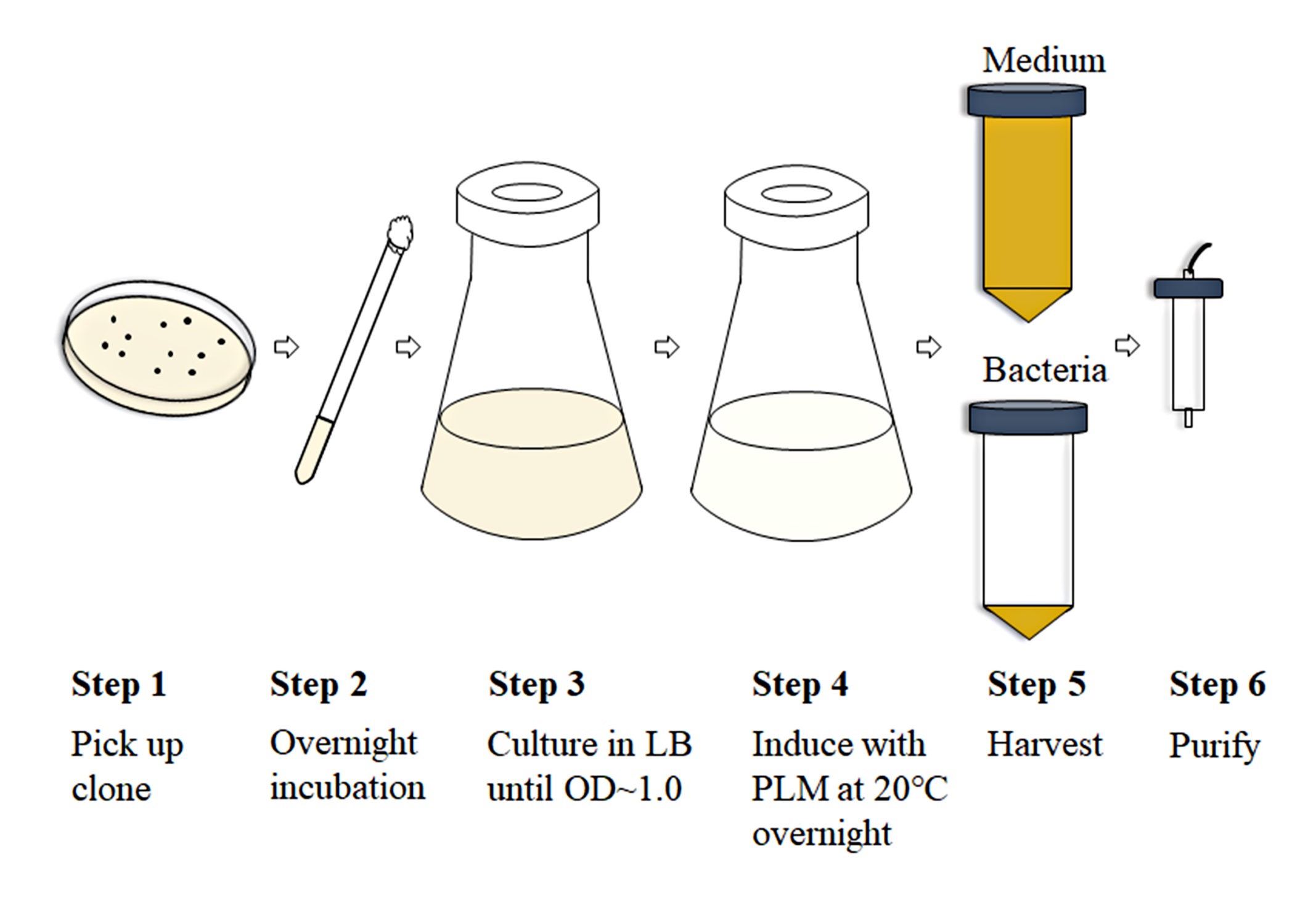
Figure 2. Scheme of the expression and purification processes for Fab production (Luo et al., 2019a)
Data analysis
- Take one aliquot of the collected sample ‘Ind’ and centrifuge at 3,500 x g for 10 min to separate medium (marked as ‘Medi’), and use B-PER to incubate the pellet for 15 min at room temperature to release the intracellular soluble proteins.
- Centrifuge at 12,000 x g for 30 min at 4 °C to separate intracellular soluble (marked as ‘Sup’) and insoluble (marked as ‘IB’) fractions.
