- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Supported Cell Membrane Sheets to Monitor Protein Assembly
Published: Vol 9, Iss 18, Sep 20, 2019 DOI: 10.21769/BioProtoc.3368 Views: 5628
Reviewed by: David PaulYosuke SenjuAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jan 2019

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Studying protein-protein and protein-lipid interactions in their native environment is highly desirable, yet, the heterogeneity and complexity of cellular systems limits the repertoire of experimental methods available. In cells, interactions are often taking place in confined microenvironments where factors such as avidity, hindered diffusion, reduced dimensionality, crowding etc. strongly influence the binding kinetics and therefore it can be problematic to equate binding affinities obtained by bulk in-solution methods (e.g., Fluorescence Polarization, Isothermal titration calorimetry, Microscale thermophoresis) with those occurring in real cellular environments. The Supported Cell Membrane Sheet method presented here, addresses these issues by allowing access to the inner leaflet of the apical plasma membrane. The method is a highly versatile, near-native platform for both qualitative and quantitative studies of protein-protein and protein-lipid interactions occurring directly in or on the plasma membrane.
Keywords: Supported cell membrane sheetsBackground
Proper evaluation of the binding strength between biomolecules is pivotal for understanding cellular pathways and for rational drug development. The general understanding of both the dynamics and kinetics of biomolecular interactions often relies on experimental approaches where single protein domains of interest are isolated from their native cellular environment and tested under conditions that may deviate significantly from the specific cellular compartment in which they actually occur. In cells, various microenvironments containing a wealth of different proteins, protein-polymers, and lipid membranes with diverse properties and shapes, can contribute to anisotropic concentrations, reduced dimensionality and diffusion rates, all of which greatly affect the binding strength.
Supported Cell Membrane Sheets (SCMS) are ideal for studying interactions occurring on (or in) the native cytoplasmic cell membrane. SCMS are prepared from cultured adherent cells by firmly attaching a cover glass on top of cultured cells and subsequently separating the apical plasma membrane from the cells by lifting the cover glass. This exposes the inner leaflet of the plasma membrane for binding of fluorescently labeled ligands which can then be studied by microscopy (Figure 1).
The method was originally developed by Perez et al. (2006), to qualitatively investigate the mobility of GPCRs and their association to G-proteins, and has also been used for both FRET based stoichiometry (Miles et al., 2013) and super resolution microscopy based cluster analysis (Scarselli et al., 2012). A related study by Roizard et al. (2011) also managed to produce supported cell membrane sheets on porous beads (~80 μm in diameter) leaving both the intracellular and the solvent exposed extracellular side of the membrane accessible.
