- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

PEG-mediated, Stable, Nuclear and Chloroplast Transformation of Cyanidioschizon merolae

Published: Vol 9, Iss 17, Sep 5, 2019 DOI: 10.21769/BioProtoc.3355 Views: 4974
Reviewed by: Dennis J NürnbergAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jan 2017

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols



Abstract
The ability to achieve nuclear or chloroplast transformation in plants has been a long standing goal, especially in microalgae research. Over past years there has been only little success, but transient and stable nuclear transformation has been achieved in multiple species. Our newly developed method allows for relatively simple transformation of Cyanidioschizon merolae in both nuclear and chloroplast genome by means of homologous recombination between the genome and a transformation vector. The use of chloramphenicol resistance gene as the selectable marker allows for plate-based efficient selection of mutant colonies. Overall, the method allows the generation of mutant strains within 6 months.
Keywords: Cyanidioschizon merolaBackground
Cyanidioschizon merolae 10D is a small single-cell eukaryotic red algae, that inhabits acidic hot springs. Lately, all three of its genomes were sequenced: nucleus (Matsuzaki et al., 2004), mitochondrion (Ohta et al., 1998), and plastid (Ohta et al., 2003). Its simple cell anatomy, the presence of a single mitochondrion and chloroplast, a highly reduced spliceosome (Stark et al., 2015), almost deprived of introns nuclear genome make this organism a suitable candidate for a model organism in broad range of plant research, from cell and plastid anatomy, through physiology and up to genetic engineering (Kuroiwa et al., 1998; Nozaki et al., 2003). Its simplicity makes it particularly useful for genetic manipulations. Thus, many genetic techniques have been applied and optimized, for this organism, including antisense suppression, transient expression, gene disruption, and stable insertion into the nuclear genome (Newell, 2000; Walker et al., 2005; Hallmann, 2007; Minoda et al., 2004; Ohnuma et al., 2008; Imamura et al., 2009; Ohnuma et al., 2009; Imamura et al., 2010; Watanabe et al., 2011, Zienkiewicz et al., 2017a). Recently, also stable transformation of the chloroplast genome has been achieved (Zienkiewicz et al., 2017b; Zienkiewicz et al., 2018) in our laboratory. The developed method takes advantage of double homologs recombination in C. merolae, allowing for integration of foreign DNA into the genome in specific regions. A gene knock-out mutation replaces or disrupts the gene of interest with the cat (chloramphenicol acetyltransferase; Zienkiewicz et al., 2017a) gene under the original promoter or any familiar nuclear or chloroplast promoter (Krupnik et al., 2018). Promoters driving the transcription of the cat gene should exhibit at least moderate to significant constitutive activity; therefore application of promoters already tested is advised (Krupnik et al., 2018). By introducing Diphtheria toxin (fragment A-DTA) under a constitutive promoter into the transformation vector we managed to virtually exclude the possibility of random, illegitimate recombination events (Zienkiewicz et al., 2018). A single recombination event, random integration or otherwise unspecific incorporation of the transformation vector into the nuclear genome would likely cause expression of the A-DTA protein and in effect results in cell lysis. However, the heterogeneity of chloroplast transformation still remains a challenge. We have shown that continued growth of chloroplast mutants in increasing concentration of chloramphenicol allows in principal for reduction of heterogeneity and selection of homogenous mutants. Chloramphenicol is the only antibiotic that is toxic to C. merolae (out of c.a. 10 tested, Zienkiewicz et al., 2017a). This method is just as simple as application of lipofectamine for DNA delivery and is as efficient as the gene-gun method but without its drawbacks and complexity (Zienkiewicz et al., 2017b).
