- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Cryo-transmission Electron Microscopy of Outer-inner Membrane Vesicles Naturally Secreted by Gram-negative Pathogenic Bacteria


Published: Vol 9, Iss 18, Sep 20, 2019 DOI: 10.21769/BioProtoc.3367 Views: 6777
Reviewed by: Alba BlesaKristin L. ShinglerBeatrice Li
Original research article
The authors used this protocol in:

Jan 2015
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
A protocol was developed to visualize and analyze the structure of membrane vesicles (MVs) from Gram-negative bacteria. It is now accepted that these micrometric spherical vesicles are commonly produced by cells from all three domains of life, so the protocol could be useful in the study of vesicles produced by eukaryotes and archaea as well as bacteria. The multiplicity of functions performed by MVs, related to cell communication, interaction with the immune system, pathogenesis, and nutrient acquisition, among others, has made MVs a hot topic of research.
Due to their small size (25-300 nm), the observation of MVs requires electron microscopy and is usually performed by transmission electron microscopy (TEM) of negatively stained MVs. Other protocols applied for their visualization include scanning electron microscopy, TEM after fixation and embedding of vesicles, or even atomic force microscopy. In some of these techniques, vesicle structure is altered by drying, while others are time-consuming and most of them can generate artifacts. Cryo-TEM after plunge freezing allows the visualization of samples embedded in a thin film of vitreous ice, which preserves their native cellular structures and provides the highest available resolution for the imaging. This is achieved by very high cooling rates that turn the intrinsic water of cells into vitreous ice, avoiding crystal formation and phase segregation between water and solutes. In addition to other types of characterization, an accurate knowledge of MV structure, which can be obtained by this protocol, is essential for MV application in different fields.
Background
In recent years, many studies have been conducted on membrane vesicles (MVs) produced by Gram-negative bacteria (Kulp and Kuehn, 2010). MVs are spherical membranous structures with diameters ranging between 20 and 300 nm, and they enable a protected secretion of proteins, lipids, RNA, DNA and other effector molecules (Beveridge, 1999; Mashburn-Warren and Whiteley, 2006; Ellis and Kuehn, 2010). The ability of bacterial MVs to interact with and enter host cells has prompted their exploration for novel clinical and biotechnological applications (Chen et al., 2010; Gujrati et al., 2014; Robbins and Morelli, 2014; van der Pol et al., 2015).
Different methods have been used to characterize and quantify bacterial MVs such as protein or lipopolysaccharide quantification, nanoparticle tracking analysis, flow-cytometry or proteomic analysis among others, but none of them allow MV visualization or clarify MV structure (Kulp and Kuehn, 2010). In many studies, MV-producing cells and vesicle morphology have been visualized using conventional electron microscopy techniques such as scanning electron microscopy or thin-section transmission electron microscopy (TEM) of samples chemically fixed and dehydrated at room temperature (Fiocca et al., 1999; McBroom and Kuehn, 2007; Deatherage et al., 2009; Alves et al., 2015; Ercoli et al., 2015). However, in both methods the extracellular matter in which MVs are found has a propensity to collapse and be removed during sample preparation (Beveridge, 1999; Fiocca et al., 1999; Nevot et al., 2006; Mashburn-Warren and Whiteley, 2006; McBroom and Kuehn, 2007; Deatherage et al., 2009; Ellis and Kuehn, 2010; Chen et al., 2010; Gujrati et al., 2014; Robbins and Morelli, 2014; Alves et al., 2015; Ercoli et al., 2015; van der Pol et al., 2015). TEM observation of negatively stained MVs is the most commonly used technique to assess MV presence and morphology. In this case, MVs are dried on a grid at room temperature and most of them are visualized as irregular and wrinkled round structures (Figure 1A). Although useful for confirming the presence of MVs, this technique does not provide enough resolution to visualize their exact shape or the presence of atypical MVs or artifacts, which may arise from the preparation process or when working with new or genetically manipulated strains (Bernadac et al., 1998; Kolling and Matthews, 1999; Wai et al., 2003; Lee et al., 2007; McBroom and Kuehn, 2007; Roier et al., 2016). The variability created by these features can hamper studies evaluating the application of MVs in different fields (Gujrati et al., 2014; van der Pol et al., 2015). Furthermore, in conventional electron microscopy techniques, samples have to be observed under a high vacuum and the electron beam leads to the destruction of native biological structure (Nevot et al., 2006).
In cryo-transmission electron microscopy (cryo-TEM), biological specimens are immobilized or fixed using physical procedures based on freezing. The goal is to obtain vitreous or amorphous ice from the sample water after ultra-rapid freezing, avoiding ice crystal formation and thus preserving the cellular structures. In contrast with conventional methods, ultra-rapid freezing immobilizes all molecules in a sample within milliseconds. After this instantaneous cryo-immobilization, it is very important to maintain this state throughout the imaging acquisition in the cryo-electron microscope (Cavalier et al., 2008).
In recent years, improvements in cryo-TEM techniques have enabled the imaging of biological specimens with greatly enhanced resolution and close to their native state, which has refined our knowledge of bacterial structures, including bacterial MVs (Renelli et al., 2004; Jensen and Briegel, 2007; Studer et al., 2008; Palsdottir et al., 2009; Frias et al., 2010; Perez-Cruz et al., 2013 and 2015).