- To investigate the distribution of anti-VEGF Fab expression, analyze all samples Pre, Ind, Medi, Sup, and IB by SDS-PAGE (Figure 3A) and WB (Figure 3B). Use a goat anti-human kappa light chains antibody as the capture antibody, and rabbit anti-goat IgG antibody conjugated with HRP as the second antibody.
- The purification process (Figure 4A) can be analyzed by SDS-PAGE of samples Medi, FL, and E (Figure 4B).
- For the purity estimation of the Fab antibody, the product can be determined by size exclusion high performance liquid chromatography (SEC-HPLC) using TSK G2000SWXL column (5 μm, 0.78 x 300 mm) at an Agilent 1260 HPLC system. The purity of each Fab can be calculated as the area percentage of the corresponding peak detected.
- The molecular weight of the Fab can be accurately measured using an ultraperformance liquid chromatography-quadrupole time-of-flight mass spectrometer (MALDI-TOF MS).
- The affinity between the Fab and the specific antigen can be tested using the ForteBio Octet RED96e system.
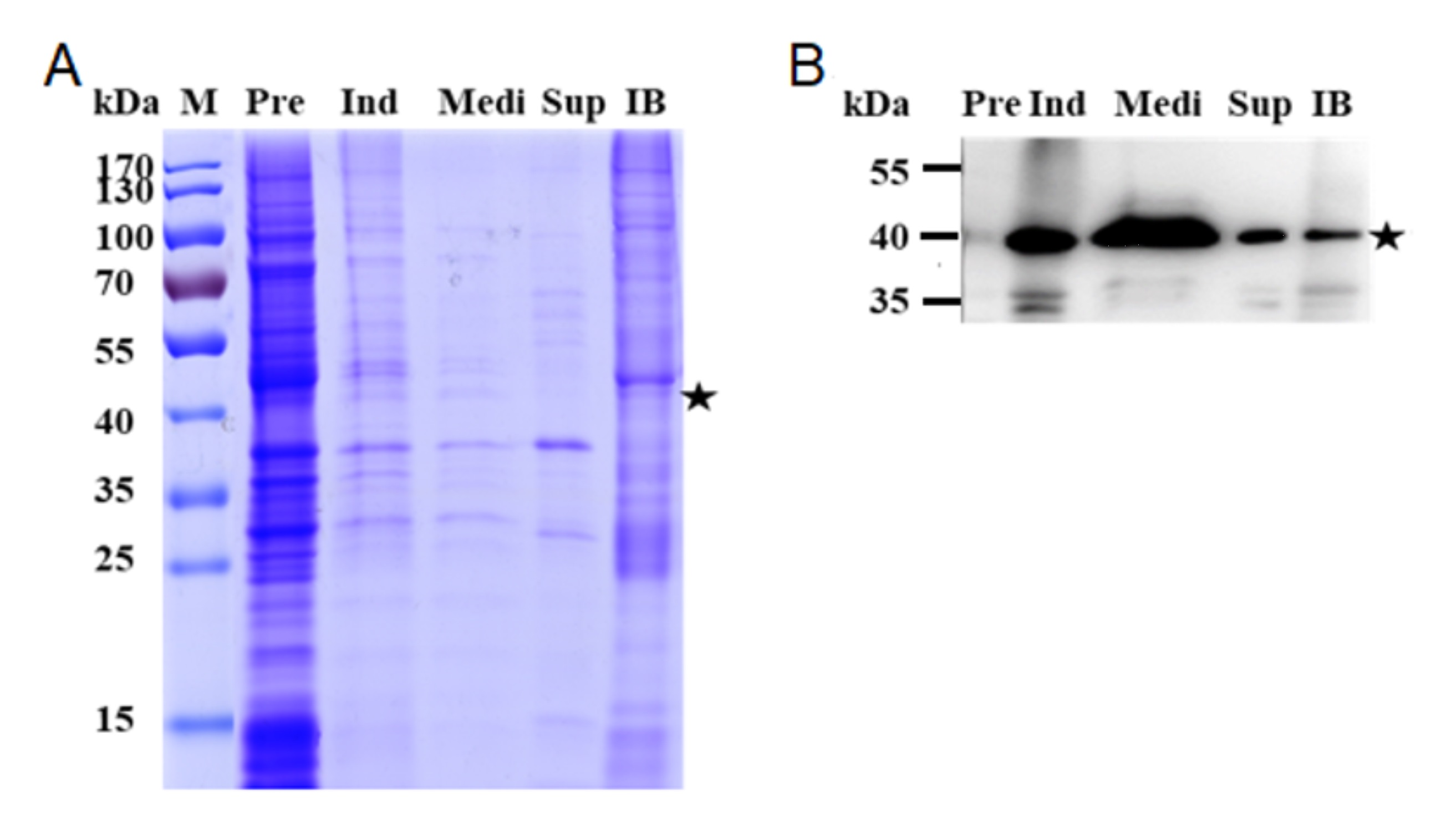
Figure 3. Expression of anti-VEGF Fab in E. coli. A. SDS-PAGE; B. Western blot. Most of anti-VEGF Fab was secreted into the culture medium. Lane Pre: bacteria before induction; Ind: bacteria after induction; Medi: Medium; Sup: intracellular soluble fraction; IB: Inclusion bodies. The samples charged in the same volume and all samples were non-reducing. The star (★) indicates the predicted size of Fab at about 40 kDa (Luo et al., 2019b).