Here, we have adapted the SCMS method for also measuring protein-protein binding affinities, which is particularly useful in cases where one or more interaction partners are membrane bound or embedded. The method is highly applicable for studying scaffolding protein where several interaction domains in one protein, formation of higher order structures and direct interaction with the lipid membrane may induce significant avidity effects improving the effective binding strengths and change kinetics by several orders of magnitude. Please use Erlendsson et al. (2019) as a practical and theoretical case study.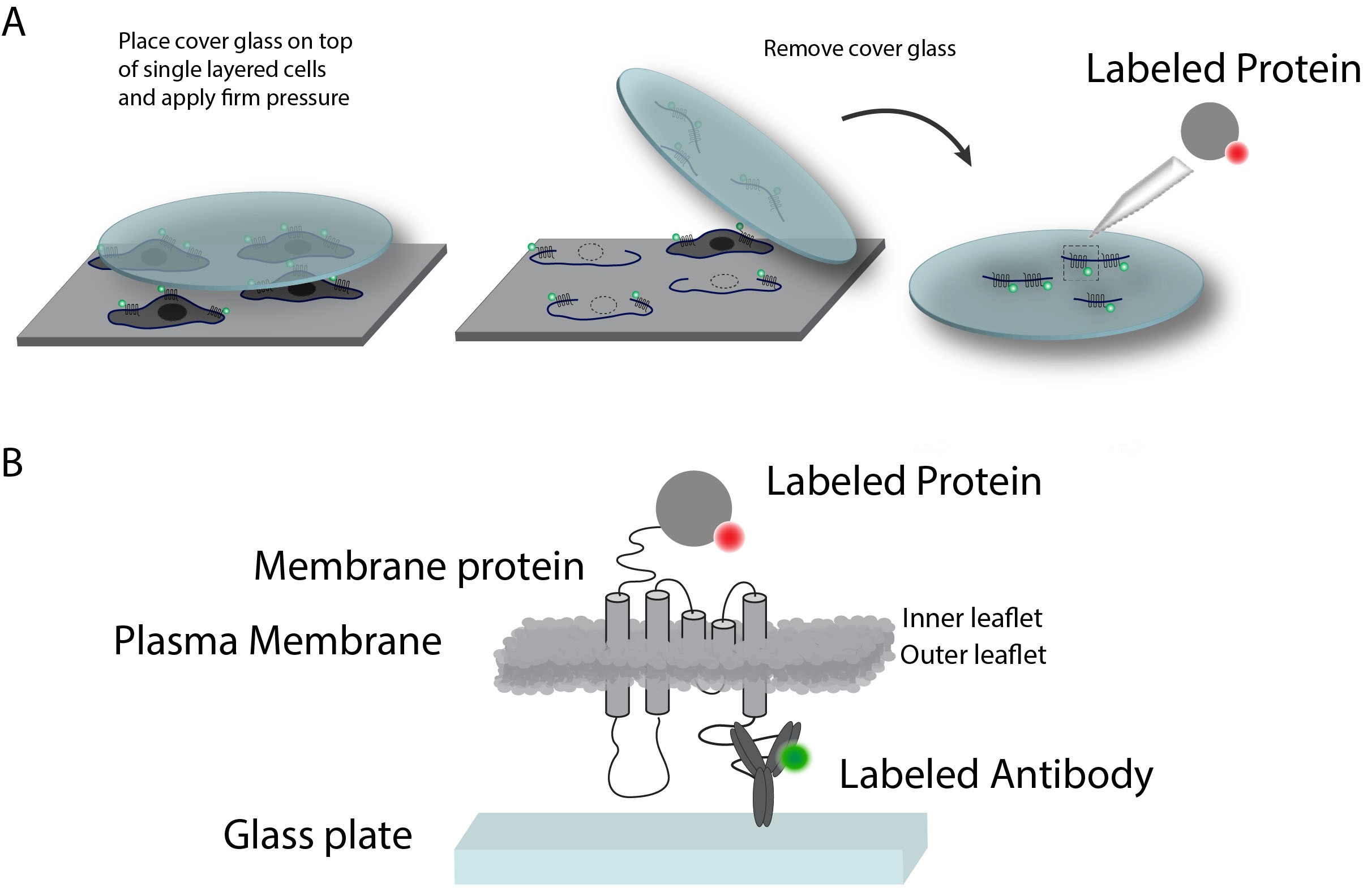
Figure 1. General principle for preparation of Supported Cell Membrane Sheets. A. SCMS are prepared from transfected HEK293 Grip Tite cells. The membrane protein is labeled with Alexa Fluor 488 in this case (green). Pressing a coated glass coverslip onto the cells and subsequently removing it, will detach the apical plasma membrane from the remains of the cells, and leave the inner leaflet exposed for binding experiments. B. A closer schematic view of the principle. The Antibody labeling can be replaced with a fluorescent protein. Figures are adapted from Erlendsson et al. (2019).
Materials and Reagents
Note: Listed materials and reagents with specific catalog numbers are generic and can be replaced with similar products from other vendors. Materials and reagents marked by an asterisk (*) should preferably not be replaced with other brands or vendors.
- P1000 pipette
- Object Glass (various vendors)
- Forceps for picking up Cover glass (various vendors) (see Figure 2A)
- Ultracentrifuge Beckman Coultier Optima or similar
- Harric plasma cleaner (or similar) suitable for cover glass slides
- 400 ml glass beaker
- Empty PD-10 column (GE Healthcare, catalog number: 17085101)
- *Cover Glass Round 25 mm, thickness #1, (VWR, catalog number: 631-1346)
- Cell culture flasks surface area 25 cm2, canted neck, cap (vented) (Corning, Sigma-Aldrich, catalog number: CLS430639)
- 1.5 ml Eppendorf tubes
- Cell culture plates 6-well flat bottom (various vendors, Polystyrene, Sterile)
- 50 ml Falcon tubes (various vendors)
- 15 ml Falcon tubes
- Whatman paper
- Parafilm 10 cm wide (various vendors)
- Syringe Needle, bend the tip (!) for picking up cover glass (various vendors) (see Figure 2A)
- 10 ml syringe top (flat plunger) (see Figure 2A)
- Square BioAssay Dish (Nunc, Thermo Fischer, catalog number: 240835) (see Figure 2A)
- Soft rubber pipettes
- Constructs (Please see Erlendsson et al. 2019) for examples:
- FLAG-tagged plasma membrane-localized receptor or YFP tagged protein (no labeling necessary in that case)
- SNAP-tagged scaffolding protein
- *ANTI-FLAG® M1 antibody (Millipore, Sigma-Aldrich, catalog number: F3040)
- *HEK 293 Grip Tite cells (Thermo Fisher, catalog number: A14150)
- KCl (Sigma-Aldrich, catalog number: P9333)
- MgCl2 (Sigma-Aldrich, catalog number: M8266)
- CaCl2 (Sigma-Aldrich, catalog number: C1016)
- HEPES (Sigma-Aldrich, catalog number: H3375)
- Glucose (Sigma-Aldrich, catalog number: G8270)
- Bovine Serum Albumin (Sigma-Aldrich, catalog number: A2153)
- Trizma-base (Sigma-Aldrich, catalog number: T1503)
- NaCl (Sigma-Aldrich, catalog number: S7653)
- NaHCO3 (Sigma-Aldrich, catalog number: S8875)
- 1,4-Dithiothreitol (Sigma-Aldrich, catalog number: DTT-RO)
- Paraformaldehyde Crystalline (Sigma-Aldrich, catalog number: P6148)
- *ProLongTM Gold Antifade Mountant (ThermoFisher, catalog number: P36930)
- poly-L-ornithine hydrobromide (Sigma-Aldrich, catalog number: P-8638)