There are several methods for freezing samples. Plunging samples into a pre-cooled cryogen, such as ethane or propane, is a common technique for freezing suspensions (viruses, liposomes, bicelles, micelles...), molecular assemblies (proteins, nucleic acids...), isolated organelles and cell structures and even small cells such as bacteria (Dobro et al., 2010). This method, known as plunge freezing, has been used to visualize bacterial MVs (Renelli et al., 2004), and has revealed, for example, the existence of a new type of Gram-negative MVs produced by environmental and pathogenic bacteria, named outer-inner membrane vesicles (O-IMVs) (Figures 1B, 2C and 2) (Perez-Cruz et al., 2013 and 2015).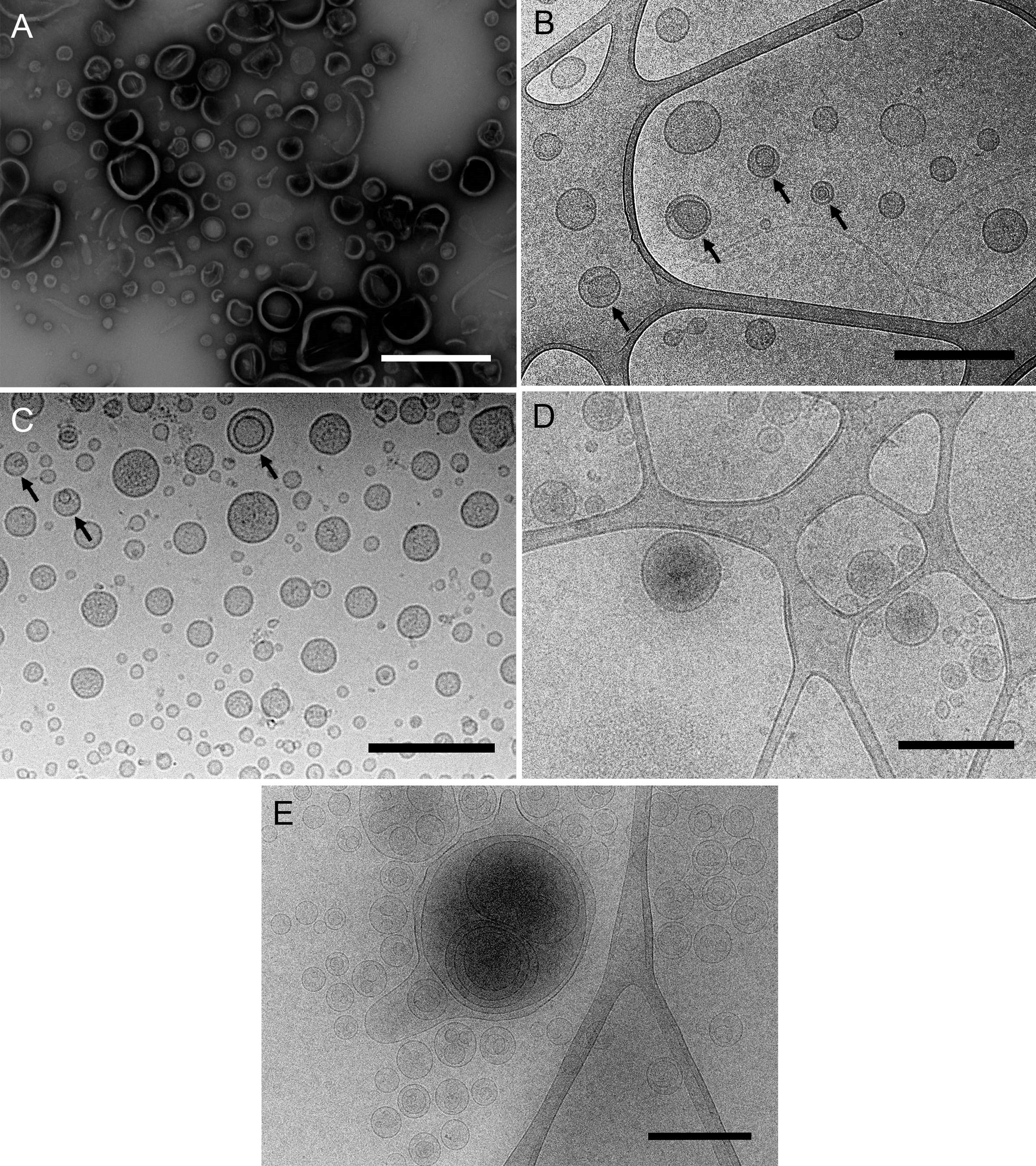
Figure 1. Different samples of isolated MVs observed by (cryo-)TEM. A. MVs from Shewanella vesiculosa M7T prepared by negative staining and observed by TEM. Vesicles appear collapsed and as irregular and wrinkled round structures. B and C. MVs from Shewanella vesiculosa M7T and (C) Acinetobacter baumannii AB41 prepared by plunge freezing and observed by cryo-TEM. MVs appear as regular round structures and different types can be observed: conventional bacterial outer membrane vesicles with one membrane layer and a new type called outer-inner membrane vesicles (O-IMV) with two membrane layers (black arrows). D and E. Exosomes produced by melanoma cancer cells and (E) liposomes synthesized from lipids. All types of vesicles appear as regular round structures and membrane layers are clearly visualized. Scale bars = 500 μm.