Materials and Reagents
- Syringe filters (0.45 μm, sterile, PTFE, VWR, Germany, catalog number: 514-0073)
- 1.5 ml microtubes (GenoPlast, Poland, GP30623B/80-1500)
- 250 ml Erlenmeyer glass flasks (Simax, Germany)
- 50 ml centrifuge tubes FALCON type (GenoPlast, Poland, GP440PSK)
- Petri dishes (12 cm diameter)
- Parafilm
- Cell culture of C. merolae, 10D (NIES-3377 Unialgal, Clonal and axenic or NIES-1332, Unialgal, Clonal and Non-axenic) strain, was obtained from Microbial Culture Collection (mcc.nies.go.jp, Tsukuba, Japan) and was used throughout this study
Note: Cells were grown in MA2 liquid medium (Minoda et al., 2004) in a glass vessel under continuous white light (50 μmol photon m-2 s-1; measured with Quantitherm PAR/Temp Sensor ) at 42 °C or on Petri dishes filled with MA2 medium, solidified by addition of 1% (w/v) agar (Blirt, Poland). - Tansformation vector
Construction of the transformation vector depends entirely on the method of DNA integration or the general construction of the intended experiment. For example, a gene can be knocked out by means of double homologous recombination. For that purpose, a transformation vector should consist of two homologous flanks of 500-1000 bp length, at both 3’ and 5’ ends of the sequence of the gene of interest. Both flanks should be connected with a sequence of a selectable marker gene (cat–Chloramphenicol acetyltransferase) under a constitutive promoter, e.g., (Papc) (Zienkiewicz et al., 2018). - Agar (LE Standard Blirt, Poland, catalog number: AG41)
- PEG (Polyethylene glycol, Fluka, Germany, catalog number: BCBC0873)
- (NH4)2SO4 (Chempur, Poland, CAS: 7783-20-2, WE: 231-984-1)
- MgSO4·7H2O (Chempur, Poland)
- KH2PO4 (Chempur, Poland, CAS: 7778-77-0, WE: 231-913-4)
- CaCl2·2H2O (Chempur, Poland CAS: 10035-04-8, WE: 233-140-8)
- EDTA·2Na (Carl Roth, Germany, CAS: 6381-92-6, EG-Nr. 205-358-3)
- FeCl3·6H2O (Chempur, Poland, CAS: 10025-77-1, WE: 231-729-4)
- H3BO3 (Chempur, Poland, CAS: 10043-35-3, WE: 233-139-2)
- MnCl2·4H2O (Chempur, Poland, CAS: 13446-34-9, WE: 231-869-6)
- ZnCl2 Chempur (Poland, CAS: 7646-85-7, WE: 231-592-0)
- Na2MoO4·2H2O (Chempur, Poland, CAS: 10102-40-6, WE: 231-551-7)
- CoCl2·6H2O (Chempur, Poland CAS: 7791-13-1, WE: 231-589-4)
- CuCl2·2H2O (Chempur, Poland CAS: 10125-13-0, WE: 231-210-2)
- PEG 4000 (Fluka, Germany, CAS: 25322-68-3)
- Chloramphenicol (BioShop, Canada, CAS: 56-75-7, Lot No.:8E55095)
- Chloramphenicol (see Recipes)
- MA-1 (see Recipes)
- MA2 (Allen medium) and 10x MA2 (Minoda et al., 2004, see Recipes)
- Agar-solidified plates (see Recipes)
- A2 Fe-EDTA solution (see Recipes)
- A2 trace element stock solution (see Recipes)
- PEG 60% (see Recipes)
Equipment
- Temperature-regulated centrifuges (e.g., Eppendorf Centrifuge, Germany, models: 5810 R and 5415 R)
- Spectrophotometer (e.g., Shimadzu, Japan, model: UV-1800)
- NanoDrop (e.g., NanoDrop Lite, Thermo, USA)
- Quantitherm PAR/Temp Sensor (Hanzatech, UK)
- Growth chamber/incubator (sustainable conditions of 42 °C and 50 μmoles m-2 s-1 of light)
- Orbital shaker
Procedure
- C. merolae standard growth conditions
- Standard growth medium is MA2 Allen medium (Recipe 2). We suggest preparation of 10x concentrated stock solution of MA2 with A2 trace elements, without A2 Fe-EDTA and autoclave it. For every new preparation of culture medium, dilute an appropriate volume. After autoclaving add A2 Fe-EDTA (Recipe 4).