- *Alexa FluorTM 488 NHS Ester (Succinimidyl Ester) (Invitrogen, ThermoFisher, catalog number: A20000)
- *SNAP-surface dye 568/647 (New England Biolabs, catalog numbers: S9112S, S9137)
- D-MEM (Gibco, Sigma-Aldrich, catalog number: 11965-092)
- Fetal Bovine Serum (Gibco, catalog number: 16000044)
- Opti-MEM Reduced Serum Medium (Gibco, Sigma-Aldrich, catalog number: 31985062)
- LipofectamineTM 2000 Transfection Reagent (Invitrogen, Thermo Fischer, catalog number: 11668027)
- Dulbecco’s Phosphate Buffered Saline (Sigma-Aldrich, catalog number: D8537)
- GibcoTM Trypsin-EDTA (0.25%), Phenol red (Gibco, catalog number: 25200056)
- Geneticin Selective Antibiotic (G418 Sulfate) (Gibco, catalog number: 10131027)
- Penicillin-Streptomycin (Roche, catalog number: 11074440001)
- Trypan Blue solution (Sigma-Aldrich, catalog number: T8154)
- Cell Culture Contamination Detection Kit (Invitrogen, catalog number: C7028)
- TBS Buffer (see Recipes)
- DMEM media (see Recipes)
- Sheet Buffer (see Recipes)
Equipment
- Invitrogen CountessTM Automated Cell Counter (or similar)
- CountessTM Cell Counting Chamber Slides (Invitrogen, catalog number: C10228)
- NAP-5 columns (GE Healthcare, Sigma-Aldrich, catalog number: GE17-0853-01)
- NanoDrop or similar for measuring protein concentration and degree of labeling
- Suction or large syringe for quickly removing media and buffers
- TECAN M200 Infinite Pro
- Confocal laser-scanning microscope (Zeiss, model: LSM510)
We used, LSM 510, Carl Zeiss with a 63x/1.3 oil immersion objective or similar:- 488 nm laser line from an argon-krypton laser, and a 505-550 nm bandpass filter
- 568 was excited at 543 nm with a helium-neon laser, detected using a 585 nm long-pass filter
- 647 was excited at 642 nm with a helium-neon laser, detected using a 650 nm long-pass filter
Software
- Microsoft Excel (Microsoft)
- ImageJ (NIH, USA; RRID:SCR_003070)
- GraphPad Prism (GraphPad Software Inc., San Diego, USA; RRID:SCR_002798)
Procedure
- Labeling
A1. Protein labeling
The protein used for the binding studies below should be pure and stable at concentrations 10x above the expected KD for at least 1 day at 10 °C after purification. In our experiments, we purify and label a new batch of protein for each independent experimental repetition. As maleimide or NHS labeling might interfere with membrane binding, we suggest using SNAP-tagged constructs for the initial binding studies. We use an N-terminal GST-SNAP-tag with a thrombin cleavage site between the GST and SNAP tag.- For labeling of GST tagged protein (concentration should be 2-4 mg/ml and volume around 1 ml) bound to roughly 500 μl Glutathione-sepharose bead slurry (Total volume ~1500 μl), add SNAP-surface dye (2.5 mM stock in DMSO–use concentrations of 10-20 μM final concentration for labeling) and incubate for two hours at 4 °C. The beads/protein/dye slurry can be contained in a PD-10 column.
- After labeling, excess dye is removed by simply washing the labeled protein beads on the column. Use 5-10 bead volumes TBS buffer (or similar) for complete removal of free dye. Leave in at least 3 bead volumes of TBS buffer (or similar).
- You can now add your protease (in our case thrombin ~5 U) to the stained protein (~2-4 mg) still attached on the beads and leave at gentle agitation at 4 °C until cleaved–overnight (10-16 h are usually sufficient for complete cleavage).