Plunge freezing of MVs allows us to observe their exact shape, size and integrity, the presence of one or more membrane layers, and attached surface-associated structures like viruses, among other features (Perez-Cruz et al., 2016).
Bacterial MVs are not the only focus of investigation. Another expanding research area is the study of extracellular vesicles (EVs), which are released by cells from all domains of life-Eukarya, Archaea and Bacteria-and are considered intercellular communicasomes, acting as a mechanism for distance delivery of active compounds between cells (Yoon et al., 2014). EVs have demonstrated great promise as natural drug delivery systems loaded with therapeutic molecules (Armstrong and Stevens, 2018). Other candidates for the improvement of drug delivery systems are liposomes and other lipidic formulations (Alavi et al., 2017), varying in size and lamellar number, and cryo-TEM observation could also be applied to advance their development. The described protocol therefore has potential application for other types of vesicles currently under active research (Figures 1D and 1E).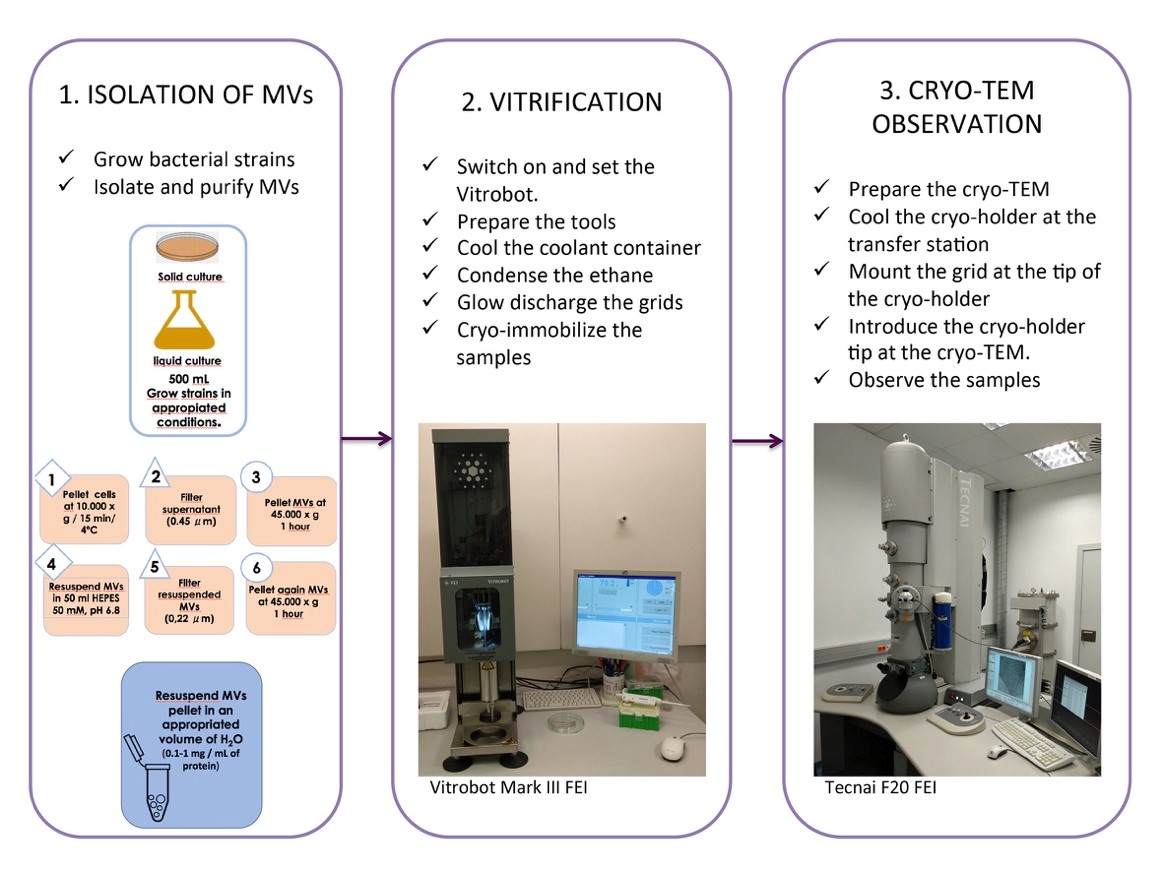
Figure 2. Cryo-TEM of isolated MVs, a general overview of the whole protocol
Materials and Reagents
- Tweezers No. 5 (Dumont, catalog number: 21974-1)
- Safe tweezers (Wiha, catalog number: 44518)
- Vitrobot forceps (Ted Pella, catalog number: 47000-500)
- Micropipettes 20, 100 and 1000 (Eppendorf, model: Research)
- Membrane filters, white, individually packed. 0.45 µm, diameter 47 mm, sterile (ME 25 ST, Whatman®, catalog number: 10401670)
- Disposable filter device, 0.22 µm, sterile and non-pyrogenic polyethersulfone membrane with polypropylene housing (PURADISC 25 AS, Whatman®, catalog number: 6780-2502)
- 250 ml centrifuge polypropylene bottles (Beckman Coulter, catalog number: 356011)
- 50 ml centrifuge polypropylene tubes (Nalgene, catalog number 3139-0050)
- 500 ml and 1000 ml screw neck glass bottles (DURAN®)
- Vacuum filtration unit (DURAN® , catalog number: XT09.1)
- Chocolate agar plates (Beckton Dickinson, catalog number: PA-254035.05)
- Lacey Carbon 300 mesh grids (Ted Pella, catalog number: 01895-F)
- Quantifoil® R 2/2 Cu 200 mesh grids (Quantifoil Micro Tools GmbH, catalog number: Q23209)
- Holey Carbon 300 mesh grids (Agar, catalog number: AGS147-3)
- Filter paper for Vitrobot (Ted Pella, catalog number: 47000-100)
- Cryo Grid Box base only (Ted Pella, catalog number: 160-41)
- Cryo Grid Box Handling Rod (Ted Pella catalog number: 160-46)
- Neoprene gloves (Seton, catalog number: 10STA002)
- Parafilm (Sigma-Aldrich, catalog number: P7793-1EA)
- Petri dish (Fisher Scientific, catalog number: 11812532)
- Yellow pipette tips (Sigma-Aldrich, catalog number: EP4925000111)
- Blue pipette tips (Sigma-Aldrich, catalog number: EP3124000121)
- Neisseria gonorrhoeae (ATCC 43069)
- Pseudomonas aeruginosa PAO1 (Own collection)
- Acinetobacter baumannii AB41 (Clinical isolate)
- Ringer ¼ (Sigma-Aldrich, catalog number: 96724-100TAB), prepare the solution as per the manufacturer’s instructions (Reference 1)
- Trypticase soy broth (Oxoid, catalog number: CM129), prepare the solution as per the manufacturer’s instructions (Reference 10)
- Müeller-Hinton broth (Oxoid, catalog number: CM0405), prepare the solution as per the manufacturer’s instructions (Reference 11)