- The cultures should be grown in a transparent sterile vessel, of at least five times larger volume than the cell culture (e.g., 50 ml cultures in 250 ml flasks).
- Cultures must be gently agitated (30-50 rpm on an orbital shaker).
- Cultures must be sterile so any culture vessel must be closed off but it must allow for gas exchange. Atmospheric CO2 is sufficient for growth, but up to 5% CO2 in air can be applied.
- The light intensity should be in the range between 50 and 100 µmoles photons m-2 s-1 of continuous illumination.
- Growth rates are similar in the temperature range of 37-42 °C.
- OD of C. merolae should be kept between 0.01 and 1 at most. We advise to refresh every culture weekly or biweekly by inoculation of fresh culture out of 1/100 of the volume of the old culture.
- PEG transformation of C. merolae cells
- Set up at least (work in duplicates) 2x 50 ml culture of OD750 = 0.1 a week prior to transformation and allow it to grow in the standard conditions up to OD750 = 0.4.
- Two days before transformation, introduce a photoperiod of 8/16 ho light/dark.
- One hour before transformation expose to standard light conditions (if the dark period isn’t completed).
- Spin down 100 ml of culture at 40 °C and 2,000 x g for 5 min.
- Resuspend the pellet in 50 ml of MA-1 solution at 40 °C and spin down again at 40 °C and 2,000 x g for 5 min.
- Resuspend the pellet in 0.5 ml of MA-1 solution at 40 °C (now ready for transformation).
- Produce large quantity (above 50 μg and high concentration (above 200 ng/μL, measure precisely on NanoDrop) of transformation vector.
- Dilute the transformation vector to 20 μg in total 400 μl MA-1 buffer.
- Add 100 μl of the cell suspension to 400 μl of DNA and gently mix it by pipetting it up and down 2-3 times.
- Add 500 μl of 60% sterile PEG and gently mix it with the tip (the final concentration of PEG should be 30%).
- Incubate for 5 min at room temperature.
- Transfer the cell suspension to 50 ml MA2 and spin down at 40 °C and 2,000 x g for 5 min, repeat one more time.
- Resuspend in 50 ml of MA2 medium and apply standard growth conditions.
- Allow growing under continuous light for 3 days, after that introduce selectable conditions–chloramphenicol at 150 μg/ml form freshly prepared stock solution of 100 mg/ml.
- Culture should fade (slowly turn yellow/white) and then regain growth (by appearance of green color).
- After 2-3 weeks, transfer 500 μl of the culture into 50 ml of MA2 medium in 250 ml flask.
- Plate 50 μl of culture on 1% (w/v) agar-solidified MA2 Petri dishes, seal 3 times with parafilm to avoid excessive evaporation and drying out of the medium matrix.
- Allow growing in standard conditions until colonies appear.
- Pick 20-50 colonies and transfer into a 250 ml flask with 50 ml of MA2 containing 100 μg/ml chloramphenicol.
- After it grows up to OD750 = 0.4, then inoculate into MA2 media with150, 200 and 250 μg/ml of chloramphenicol, respectively.
- Allow it to grow for ~2 weeks up to OD750 ~0.5.
- Inoculate new cultures from the successful, highest-concentration-of-chloramphenicol-grown surviving culture.
- Repeat as long as necessary to achieve and confirm homogeneity by appropriate genetic means (e.g., by qPCR specific to WT and modified genomes, Southern Blott The time needed for the entire procedure may vary and typically takes about 6 months).