- Elute the cleaved and stained protein from the beads using gravity flow, and centrifuge samples at 100,000 x g for 1 h to remove all potential aggregates. When the centrifugation of the protein is done supernatants are immediately moved to clean tubes on ice for determination of protein concentration and labeling degree.
Primary dye conjugation of antibodies is performed using Invitrogen Alexa FluorTM NHS Ester (Succinimidyl Ester) dyes and monoclonal ANTI-FLAG® M1 antibody produced in mouse. In principle the dyes and antibody (if you have other tags on your protein) can be fitted to your needs and microscope setup).- Dilute antibody stock to 1 μg/μl in a total volume of 100 μl–use PBS buffer (no Tris or other buffer containing free amines).
- Add 10 μl 1 M NaHCO3 to ensure alkaline reaction conditions.
- Dissolve ~200 μg dye (standard aliquot amount from manufacturer) in the 110 μl alkaline antibody solution, then wrap tube in Alufoil and incubate at 4 °C under slow agitation overnight.
- Next day, equilibrate Nap-5 gel filtration columns using PBS buffer (three column volumes).
- Bring the reaction volume up to 200 μl with PBS (add 90 μl) and gently mix.
- Add the entire volume to the center of the column.
- Allow the sample to enter the column and after the last drip, add 550 μl PBS to wash.
- Add 300 μl PBS and collect the eluate into 1.5 ml micro-centrifuge tubes by drop counting (3-4 drops per tube).
A3. Degree of labeling
Determine protein concentration and degree of labeling of all constructs. We use a TECAN M200 Infinite Pro instrument for this. The degree of labeling is determined as follows:
First, calculate the protein concentration considering the contribution from the fluorophore: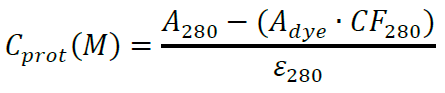
Then calculate the moles of dye per mole protein: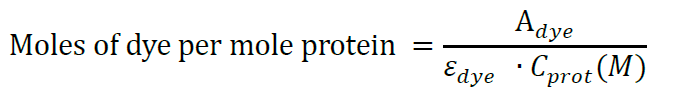
A280 and Adye represent the sample absorbance at 280 nm and absorbance at the dye excitation wavelength, respectively, e280 and edye are the extinction coefficients of the protein at 280 nm and of the dye at the appropriate wavelength, respectively. CF280 is the correction factor indicating how much the dye contributes to the Absorbance at 280 nm. For Alexa Flour 488 labeled antibodies use e280 = 203,000 and CF280 = 0.11. For SNAP-surface 549 (DY-549) use edye = 150,000 and CF280 = 0.08. For SNAP-surface 647 (Alexa Fluor 647) use edye = 239,000 and CF280 = 0.03. If other dyes are found more suitable for your system, use extinction coefficients and correction factors provided by the dye vendor. In our experiments, we aim for labeling efficiencies 5 moles of dye per mole protein for the antibody and above 0.7 moles of dye per mole protein for the protein. - Cell cultures
HEK293 Grip Tite 293 cells should be maintained at 37 °C under 5% CO2 in sterile T25 cm2 flasks. Cells should be regularly split 1/10 Monday and Friday and never reach more than 90% confluency. If you are using different-sized flasks, scale the reagent volumes up or down accordingly. Cell cultures should be routinely tested for mycoplasma contamination by using Cell Culture Contamination Detection Kit (Invitrogen), or similar.- Remove media from the flask. Rinse the cells with 5 ml of Dulbecco’s Phosphate-Buffered Saline (D-PBS) without Ca2+ or Mg2+.
- Add 1.5 ml of pre-warmed, 0.25% trypsin-1 mM EDTA solution to the flask.
- Incubate at 37 °C until cells have detached (about 2 min). Check the cells under a microscope and confirm that most of the cells have detached. If cells are still attached, incubate a little longer until most of the cells have detached.
- Add 8.5 ml pre-warmed D-MEM medium (see materials) containing 10% FBS and penicillin-streptomycin. Carefully pipette cells suspension up and down to break cell clumps.
- For cell passage add 1.5 ml cell suspension to 13.5 ml fresh pre-warmed D-MEM medium containing 10% FBS and Penicillin-Streptomycin to a new T25 cm2 flask. Add 600 µg/ml Geneticin and place in an incubator.
- For transfections mix 10 μl cell suspension with 10 μl Trypan Blue and add 10 μl to the cell counting chamber and count cells. Add 4 x 106 cells in a total volume of 15 ml fresh pre-warmed D-MEM medium containing 10% FBS and penicillin-streptomycin to a new T25 cm2 flask. Add 600 µg/ml Geneticin and incubate overnight.
- The following day prepare transfections by adding 10 μl Lipofectamine to 1.5 ml Opti-MEM medium in a 15 ml Falcon tube and shake for 2 s. In another tube add 3 μg of vector DNA containing the FLAG-tagged construct to 1.5 ml Opti-MEM and flip a couple of times. Let both stand for 30 min. After 5 min, mix and incubate for additional 30 min.