- HEPES (Sigma-Aldrich, catalog number: 7365-45-9), prepare the solution as per the manufacturer’s instructions (Reference 18)
- Modified Lowry Protein Assay kit (Thermo Scientific, catalog number: 23240)
- Ethanol 96% (Sigma-Aldrich, catalog number: 16368)
- Ethane (AIR liquide)
- Liquid nitrogen (AIR liquide)
Equipment
- Fume hood (Flores Valles, model: LVG 190)
- Glow Discharge unit (BALTEC, model: CTA005)
- Minishaker (IKA®, model MS2)
- Vitrification Robot (FEI, model: VitrobotTM Mark III)
- Cryo-holder and cryo-transfer station (Gatan, model: 626)
- Cryo-electron microscope (FEI, model: Tecnai F20 200 kV) equipped with a CCD camera (FEI, model: Eagle 4kx4k)
- Orbital shaker (Innova® 44, Incubator Shaker Series, New Brunswick Scientific)
- Centrifuge (Beckman Coulter, model: Avanti J-20 XP)
- Centrifuge (Beckman Coulter, model: Allegra 25R)
Software
- Vitrobot V1.05B051 (FEI)
- Tecnai version 4.3 (FEI)
- TEM Imaging & Analysis version 4.4 (FEI)
Procedure
- Isolation of MVs
- Grow the bacterial strains.
- Grow Neisseria gonorrhoeae ATCC 43069 to confluence by spreading 0.1 ml of a 109 cell/ml suspension on the surface of chocolate agar plates using a sterile glass spreader and rotating the plate. Incubate the plates for 65 h, at 37 °C in a 5% CO2 incubator. Seeding 10 to 20 Petri dishes should provide enough MVs.
- Grow Pseudomonas aeruginosa PAO1 in 2 L flasks with 500 ml of trypticase soy broth at 37 °C for 5 h, and Acinetobacter baumannii AB41 in 2 L flasks with 500 ml of Müeller-Hinton broth at 30 °C for 15 h. Incubate both liquid cultures in an orbital shaker at 100 rpm.
- Isolate MVs from Pseudomonas aeruginosa PAO1 and Acinetobacter baumannii AB41 from liquid media culture.
- Recover broth cultures of A. baumannii AB41 and P. aeruginosa PAO1 in the late logarithmic phase (OD1.8).
- Place cultures in 250 ml sterile polypropylene centrifuge bottles.
- Pellet the cells by centrifugation at 10,000 x g for 15 min at 4 °C (Allegra 25R, Beckman Coulter).
- Discard cells and place the supernatants in a sterile screw neck glass bottle.
- Filter the supernatant through 0.45-μm-pore-size membrane filters with a vacuum filtration unit to remove remaining bacterial cells.
- Place the filtered supernatants in 50 ml sterile polyprolylene centrifuge tubes and centrifuge at 45,000 x g for 1 h at 4 °C (Avanti J-20 centrifuge, Beckman Coulter).
- Carefully discard the supernatant and save the pellet (normally a very small pellet is obtained).
- Resuspend the pellet in 50 ml of 50 mM HEPES pH 6.8 and filter it through a 0.22-μm-pore-size disposable filter device using a syringe and saving the filtrate in another 50 ml sterile polyprolylene centrifuge tube.
- Pellet the vesicles again at 45,000 x g for 1 h at 4 °C and resuspend the pellet in 500 µl of H2O.
- Measure the concentration of MVs obtained with the Lowry method. For a correct visualization of the MVs by cryo-TEM, a final concentration of 0.1-1 mg/ml of protein is suitable.
- The final volume used to resuspend the MV pellet can be adjusted depending on the yield of MVs obtained with each particular strain. 20-50 µl should be enough to prepare grids for cryo-TEM observation.
- Isolate and purify MVs from Neisseria gonorrhoeae ATCC 43069 from confluent solid cultures.