Data analysis
Due to 2-3 week-long period of selectable growth, followed by plating of the cells on solid medium it is impossible to approximate the efficiency of transformation. However, in our earlier attempts (Zienkiewicz et al., 2017a) we have obtained viable colonies in 10 out of 11 attempts. The introduced mutation supplied the cell with the resistance gene, without disrupting any native gene. When introducing any mutation of a functional gene, a lower efficiency should be expected, depending on the gene of interest. Homogeneity may be achieved by multiple passaging of cultures in stepwise increasing (typically, increase the chloramphenicol concentration by 50 µg/ml for every passage) concentration of the selectable marker. Due topresence of multiple copies of the chloroplast genome, it is more difficult to produce homogenous chloroplast mutants. In our experience, it takes 3 to 6 months longer and it requires higher concentrations of chloramphenicol. Moreover, the presence of WT and modified multiple copies of the chloroplasts genomes may lead to optimum heterogeneity, where a cell must maintain both versions to endure specific maximal concentration of chloramphenicol. The more important is the gene of interest, the more likely it is to occur.
Recipes
- MA-1 (100 ml) (Table 1)
Table 1. MA-1 buffer components
Note: Don’t adjust pH. Autoclave and store sterile at RT, before transformation warm up to 40 °C.
- MA2 (Allen medium) (Table 2)