The samples charged in the same volume and all samples were non-reducing. The star (★) indicates the predicted size of Fab at about 40 kDa (Luo et al., 2019b).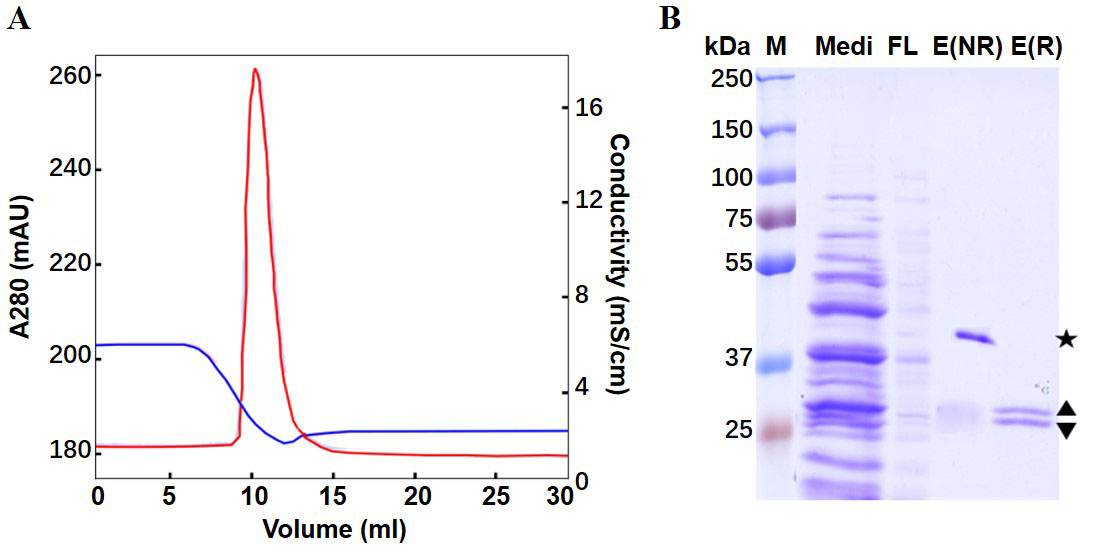
Figure 4. Purification of anti-VEGF Fab by Capto L affinity chromatography. A. Eluted chromatography spectrum. Red curve: UV absorbance at 280 nm; Blue curve: Conductivity. B. SDS-PAGE. Lane Medi: Pre-loading sample (medium); FL: flow through; E(NR): non-reducing elutant; E(R): reduced elutant. The star (★) indicates the non-reducing Fab; the diamond (◆) indicates the educed fragments of LC(▲) and HC(▲) (Luo et al., 2019b).
Recipes
- PLM medium
7.5 mM (NH4)2SO4
0.4 mM MgSO4
11 mM glucose
111 mM PIPES
Adjust to pH 7.0
Note: PLM medium should be steam-sterilized or filter-sterilized immediately after preparation. - Buffer A
50 mM citric acid
50 mM sodium citrate
Adjust to pH 6.5
Note: The buffer should be filtered through a 0.22 μm filter and used within one week. - Buffer B
50 mM citric acid
50 mM sodium citrate
Adjust to pH 2.3
Note: The buffer should be filtered through a 0.22 μm filter and used within one week. - TAE buffer
40 mM Tris-acetate, pH 8.5
1 mM EDTA
Acknowledgments
This work was in collaboration with Jecho Labs, and a US patent was applied jointly (CAGLIERO, Cedric; BURNETTE, Andrew; XIE, Yueqing; JIANG, Hua; ZHU, Jianwei; LU, Huili; LUO, Manyu. Method for Preparing Recombinant Protein from Bacterium and Composition Containing the Same. United States Patent Application, Date: Dec. 31, 2018, Application No. 16/237265). The work was also supported in part by the National Natural Science Foundation of China (No. 81773621 to Zhu J.), the Science and Technology Commission of Shanghai Municipality (No. 17431904500 & 17ZR1413700 to Lu H.). We thank all the authors of our original research article (Luo et al., 2019a Appl Microbiol Biotechnol).