- Resuspend cells and MVs from each agar plate by adding 15 ml of Ringer ¼ per plate and using a cell scraper. Place all the volumes together in a screw neck glass bottle.
- Place resuspended cells in 250 ml sterile polypropylene centrifuge bottles.
- Follow the previously described steps from A2c to A2k.
- Grow the bacterial strains.
- Vitrification
- Switch on the Vitrobot by the toggle switch at the back (Figure 3A and Video 1).
 Video 1. Cryo-TEM of bacterial outer membrane vesicles
Video 1. Cryo-TEM of bacterial outer membrane vesicles - Fill the humidifier with distilled water.
Fill the syringe with 60 ml distilled water and inject through the plastic tube located at the bottom (Figure 3A and Video 1). - Switch on the chamber light using the “console” page of the Vitrobot User Interface.
- Change the blotting pads.
Manually pull out the front door of the Vitrobot chamber and remove the plastic rings from the blotting pads, attach new pieces of filter paper to the plastic rings and clip them to the blotting pads (Video 1). - Set the conditions using the Vitrobot User Interface.
- On the “console” page, set the conditions of the chamber to 25 °C and 100% humidity (Figure 3B and Video 1).
- Go to the “options” page and set the blotting and freezing conditions as follows: blot offset 0, blot total 1, blot time 2.5, wait time 120 s, plunge time 0 and drain time 0 (Figure 3C and Video 1).
- Also set other user specifics: tick “using footpedal” and “turning off the semi-automatic grid” (Figure 3C and Video 1).
- Prepare the tools.
Apply some ethanol to the tip of the forceps as well as nitrogen air at a pressure of 7 bars. Also apply nitrogen air to the metallic and plastic parts that will be in contact with liquid nitrogen to minimize dust contamination. The metallic parts consist of the container for liquid ethane, the support for the grid boxes and the spindle; the plastic parts consist of the grid box and the grid box container (in our case, modified Falcon tubes) (Figure 3D and Video 1).
Figure 3. Vitrification. A. Vitrification Robot Vitrobot Mark III FEI. The black arrows point the entry pots to the climate chamber, the white arrow points the humidifier and the white arrowhead points the sample injection window. B. Vitrobot settings in “console page”. C. Vitrobot settings in “options page”. D. Vitrobot accessories: 1, modified Falcon tubes; 2, anticontamination ring; 3, coolant container; 4, Cryo-grid box handling rod; 5, cryo-grid box base; 6, grid-box support; 7, spindle; 8, ethane container; 9, Vitrobot tweezers. E. Vitrobot tweezers secured on the connection groove at the central axis. The black arrow points the grid and the white arrow points the connection groove. F. Vitrobot tweezers inside the climate chamber. The black arrows point the blotting pads. G. Cryo-immobilized grid inside the liquid ethane. The white arrow points the ethane container and the black arrow points the cryo-grid box base. - Cool the coolant container.
Mount the ethane container, the grid box support, the grid box, the spindle and the anti-contamination ring in the coolant container and fill the outer ring of the container with liquid nitrogen until all the components are cooled. All the components have been cooled when the liquid nitrogen stops boiling (Video 1). - Condense the ethane.
Position the exit tip of the ethane cylinder inside the ethane container, open the main valve of the gas cylinder and slowly liquefy the ethane. When the bubbles on the surface of the liquid ethane reach the bottom brim of the spindle, close the main valve of the ethane cylinder and remove the spindle (Video 1). - Prepare the grids.
Place the grids for the experiment on a piece of Parafilm fixed inside a Petri dish plate and bring the grids to the Glow Discharge unit (Figure 4A).
Figure 4. Glow Discharge. A. Glow Discharge unit BALTEC CTA 005. The black arrow indicates the position of the grid inside the chamber. B. Glow Discharge settings. - Glow-discharge the grids.
- Remove the lid of the Petri dish plate and ensure the carbon faces of all the grids are facing upwards.
- Manually pull up the chamber door of the Glow Discharge device, place the opened Petri dish plate containing the grids inside and pull down the door (Video 1).
- Set the timer at 30 s and the current to a value between 2 and 6 mA (Figure 4B).
- Press the toggle switch at the back and wait for the vacuum chamber to reach about 10-1 mbar (the red LED indicating the pressure on the panel will change to yellow) and press “start” to switch on the UV light (Video 1).
- Wait for the set time and the UV light will switch off automatically (Video 1).
- Remove the grids from the Glow Discharge unit.
- Switch the toggle off and wait for the chamber to be vented (the sound will stop) (Video 1).
- Manually pull up the door of the chamber, take out the Petri dish plate containing the grids, close the door of the chamber and transfer the grids to the Vitrobot (Video 1).
After the glow discharge, liquid samples will spread over the grid surface more easily. Use the grids within one hour after charging, otherwise they will need to be charged again.
- Press the foot pedal or select “place new grid” in the Vitrobot interface.
- Mount one grid on the Vitrobot tweezers (Video 1).
Place the slider of the Vitrobot forceps in the upper position, carefully attach one of the grids by its edge and lock the slider on the nearest groove of the forceps. - Secure the tweezers on the connection groove at the central axis.
The sample must be deposited on the carbon side of the grid. When securing the Vitrobot tweezers, the carbon side of the grid should be facing left or right, depending on which side entry port the operator is using to deposit the sample (Figure 3E and Video 1). - Move the tweezers into the climate chamber. Press the foot pedal or select “start process” (Figure 3F and Video 1).