Table 2. MA-2 buffer components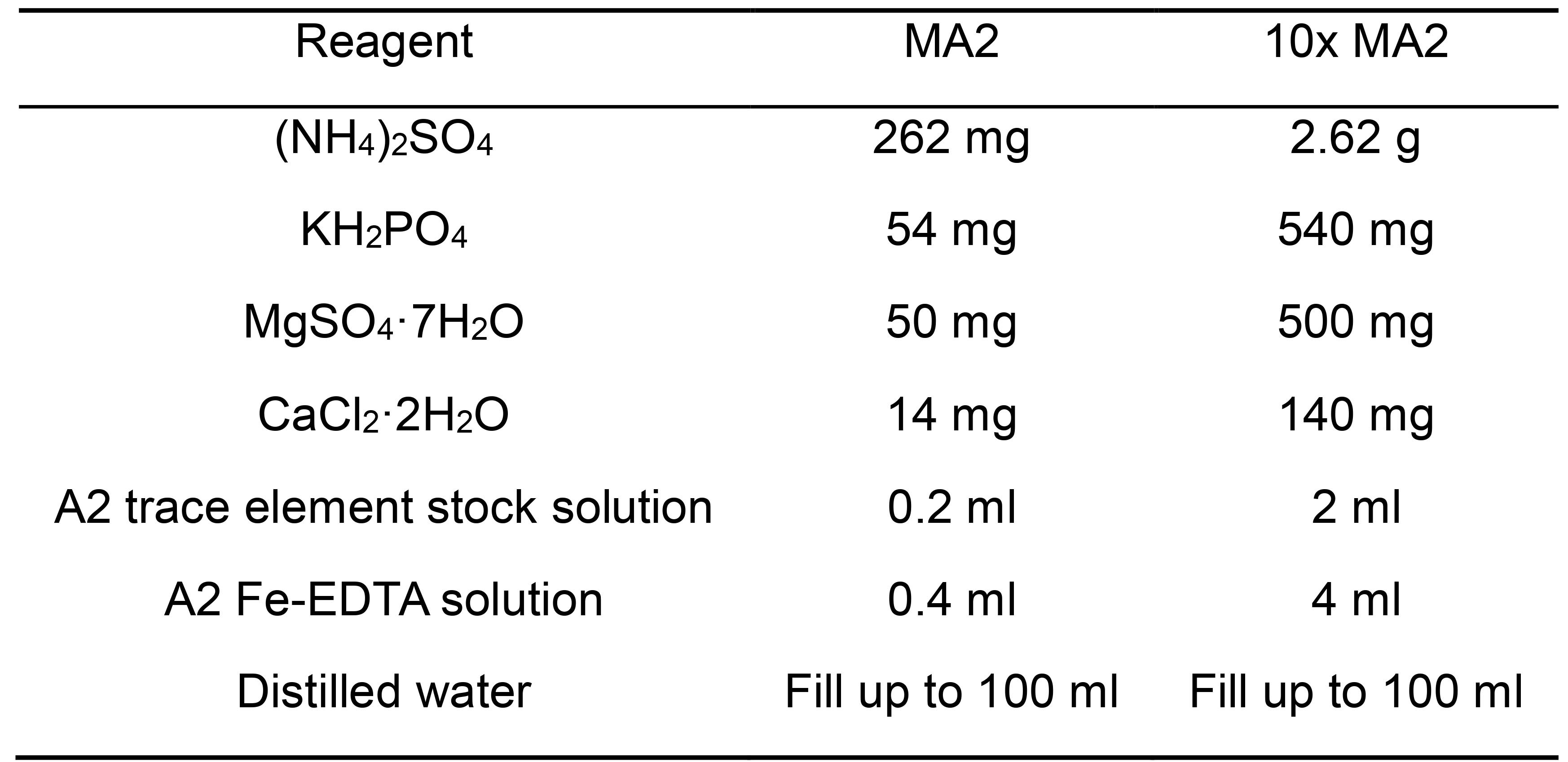
Note: Set pH to 2.5, autoclave and store sterile at RT. - Agar-solidified plates (Table 3)
Table 3. Agar-solidified plates components
Note: Autoclave agar separately in water, combine with MA2 solution in sterile conditions when both solutions are cooled down to ca. 60 °C.
- A2 Fe-EDTA solution (Table 4)
Table 4. A2 Fe-EDTA solution components
Note: First dissolve EDTA and set pH to 7, then add FeCl3, filter sterilize, store at 4 °C. - A2 trace element stock solution (Table 5)
Table 5. A2 trace element stock solution components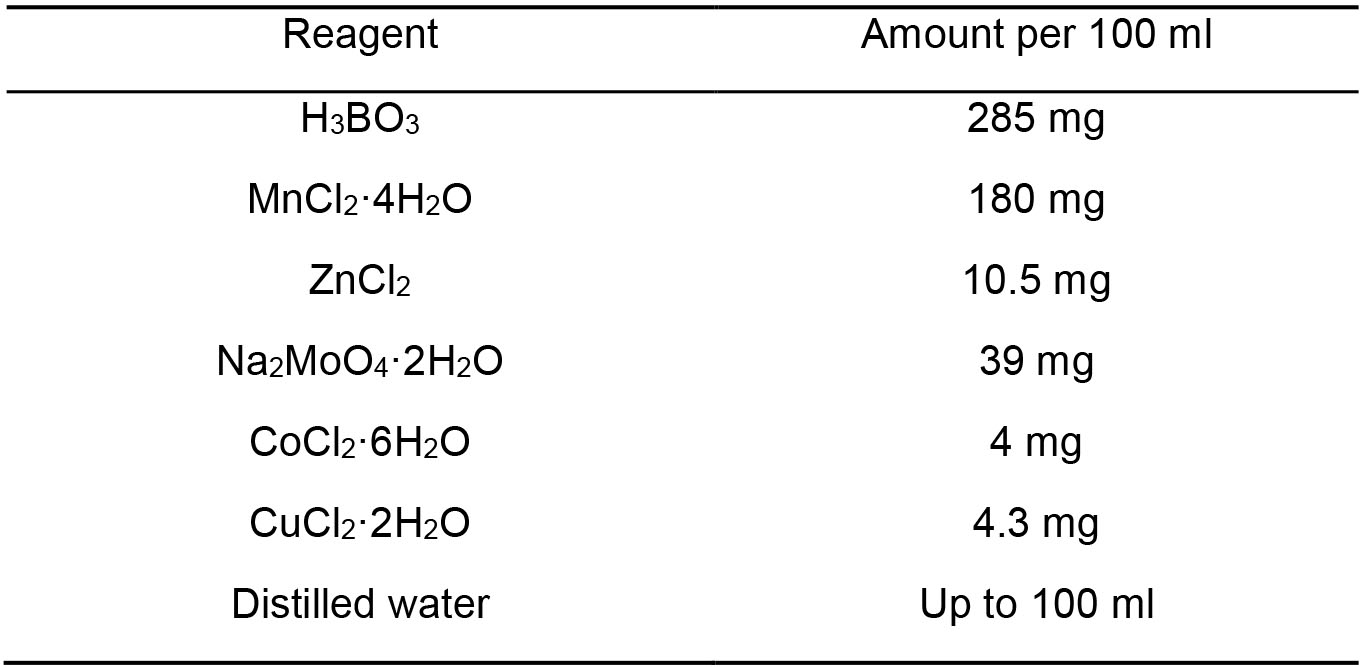
Note: Autoclave, store at 4 °C. - PEG 60% (w/v) (Table 6)
Table 6. PEG 60% component
Note: Prepare PEG at least a day earlier and allow to dissolve overnight, filter the solution with a syringe filter (0.2 µm). Store at RT. - Chloramphenicol (100 mg/ml) stock solution (Table 7)
Table 7. Chloramphenicol (100 mg/ml) stock solution components
Note: Prepare fresh every time, do not store.
Acknowledgments
This protocol was adapted from Zienkiewicz et al. (2017a and 2017b). This study was supported by grant Opus 5 (DEC-2013/09/B/NZ1/00187) awarded by the Polish National Science Centre to M.Z.
Competing interests
No competing interests declared.
References
- Hallmann, A. (2007). Algal transgenics and biotechnology. Transgenic Plant J 1(1): 81-98.