- Remove media from T25 flasks containing cells for transfection. Wash with D-PBS without Ca2+ or Mg2+ and add 1 ml Opti-MEM.
- Add transfections mixtures to the T25 flasks and incubate overnight. The flask should contain 4 ml total (1 ml from Step 8 and 3 ml from Step 7).
- Next day repeat Steps 1-4 but this time dilute transfected cells to 4 x 105 cells/ml.
- Add 1 ml prewarmed D-MEM medium to each well in a six-well plate and add 4 x 105 cells to each well (2 x 105 cells in 2 ml per well) incubate overnight for cells to attach. Cells should be flattened out and between 40% and 60% confluent.
- For labeling of FLAG-tagged plasma membrane expressed target, remove 1 ml of medium from each cell well leaving 1 ml. Add 1 μg/ml of Alexa Fluor 488 labeled ANTI FLAG M1 antibody (see A2) to each well and keep on ice for an hour.
- Supported membrane sheets
C1. Preparations for making inverted membrane sheets- Initiate preparation of cover glass by cleansing for 20 min at max. power in a Harric plasma cleaner (or similar) and then coat in 1-2 ml 0.3 mM poly-L-ornithine hydrobromide for 30 min followed by washing twice in water: Eight glasses are cleaned at a time by placing these on object glasses. Immediately after cleaning the glasses are transferred to six well plate wells and covered with 0.3 mM poly-L-ornithine hydrobromide. After 30 min, the poly-L-ornithine hydrobromide solution is removed and cover glasses are washed once in approximately 2 ml water in the well and finally 1 ml water is added to keep glasses covered.
- Typically, 24 glasses are prepared. Make a few more than needed–some will break.
- Prepare ‘Sheet buffer’ (see Recipes) by adding 50 mg of fresh BSA to 50 ml buffer in a 50 ml Falcon tube and keep it on ice. Also, put 2 x 50 ml of MilliQ water on ice.
- Prepare tissue paper box by wetting Whatman paper and cover it with parafilm. Make grids and write labels with a permanent marker. Do not use a red marker as this will dissolve during PFA fixation below (Figure 2).
- Put cells M1 labeled cells on ice and start ultracentrifugation of protein preps (see B12).
- Fluorescently labeled protein.
- Cold M1 labeled cells (with labeled M1 still on).
- Cleaned and poly-L-ornithine hydrobromide treated cover glasses in water approx. 24 pieces depending on desired curves and sample points.
- BioAssay dish with moistened Whatman paper and marked up parafilm (Figure 2A).
- 50 ml cold sheet buffer with 1 mg/ml BSA.
- 2x 50 ml cold water.
- In addition, you need tweezers, flat syringe pistil and bend needle (Figure 2A). Also, you need a P1000 pipette, a clock and suction.
- At this point also put ~700 μl per sheet 4% PFA and 100 ml PBS on ice for later use.
To prepare sheets, do as follows for each well with cells:- Aspirate medium containing M1 antibody (Figure 2B).
- Add approximately 2 ml of cold water to wash cells and start clock (Figure 2B).
- Replace water with clean water to allow osmotic swell.
- After 60 s, remove water, pick up cover glass and put on top of the cells with poly-L-ornithine hydrobromide treated surface facing the cells (Figures 2D-2E). Now press hard on the cover glass with the pistil (Figure 2F). Apply constant pressure while moving around the pistil on the glass for 1 min. It might take a few tries to get the pressure right without breaking the cover glass. To get the pressure right, make an initial series in which the applied pressure is gradually increased and assess the number of sheets using the confocal microscope (see below).
The pressure required is more than you think. An alternative way to produce the inverted membrane sheets is to firmly press down the cover glass on the cells for 10 s and then remove the water completely. Make sure the cover glass is fully attached to the cells in the bottom of the well. Then wait for 120 s–the air pressure/surface tension between the cover glass and the well surface will also be sufficient to produce inverted membrane sheets. - Next, again use the bended needle and tweezers to pick up the cover glass and transfer it to the BioAssay dish with the surface containing the sheets facing upwards. Be patient, it might take a few tries to learn how to pick up the cover glass with the needle. If sufficient pressure has been applied the cover glass will be sticking quite well to the cell surface.
- Immediately after transfer, cover the sheets with approximately 700 μl sheet buffer with BSA to prevent drying and to coat surface with BSA (Figure 2G).
- Repeat process for all sheet preps.
- Leave last prepared sheets in the cold and dark for 20 min to allow BSA to coat the sheets.
- Meanwhile, prepare protein dilutions as calculated above by diluting protein in sheet buffer. Initial dilution in protein buffer is done for the lower conc. Prepare 1 ml per condition.