- Place the coolant container on the platform ring (Video 1).
- Raise the coolant container towards the climate chamber.
Press the foot pedal or select “continue” (Video 1). - Lower the Vitrobot forceps to the sample application position.
Press the foot pedal or select “continue” (Video 1). - Apply the sample to the grid.
Open the side port of the climate chamber and apply 3 μl with a micropipette (Video 1). - Activate the blotting and the plunge freezing.
Press the foot pedal or select “continue” and wait for the coolant container and forceps to be lowered (Video 1). - Carefully remove the forceps from the groove of the central axis of the Vitrobot.
Transfer the coolant container and the forceps to the bench (Video 1). Be careful to keep the grid below the liquid ethane surface during this step. - Wait until the ethane cloud has dispersed and slowly lift the grid through the ethane surface.
Once the grid is completely out, quickly insert it in the outer ring containing liquid nitrogen and transfer the grid to a position of the grid box (Figure 3G and Video 1). - Repeat Steps B16-B26 until all the samples have been cryo-immobilized.
When necessary, thaw the liquid ethane by covering the ethane container with another empty container for a few seconds. - Store the grids.
- When all frozen grids are inside the grid box, cool the handling rod and screw it to the center of the grid box (Video 1).
- Transfer the grid box to a precooled grid box container under liquid nitrogen and close it (Video 1).
- Switch off the Vitrobot.
Place the coolant container in the fume hood for the evaporation of the liquid nitrogen and liquid ethane and select “exit” in the Vitrobot User Interface. When the lights are turned off, the central axis is in the parking position and the interface has shut down, turn off the toggle switch at the back of the device.
- Switch on the Vitrobot by the toggle switch at the back (Figure 3A and Video 1).
- Cryo-TEM observation
- Prepare the cryo-microscope.
Transfer the grids in the Dewar with liquid nitrogen to the cryo-microscope room and cool the anti-contaminator of the microscope for at least 1 h (Video 1). - Ensure the valves are closed.
Go to the “Set up” tab. If the “Col. valves closed” button is not yellow, click on it (Video 1). - Press “Turbo on” (Video 1).
- Prepare all the accessories (Figure 5A) and cover the desk of the microscope with cold insulating materials (cork plates, bubble wrap…).

Figure 5. Mounting the grid on the Cryo-holder. A. Accessories: 1, lid; 2, funnel; 3, cryo-transfer station; 4, platform; 5, clip-ring tool. B. Upper view of the Cryo-holder GATAN 656 in the cryo-transfer station. The black arrow points the cryo-transfer station and the white arrow points the cryo-holder. C. Detail of the cryo-transfer station. 1, cryo-holder tip; 2, cryo grid box base. The black arrow points the position of the grid at the tip of the cryo-holder. - Cool down the cryo-holder.
Place the cryo-holder into the transfer station and cool it down by pouring liquid nitrogen into the Dewar of the cryo-holder and into the cryo-holder station. Connect the cable of the control station to the cryo-holder and wait till the temperature is below -165 °C (Video 1). - Prepare the cryo-holder tip.
- Transfer the grid box to its place on the platform of the transfer station and carefully unscrew and remove the rod (Figures 5B and 5C and Video 1).
- Open the cryo-shield by sliding back the circular piece at the end of the central axis of the cryo-holder (just behind the Dewar), remove the clip ring with its tool and keep it under liquid nitrogen (Video 1).
Note: The clip-ring tool has two tabs at the tip that can be placed oppositely or contiguously by turning the two wheels of the handle. To remove the clip-ring from the cryo-holder tip, put the two tabs together, place them in the groove around the clip-ring, move the wheels to position the tabs oppositely and pull up.
- Mount the grid in the cryo-holder tip.
Cool the tip of the forceps, transfer one of the grids to the slot in the cryo-holder tip, secure it with the clip-ring tool and close the cryo-shield over the sample to prevent ice contamination (Figure 5C and Video 1). To secure the grid with the clip ring, place the clip ring over the grid, press down the clip-ring tool, turn the wheels of the handle until the tabs are contiguous and remove the tool. - Disconnect the cable of the control station from the cryo-holder (Video 1).
- Prepare a polystyrene box under the compustage (Video 1).
- Press “Pre-pump air lock” (Video 1).
- Quickly remove the cryo-holder from the cryo-transfer station, and carefully introduce the tip of the cryo-holder through the hole of the microscope as far as it will go (Video 1).
- Rotate the cryo-holder to the right until its pivot matches the compustage groove. Press the cryo-holder lightly against the microscope and wait until the end of the count-down in the “vacuum overview” (Video 1). Be careful when rotating the cryo-holder to the right to avoid the liquid nitrogen falling onto the polystyrene box prepared in Step C9.
- Select “Cryo-holder” as the specimen holder.
- Once the count-down in the “vacuum overview” has ended, turn the cryo-holder to the left as far as it will go, and introduce it completely into the microscope (Figure 6A and Video 1).
- Quickly refill the Dewar of the cryo-holder with liquid nitrogen (Video 1).
- Connect the cable of the control station to the cryo-holder and check the temperature does not exceed -170 °C (Video 1).
- Click “Col. valves closed” and go to the “LowDose” tab (Video 1).
- Record images in low dose conditions and spot size 5.