Competing interests
The authors declare no conflicts of interest with the contents of this article.
References
- Augustine, R. C., York, S. L., Rytz, T. C. and Vierstra, R. D. (2016). Defining the SUMO system in maize: SUMOylation is Up-Regulated during endosperm development and rapidly induced by stress. Plant Physiol 171(3): 2191-2210.
- Danyliv, A., Glanville, J., McCool, R., Ferreira, A., Skelly, A. and Jacob, R. P. (2017). The clinical effectiveness of ranibizumab treat and extend regimen in nAMD: systematic review and network Meta-Analysis. Adv Ther 34(3): 611-619.
- Ellis, M., Patel, P., Edon, M., Ramage, W., Dickinson, R. and Humphreys, D. P. (2017). Development of a high yielding E. coli periplasmic expression system for the production of humanized Fab' fragments. Biotechnol Prog 33(1): 212-220.
- Gupta, S. K. and Shukla, P. (2017). Microbial platform technology for recombinant antibody fragment production: A review. Crit Rev Microbiol 43(1): 31-42.
- Jalalirad, R (2013). Production of antibody fragment (Fab) throughout Escherichia coli fed-batch fermentation process: Changes in titre, location and form of product. Electron J Biotechn 16(3). DOI: 10.2225/vol16-issue3-fulltext-15.
- Lobstein, J., Emrich, C. A., Jeans, C., Faulkner, M., Riggs, P. and Berkmen, M. (2012). SHuffle, a novel Escherichia coli protein expression strain capable of correctly folding disulfide bonded proteins in its cytoplasm. Microb Cell Fact 11: 56.
- Luo, M., Zhao, M., Cagliero, C., Jiang, H., Xie, Y., Zhu, J., Yang, H., Zhang, M., Zheng, Y., Yuan, Y., Du, Z. and Lu, H. (2019a). A general platform for efficient extracellular expression and purification of Fab from Escherichia coli. Appl Microbiol Biotechnol 103(8): 3341-3353.
- Luo, M., Gao, Y., Zhu, J. and Lu, H. (2019b). Extracellular expression and purification of ranibizumab in E. coli and its structural characterization. Chinese Journal of Pharmaceuticals . Submitted.
- Nelson, A. L. and Reichert, J. M. (2009). Development trends for therapeutic antibody fragments. Nat Biotechnol 27(4): 331-337.
- Panda, A. K. (2003). Bioprocessing of therapeutic proteins from the inclusion bodies of Escherichia coli. Adv Biochem Eng Biotechnol 85: 43-93.
- Rezaie, F., Davami, F., Mansouri, K., Agha Amiri, S., Fazel, R., Mahdian, R., Davoudi, N., Enayati, S., Azizi, M. and Khalaj, V. (2017). Cytosolic expression of functional Fab fragments in Escherichia coli using a novel combination of dual SUMO expression cassette and EnBase((R)) cultivation mode. J Appl Microbiol 123(1): 134-144.
- Ulmer, N., Ristanovic, D. and Morbidelli, M. (2019). Process for Continuous Fab Production by Digestion of IgG. Biotechnol J: e1800677.
- Walsh, G. and Jefferis, R. (2006). Post-translational modifications in the context of therapeutic proteins. Nat Biotechnol 24(10): 1241-1252.
- Wang, Y. G., Ding, H. Z., Du, P., Gan, R. B. and Ye, Q. (2005). Production of phoA promoter-controlled human epidermal growth factor in fed-batch cultures of Escherichia coli YK537 (pAET-8). Process Biochem 40(9): 3068-3074.
- Zhou, Y., Lu, Z., Wang, X., Selvaraj, J. N. and Zhang, G. (2018). Genetic engineering modification and fermentation optimization for extracellular production of recombinant proteins using Escherichia coli. Appl Microbiol Biotechnol 102(4): 1545-1556.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Wang, Z., Gao, Y., Luo, M., Cagliero, C., Jiang, H., Xie, Y., Zhu, J. and Lu, H. (2019). A PhoA-STII Based Method for Efficient Extracellular Secretion and Purification of Fab from Escherichia coli. Bio-protocol 9(18): e3370. DOI: 10.21769/BioProtoc.3370.
Category
Molecular Biology > Protein > Expression
Biochemistry > Protein > Isolation and purification
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.