- Aspirate sheet buffer from sheets and replace it with protein containing sheet buffer. Preferably approx. 700-1,000 μl, no less than 500 μl. Put on plastic cell dishes as lids in addition to box lids and incubate for typically 2 h or more for concentration series and leave at desired temperature in the dark.
- After incubation, remove buffer and wash sheets on the parafilm surface with 1x 700 μl sheet buffer with BSA and two times with cold PBS (no incubation in wash buffer, just start removal in the same order after the last sheet has been covered). Use soft rubber pipettes.
- Next, fix sheets for 40 min in 700 μl 4% cold PFA and in the dark. At this point take out antifade mounting media to allow this to reach room temp.
- After fixation, wash sheets three times in PBS and leave in the last wash. For a few coverslips at the time, remove PBS and wash glasses in a beaker containing ~400 ml water. Remove drops from edge using paper towels and mount glasses on the object glass on a drop of antifade (Figure 2H). Excess antifade can be sucked away. Preps. are kept in the dark at room temperature. After drying they should be stored at 4 °C.

Figure 2. Workstation and workflow. A. Work station overview. If your protein is unstable at room temperature or you wish to probe binding at lower (or higher) temperature, you can place the BioAssay Dish in ice or any suitable heating device. The Insert shows the needle with the bent tip for picking up the cover glasses. B-I. Work flow for producing SCMS–detailed description in the main text.
- Confocal Imaging
The confocal microscope, filter cubes, cameras, lasers etc. available to you might differ from our setup. If in doubt, please consult your local expert microscopist. Below we have briefly described the setup used for the Inverted membrane sheets produced for analyses in Erlendsson et al. (2019).- Confocal microscope: Zeiss LSM 510 confocal laser-scanning microscope equipped with an oil immersion numerical aperture 1.4 63x objective (Zeiss, Jena, Germany).
- The Alexa Fluor 488 dye conjugated antibody and YFP were excited with the 488 nm laser line from an argon-krypton laser, and the emitted light was detected using a 505-550 nm bandpass filter.
- 547 SNAP-surface labeled species were excited at 543 nm with a helium-neon laser, and the emitted light was detected using a 560-615 nm band pass filter. The Alexa Fluor 647 was excited at 633 nm with another helium-neon laser, and the emitted light was detected using a 650 nm long pass filter.
- The samples will contain mostly single layer sheets but also undisrupted cell and cell debris. The membrane sheets can be differentiated from cells and debris by being very flat and thin. Cells and debris are often visible in the direct transmission, whereas single layer membrane sheets are not (Figure 3A).
Data analysis
Resulting images are then analyzed using ImageJ and Excel (or similar). Please see Video 1. We recommend using Graphpad Prism (or similar) for subsequent plotting and fitting.
- Make sure Images from the different channels are perfectly aligned. There are microscope specific procedures for this–please consult your local expert microscopist if in doubt. The images should be stacked to ease the analysis.
 Video 1. Quantification of binding using ImageJ and Excel
Video 1. Quantification of binding using ImageJ and Excel - When a flat membrane sheet is found, define a region of interest (ROI) and use the measure tool (in ImageJ) to measure the mean intensity of the various channels. Also, measure the background intensity for all channels for all images. Areas with oversaturated pixels or mean intensities below background + 2σ (the background intensity + two times the standard deviation of the background intensity) should be discarded (Figure 3A).
- For binding curves measure only one ROI per cell from 8-10 cells for each condition.
- Next, the intensities need to be corrected for degree of labeling and in some cases laser gain settings. Correction of measured channel intensities can be calculated using the following equation:
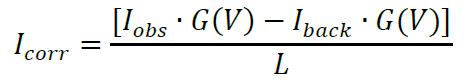
where, Iobs is mean intensity of sheet (Imembrane protein in Figure 3A) or ligand (Ilabeled protein in Figure 3A and Video 1), Iback is mean intensity of background and L is the moles of dye per mole of protein or receptor (see section A3). We do not recommend changing PMT gain settings between independent n’s, however, if necessary, this can be corrected by multiplying by G(V) (will be 1 if gain settings are similar). G(V) is calculated as follows: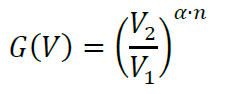
where α is Conductance of dynodes of PMT and n the number of dynodes inside the PMT. α and n are specific for the PMTs on your microscopes. - Finally, the fractional binding can calculate using:
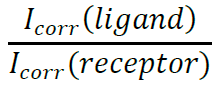
All binding curves for direct comparison should be performed in parallel, and in presence of positive and negative (non-binding) controls. - An example of a concentration series and quantified binding can be found in Figures 3B-3C.