Search mode at x7800 and 0.10 e-/Å2s, focus mode at x29000 and 0.80 e-/Å2s and exposure mode at x29000 and 1.00 e-/Å2s (Figure 6B and Video 1).
Figure 6. Cryo-TEM observation. A. Cryo-electron microscope Tecnai F20 FEI. The black arrow indicates the cryo-holder position. B. Low Dose observation.
- Prepare the cryo-microscope.
Data analysis
Reliable measurements of the vesicles were obtained from cryo-TEM images for statistical purposes. More than 7,000 vesicles were measured per strain in two independent experiments and the measurements were performed with ImageJ 1.47 software. MVs can measure between 20 and 250 nm in diameter (Perez-Cruz et al., 2015).
Notes
- A lab coat and gloves should be worn while manipulating the material (filter paper, grids, samples…) to avoid self-contamination and during the experiment to avoid skin damage from proximity to the liquefied gases.
Acknowledgments
We gratefully acknowledge the assistance of MJ Montes for the proper maintenance of the strains and the assistance of M Gimeno for her help in recording the videos.
Competing interests
The authors have declared that no competing interests exist.
References
- 96724 Ringers solution 1/4 strength tablets, Sigma-Aldrich. (Accessed Aug 19, 2019 at: https://www.sigmaaldrich.com/content/dam/sigma-aldrich/docs/Sigma-Aldrich/Datasheet/1/96724dat.pdf.)
- Alavi, M., Karimi, N. and Safaei, M. (2017). Application of various types of liposomes in drug delivery systems. Adv Pharm Bull 7(1): 3-9.
- Alves, N. J., Turner, K. B., Daniele, M. A., Oh, E., Medintz, I. L. and Walper, S. A. (2015). Bacterial Nanobioreactors-directing enzyme packaging into bacterial outer membrane vesicles. ACS Appl Mater Interfaces 7(44): 24963-24972.
- Armstrong, J. P. K. and Stevens, M. M. (2018). Strategic design of extracellular vesicle drug delivery systems. Adv Drug Deliv Rev 130: 12-16.
- Bernadac, A., Gavioli, M., Lazzaroni, J. C., Raina, S. and Lloubes, R. (1998). Escherichia coli tol-pal mutants form outer membrane vesicles. J Bacteriol 180(18): 4872-4878.
- Beveridge, T. J. (1999). Structures of gram-negative cell walls and their derived membrane vesicles. J Bacteriol 181(16): 4725-4733.
- Cavalier, A., Spehner, D. and Humbel, B. M. (2008). Handbook of Cryo-preparation Methods for Electron Microscopy. CRC Press.
- Chen, D. J., Osterrieder, N., Metzger, S. M., Buckles, E., Doody, A. M., DeLisa, M. P. and Putnam, D. (2010). Delivery of foreign antigens by engineered outer membrane vesicle vaccines. Proc Natl Acad Sci U S A 107(7): 3099-3104.
- Deatherage, B. L., Lara, J. C., Bergsbaken, T., Rassoulian Barrett, S. L., Lara, S. and Cookson, B. T. (2009). Biogenesis of bacterial membrane vesicles. Mol Microbiol 72(6): 1395-1407.
- Dehydrated Culture Media, Thermo Scientific. (Accessed Aug 19, 2019 at: http://www.oxoid.com/UK/blue/prod_detail/prod_detail.asp?pr=CM0129)
- Dehydrated Culture Media, Thermo Scientific. (Accessed Aug 19, 2019 at: http://www.oxoid.com/UK/blue/prod_detail/prod_detail.asp?pr=CM0405)
- Dobro, M. J., Melanson, L. A., Jensen, G. J. and McDowall, A. W. (2010). Plunge freezing for electron cryomicroscopy. Methods Enzymol 481: 63-82.
- Ellis, T. N. and Kuehn, M. J. (2010). Virulence and immunomodulatory roles of bacterial outer membrane vesicles. Microbiol Mol Biol Rev 74(1): 81-94.
- Ercoli, G., Tani, C., Pezzicoli, A., Vacca, I., Martinelli, M., Pecetta, S., Petracca, R., Rappuoli, R., Pizza, M., Norais, N., Soriani, M. and Arico, B. (2015). LytM proteins play a crucial role in cell separation, outer membrane composition, and pathogenesis in nontypeable Haemophilus influenzae. MBio 6(2): e02575.
- Fiocca, R., Necchi, V., Sommi, P., Ricci, V., Telford, J., Cover, T. L. and Solcia, E. (1999). Release of Helicobacter pylori vacuolating cytotoxin by both a specific secretion pathway and budding of outer membrane vesicles. Uptake of released toxin and vesicles by gastric epithelium. J Pathol 188(2): 220-226.
- Frias, A., Manresa, A., de Oliveira, E., Lopez-Iglesias, C. and Mercade, E. (2010). Membrane vesicles: a common feature in the extracellular matter of cold-adapted antarctic bacteria. Microb Ecol 59(3): 476-486.
- Gujrati, V., Kim, S., Kim, S. H., Min, J. J., Choy, H. E., Kim, S. C. and Jon, S. (2014). Bioengineered bacterial outer membrane vesicles as cell-specific drug-delivery vehicles for cancer therapy. ACS Nano 8(2): 1525-1537.