- Imamura S, Terashita M, Ohnuma M, Maruyama S, Minoda A, Weber AP, Inouye T, Sekine Y, Fujita Y, Omata T and Tanaka K. (2010). Nitrate assimilatory genes and their transcriptional regulation in a unicellular red alga Cyanidioschyzon merolae: genetic evidence for nitrite reduction by a sulfite reductase-like enzyme. Plant Cell Physiol 51(5): 707-717.
- Krupnik, T., Wasilewska, W., Drożak, A., Romanowska, E. and Zienkiewicz, M. (2018). Application of chloroplast promoters of Cyanidioschyzon merolae for exogenous protein expression. ALGAE 33(4): 351-358.
- Kuroiwa, T., Kuroiwa, H., Sakai, A., Takahashi, H., Toda, K. and Itoh, R. (1998). The division apparatus of plastid and mitochondria. Int Rev Cytol 181:1-41.
- Matsuzaki, M., Misumi, O., Shin, I. T., Maruyama, S., Takahara, M., Miyagishima, S. Y., Mori, T., Nishida, K., Yagisawa, F., Nishida, K., Yoshida, Y., Nishimura, Y., Nakao, S., Kobayashi, T., Momoyama, Y., Higashiyama, T., Minoda, A., Sano, M., Nomoto, H., Oishi, K., Hayashi, H., Ohta, F., Nishizaka, S., Haga, S., Miura, S., Morishita, T., Kabeya, Y., Terasawa, K., Suzuki, Y., Ishii, Y., Asakawa, S., Takano, H., Ohta, N., Kuroiwa, H., Tanaka, K., Shimizu, N., Sugano, S., Sato, N., Nozaki, H., Ogasawara, N., Kohara, Y. and Kuroiwa, T. (2004). Genome sequence of the ultrasmall unicellular red alga Cyanidioschyzon merolae 10D. Nature 428(6983): 653-657.
- Minoda, A., Sakagami, R., Yagisawa, F., Kuroiwa, T. and Tanaka, K. (2004). Improvement of culture conditions and evidence for nuclear transformation by homologous recombination in a red alga, Cyanidioschyzon merolae 10D. Plant Cell Physiol 45(6): 667-671.