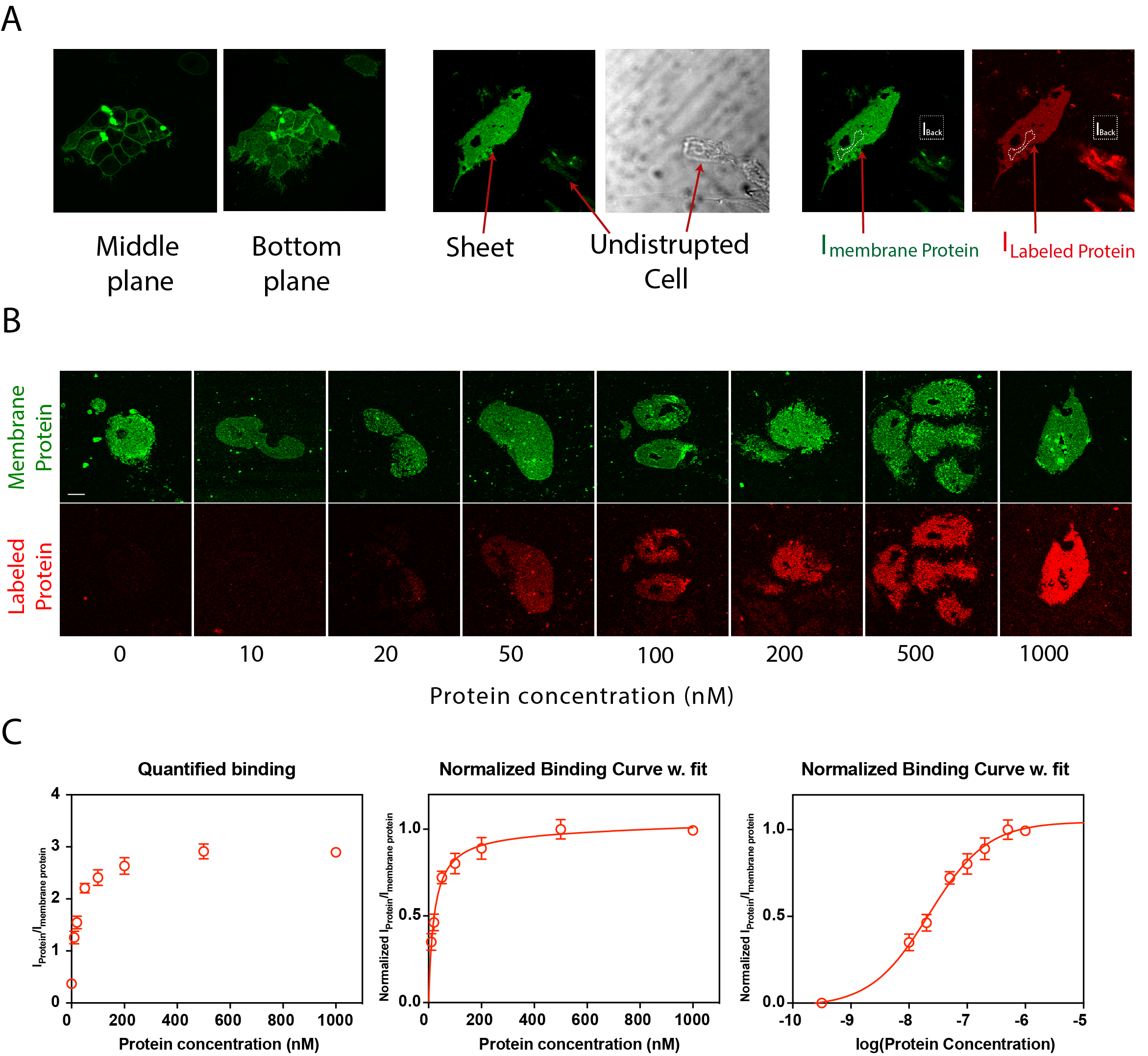
Figure 3. Identifying the SCMS and quantification. A. Single bilayers are distinguished from undisrupted cells or organelles by their lack of three-dimensional structure. These structures are clearly visible in the direct transmission. B and C. The intensities from both sheets and background for each channel is measured using ImageJ (as described in the main text) and concentration series can be made as presented in B, and quantified as shown in C. Error bars, means ± SE (n=6). For fitting these data on linear scale we use a one-site total binding model: Y = Bmax*X/(Kd + X) + NS*X, where NS is the slope of non-specific binding. On a logarithmic scale we used a sigmoidal dose-response with variable slope: Y = Bottom + [(Top-Bottom)/(1+10(Kd-X)*S)], where S is the slope steepness. Please note that forced proximity/hindered diffusion will affect the slope steepness and therefore this is not necessarily a good measure of binding cooperativity. Figures are adapted from Erlendsson et al. (2019). Scale bars: 10 μm.