- HEPES FREE ACID, Sigma-Aldrich. (Accessed Aug 19, 2019 at: www.sigmaaldrich.com/content/dam/sigma-aldrich/docs/Sigma/Product_Information_Sheet/2/h6147pis.pdf).
- Jensen, G. J. and Briegel, A. (2007). How electron cryotomography is opening a new window onto prokaryotic ultrastructure. Curr Opin Struct Biol 17(2): 260-267.
- Kolling, G. L. and Matthews, K. R. (1999). Export of virulence genes and Shiga toxin by membrane vesicles of Escherichia coli O157:H7. Appl Environ Microbiol 65(5): 1843-1848.
- Kulp, A. and Kuehn, M. J. (2010). Biological functions and biogenesis of secreted bacterial outer membrane vesicles. Annu Rev Microbiol 64: 163-184.
- Lee, E. Y., Bang, J. Y., Park, G. W., Choi, D. S., Kang, J. S., Kim, H. J., Park, K. S., Lee, J. O., Kim, Y. K., Kwon, K. H., Kim, K. P. and Gho, Y. S. (2007). Global proteomic profiling of native outer membrane vesicles derived from Escherichia coli. Proteomics 7(17): 3143-3153.
- Mashburn-Warren, L. M. and Whiteley, M. (2006). Special delivery: vesicle trafficking in prokaryotes. Mol Microbiol 61(4): 839-846.
- McBroom, A. J. and Kuehn, M. J. (2007). Release of outer membrane vesicles by Gram-negative bacteria is a novel envelope stress response. Mol Microbiol 63(2): 545-558.
- Nevot, M., Deroncele, V., Lopez-Iglesias, C., Bozal, N., Guinea, J. and Mercade, E. (2006). Ultrastructural analysis of the extracellular matter secreted by the psychrotolerant bacterium Pseudoalteromonas antarctica NF3. Microb Ecol 51(4): 501-507.
- Palsdottir, H., Remis, J. P., Schaudinn, C., O'Toole, E., Lux, R., Shi, W., McDonald, K. L., Costerton, J. W. and Auer, M. (2009). Three-dimensional macromolecular organization of cryofixed Myxococcus xanthus biofilms as revealed by electron microscopic tomography. J Bacteriol 191(7): 2077-2082.
- Perez-Cruz, C., Canas, M. A., Gimenez, R., Badia, J., Mercade, E., Baldoma, L. and Aguilera, L. (2016). Membrane vesicles released by a hypervesiculating Escherichia coli Nissle 1917 tolR mutant are highly heterogeneous and show reduced capacity for epithelial cell interaction and entry. PLoS One 11(12): e0169186.
- Perez-Cruz, C., Carrion, O., Delgado, L., Martinez, G., Lopez-Iglesias, C. and Mercade, E. (2013). New type of outer membrane vesicle produced by the Gram-negative bacterium Shewanella vesiculosa M7T: implications for DNA content. Appl Environ Microbiol 79(6): 1874-1881.
- Perez-Cruz, C., Delgado, L., Lopez-Iglesias, C. and Mercade, E. (2015). Outer-inner membrane vesicles naturally secreted by gram-negative pathogenic bacteria. PLoS One 10(1): e0116896.
- Renelli, M., Matias, V., Lo, R. Y. and Beveridge, T. J. (2004). DNA-containing membrane vesicles of Pseudomonas aeruginosa PAO1 and their genetic transformation potential. Microbiology 150(Pt 7): 2161-2169.
- Robbins, P. D. and Morelli, A. E. (2014). Regulation of immune responses by extracellular vesicles. Nat Rev Immunol 14(3): 195-208.
- Roier, S., Zingl, F. G., Cakar, F., Durakovic, S., Kohl, P., Eichmann, T. O., Klug, L., Gadermaier, B., Weinzerl, K., Prassl, R., Lass, A., Daum, G., Reidl, J., Feldman, M. F. and Schild, S. (2016). A novel mechanism for the biogenesis of outer membrane vesicles in Gram-negative bacteria. Nat Commun 7: 10515.
- Studer, D., Humbel, B. M. and Chiquet, M. (2008). Electron microscopy of high pressure frozen samples: bridging the gap between cellular ultrastructure and atomic resolution. Histochem Cell Biol 130(5): 877-889.
- van der Pol, L., Stork, M. and van der Ley, P. (2015). Outer membrane vesicles as platform vaccine technology. Biotechnol J 10(11): 1689-1706.
- Wai, S. N., Lindmark, B., Soderblom, T., Takade, A., Westermark, M., Oscarsson, J., Jass, J., Richter-Dahlfors, A., Mizunoe, Y. and Uhlin, B. E. (2003). Vesicle-mediated export and assembly of pore-forming oligomers of the enterobacterial ClyA cytotoxin. Cell 115(1): 25-35.
- Yoon, Y. J., Kim, O. Y. and Gho, Y. S. (2014). Extracellular vesicles as emerging intercellular communicasomes. BMB Rep 47(10): 531-539.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Delgado, L., Baeza, N., Pérez-Cruz, C., López-Iglesias, C. and Mercade, E. (2019). Cryo-transmission Electron Microscopy of Outer-inner Membrane Vesicles Naturally Secreted by Gram-negative Pathogenic Bacteria. Bio-protocol 9(18): e3367. DOI: 10.21769/BioProtoc.3367.
Category
Microbiology > Microbial cell biology > Cell imaging
Cell Biology > Cell imaging > Electron microscopy
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.