- Newell, C. A. (2000). Plant transformation technology. Developments and applications. Mol Biotechnol 16(1): 53-65.
- Nozaki, H., Matsuzaki, M., Takahara, M., Misumi, O., Kuroiwa, H., Hasegawa, M., Shin-i, T., Kohara, Y., Ogasawara, N. and Kuroiwa, T. (2003). The phylogenetic position of red algae revealed by multiple nuclear genes from mitochondria-containing eukaryotes and an alternative hypothesis on the origin of plastids. J Mol Evol 56(4): 485-497.
- Ohnuma, M., Misumi, O., Fujiwara, T., Watanabe, S., Tanaka, K. and Kuroiwa, T. (2009). Transient gene suppression in a red alga, Cyanidioschyzon merolae 10D. Protoplasma 236(1-4): 107-112.
- Ohnuma, M., Yokoyama, T., Inouye, T., Sekine, Y. and Tanaka, K. (2008). Polyethylene glycol (PEG)-mediated transient gene expression in a red alga, Cyanidioschyzon merolae 10D. Plant Cell Physiol 49(1): 117-120.
- Ohta, N., Matsuzaki, M., Misumi, O., Miyagishima, S. Y., Nozaki, H., Tanaka, K., Shin, I. T., Kohara, Y. and Kuroiwa, T. (2003). Complete sequence and analysis of the plastid genome of the unicellular red alga Cyanidioschyzon merolae. DNA Res 10(2): 67-77.
- Ohta, N., Sato, N. and Kuroiwa, T. (1998). Structure and organization of the mitochondrial genome of the unicellular red alga Cyanidioschyzon merolae deduced from the complete nucleotide sequence. Nucleic Acids Res 26(22): 5190-5198.
- Stark, M. R., Dunn, E. A., Dunn, W. S., Grisdale, C. J., Daniele, A. R., Halstead, M. R., Fast, N. M. and Rader, S. D. (2015). Dramatically reduced spliceosome in Cyanidioschyzon merolae. Proc Natl Acad Sci U S A 112(11): E1191-1200.
- Walker, T. L., Collet, C. and Purton, S. (2005). Algal transgenics in the genomic era. J Phycol 41(6): 1077-1093.
- Watanabe, S., Ohnuma, M., Sato, J., Yoshikawa, H. and Tanaka, K. (2011). Utility of a GFP reporter system in the red alga Cyanidioschyzon merolae. J Gen Appl Microbiol 57(1): 69-72.
- Zienkiewicz, M., Krupnik, T., Drozak, A., Golke, A. and Romanowska, E. (2017a). Chloramphenicol acetyltransferase-a new selectable marker in stable nuclear transformation of the red alga Cyanidioschyzon merolae. Protoplasma 254(1): 587-596.
- Zienkiewicz, M., Krupnik, T., Drozak, A., Golke, A. and Romanowska, E. (2017b). Transformation of the Cyanidioschyzon merolae chloroplast genome: prospects for understanding chloroplast function in extreme environments. Plant Mol Biol 93(1-2): 171-183.
- Zienkiewicz, M., Krupnik, T., Drozak, A., Wasilewska, W., Golke, A. and Romanowska, E. (2018). Deletion of psbQ' gene in Cyanidioschyzon merolae reveals the function of extrinsic PsbQ' in PSII. Plant Mol Biol 96(1-2): 135-149.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Zienkiewicz, M., Krupnik, T., Drożak, A. and Kania, K. (2019). PEG-mediated, Stable, Nuclear and Chloroplast Transformation of Cyanidioschizon merolae. Bio-protocol 9(17): e3355. DOI: 10.21769/BioProtoc.3355.
Category
Plant Science > Phycology > DNA > Transformation
Plant Science > Phycology > Nuclear transformation
Molecular Biology > DNA > Transformation
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.