Notes
The method is not restricted to any particular cell type and in principle any adherent cell type can be used. If sufficient signal-to-noise ratio can be obtained, we can also recommend using stable transfected cell lines (or even native cell lines, with a good monoclonal antibody). This will provide an even more realistic view on the effective concentrations of receptors, their clustering and potential avidity effects.
The SCMS are native cell plasma membranes and they contain most, if not all, of the native cellular receptors, transporters, ion channels etc. as well as the cytoskeleton and smaller membrane associated organelles such as vesicles. For this reason, protein ligands can have specific binding to many of these components, as well as non-specific binding to the membrane (this is referred to as the background). In transiently transfected cell cultures, overexpression of the receptor of interest will neglect the native “background” binding. In Erlendsson et al. (2019) the non-specific binding, tested by introducing a single but completely disruptive point mutation in the ligand-binding domain, is not detectable. Specific binding tested using a single but completely disruptive point mutation in the receptor binding epitope is 15-20% of binding observed with the wild-type receptor. Please note that background binding is highly system dependent and should be tested in parallel with any binding experiments.
Recipes
- TBS Buffer
150 mM NaCl
50 mM Trizma
Adjust the pH to 7.35 - DMEM media
D-MEM (high glucose: 4500 mg/L, L-glutamine: 584 mg/L)
10% Fetal Bovine Serum (FBS)
0.1 mM MEM Non-Essential Amino Acids (NEAA) - Sheet Buffer
120 mM KCl
2 mM MgCl2
0.1 mM CaCl2
10 mM HEPES
30 mM glucose
Adjust pH to 7.35
Add 50 mg of BSA to 50 ml buffer
Acknowledgments
This protocol was developed for investigating the binding affinity of scaffolding proteins to membrane embedded receptors presented in Erlendsson et al. (2019). We would like to acknowledge efforts made in the research groups lead by Karen Martinez (University of Copenhagen, Denmark) and Horst Vogel (Institute of Chemical Sciences and Engineering Ecole Polytechnique Fédérale de Lausanne, Switzerland) for development of the original SCMS protocol (Perez et al., 2006) and Thor C. Møller (University of Copenhagen, Denmark) for help with adapting the SCMS protocol for also probing binding affinities. The work was supported by the Danish Council for independent Research–Medical Sciences, University of Copenhagen BioScaRT Program of Excellence, the Lundbeck Foundation Center for Biomembranes in Nanomedicine, the UNIK Center for Synthetic Biology and the Novo Nordisk Foundation.
Competing interests
The authors declare no financial or non-financial competing interests.
References
- Erlendsson, S., Thorsen, T. S., Vauquelin, G., Ammendrup-Johnsen, I., Wirth, V., Martinez, K. L., Teilum, K., Gether, U. and Madsen, K. L. (2019). Mechanisms of PDZ domain scaffold assembly illuminated by use of supported cell membrane sheets. Elife 8. Pii: e39180.
- Miles, T. F., Dougherty, D. A. and Lester, H. A. (2013). The 5-HT3AB receptor shows an A3B2 stoichiometry at the plasma membrane. Biophys J 105(4): 887-898.
- Perez, J. B., Martinez, K. L., Segura, J. M. and Vogel, H. (2006). Supported cell-membrane sheets for functional fluorescence imaging of membrane proteins. Adv Funct Mater 16: 306-312.
- Roizard, S., Danelon, C., Hassaine, G., Piguet, J., Schulze, K., Hovius, R., Tampe, R. and Vogel, H. (2011). Activation of G-protein-coupled receptors in cell-derived plasma membranes supported on porous beads. J Am Chem Soc 133(42): 16868-16874.
- Scarselli, M., Annibale, P. and Radenovic, A. (2012). Cell type-specific β2-adrenergic receptor clusters identified using photoactivated localization microscopy are not lipid raft related, but depend on actin cytoskeleton integrity. J Biol Chem 287(20): 16768-16780.
Article Information
Copyright
![]() Erlendsson et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Erlendsson et al. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Erlendsson, S., Thorsen, T. S. and Madsen, K. L. (2019). Supported Cell Membrane Sheets to Monitor Protein Assembly. Bio-protocol 9(18): e3368. DOI: 10.21769/BioProtoc.3368.
- Erlendsson, S., Thorsen, T. S., Vauquelin, G., Ammendrup-Johnsen, I., Wirth, V., Martinez, K. L., Teilum, K., Gether, U. and Madsen, K. L. (2019). Mechanisms of PDZ domain scaffold assembly illuminated by use of supported cell membrane sheets. Elife 8. Pii: e39180.
Category
Cell Biology > Cell-based analysis > Protein interaction
Neuroscience > Cellular mechanisms > Receptor-ligand binding
Biochemistry > Protein > Interaction
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.