- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

A High-throughput and Pathophysiologically Relevant Astrocyte-motor Neuron Co-culture Assay for Amyotrophic Lateral Sclerosis Therapeutic Discovery

Published: Vol 9, Iss 17, Sep 5, 2019 DOI: 10.21769/BioProtoc.3353 Views: 7405
Reviewed by: Xi FengSunanda MarellaAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Mar 2019
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Amyotrophic lateral sclerosis (ALS) is an adult onset neurological disorder characterized by loss of motor neurons leading to progressive muscle wasting and eventually death. Astrocytes play a key role in disease pathogenesis. However, the ability to study astrocytic support towards motor neurons in ALS has been limited by a lack of sustainable high-throughput human cell models. Moreover, the ability to assess how astrocytic support of motor neurons is influenced by drug treatment or nutritional supplementation has been hampered by the lack of robust methodology. We have developed a high-throughput astrocyte motor neuron co-culture assay, which, by using Hb9-GFP+ motor neurons enables researchers to assess how ALS affects the ability of astrocytes to support motor neurons in 384-well plates. Moreover, astrocyte function can be manipulated by nutritional supplementation or drug treatment to identify possible therapeutic targets.
Keywords: ALSBackground
Amyotrophic lateral sclerosis (ALS) is a neurological disorder resulting in degeneration of both upper and lower motor neurons, resulting in the progressive failure of the neuromuscular system. Death typically occurs 2-3 years post-symptom onset, due to a lack of effective therapies. Although death of motor neurons is a key feature of ALS, the disease is non-cell autonomous with neighboring cells such as astrocytes, microglia and oligodendrocytes playing a key role (Ferraiuolo et al., 2011b; Ferraiuolo et al., 2016; Frakes et al., 2017; Vandoorne et al., 2018). Astrocytes play a crucial metabolic role in the CNS as they are the major source of brain glycogen, and astrocyte lactate can be taken up by motor neurons and used as a source of energy (Pellerin and Magistretti, 1994). Several mechanisms have been implicated in astrocyte-mediated motor neuron death, including release of nitric oxide and prostaglandin E2, altered glutamate transporter expression, reduced lactate release and reduced extracellular vesicle mediated release of miRNA (Lin et al., 1998; Ferraiuolo et al., 2011a; Ferraiuolo et al., 2011b; Allen et al., 2019; Varcianna et al., 2019).
Until recently, the ability to study how patient derived astrocytes affect motor neuronal function has been hampered by technological limitations. Initial pioneering studies (Haidet-Phillips et al., 2011, Re et al., 2014) demonstrated that post-mortem human sporadic and familial patient-derived astrocytes could induce motor neuron death in vitro. Although elegant, this methodology presents with limitations in terms of limited availability, scalability and represents end stage of disease. The development of techniques to reprogram fibroblasts into pluripotent stem cells (iPSCs) by Yamanaka and colleagues (Takahashi and Yamanaka, 2006) has radically improved our ability to study human CNS disorders in vitro, for a recent review see (Myszczynska and Ferraiuolo, 2016). This technological advancement was followed up by methodologies to directly reprogram mouse fibroblasts into induced neuronal progenitor cells (iNPCs) and concomitantly by Meyer and Ferraiuolo who developed novel methodologies to reprogram adult human fibroblasts into iNPCs from control and ALS subjects (Kim et al., 2011; Meyer et al., 2014). This advancement in direct reprogramming, has overcome the challenge of phenotypic inconsistency caused by clonal variation in iPSCs and has significantly shortened the reprogramming timescales, reducing costs and increasing throughput (Myszczynska and Ferraiuolo, 2016). Furthermore, an additional beneficial feature of the direct reprogramming approach is that the epigenetic state of the cell is not reset, so any aging phenotype inherent in the donor fibroblasts should be retained (Mertens et al., 2015). This subsequent development has enabled ALS researchers to assess the effect of disease on patient derived cells in vitro, reducing the reliance on post-mortem tissue and animal models of disease that may lack translational efficacy.
In the co-culture approach described in this paper, iNPCs were derived from ALS patient fibroblasts and healthy donor control fibroblasts as described previously (Meyer et al., 2014). iNPCs were differentiated into iAstrocytes by culturing in iAstrocyte media for at least 5 days (Figure 1). Mouse motor neurons expressing the green fluorescent protein (GFP) under the Hb9 motor neuron-specific promoter (called from now on Hb9-GFP+ MN) were differentiated from murine embryonic stem cells (mESCs) via embryonic bodies (mEBs), as previously described (Wichterle et al., 2002; Haidet-Phillips et al., 2011). The advantage of this approach over existing co-culture methods using post-mortem material or iPSCs, is that iAstrocyte support of motor neurons can be measured in a high-throughput, cost-effective fashion, under a number of conditions including drug treatment or nutritional supplementation. Furthermore, traditional cell-based high throughput screening methodologies that have been utilized in the ALS field have focused on animal or human cell models of the disease in monoculture, for a recent review see (McGown and Stopford, 2018). In the methodological approach adopted here, iAstrocytes can be reproducibly differentiated within a week from iNPCs and their effect on motor neuron survival can be measured in co-culture. Survival can be assessed by monitoring fluorescence of the motor neurons over time and counting the number of viable motor neurons (Meyer et al., 2014; Hautbergue et al., 2017; Allen et al., 2019). Moreover, the effect of treated or untreated iAstrocyte conditioned media on motor neuron survival can also be assessed (Varcianna et al., 2019). Therefore, this approach has the advantage over traditional high throughput monoculture methodologies of being more physiologically representative of the in vivo state and therefore has greater translational potential.
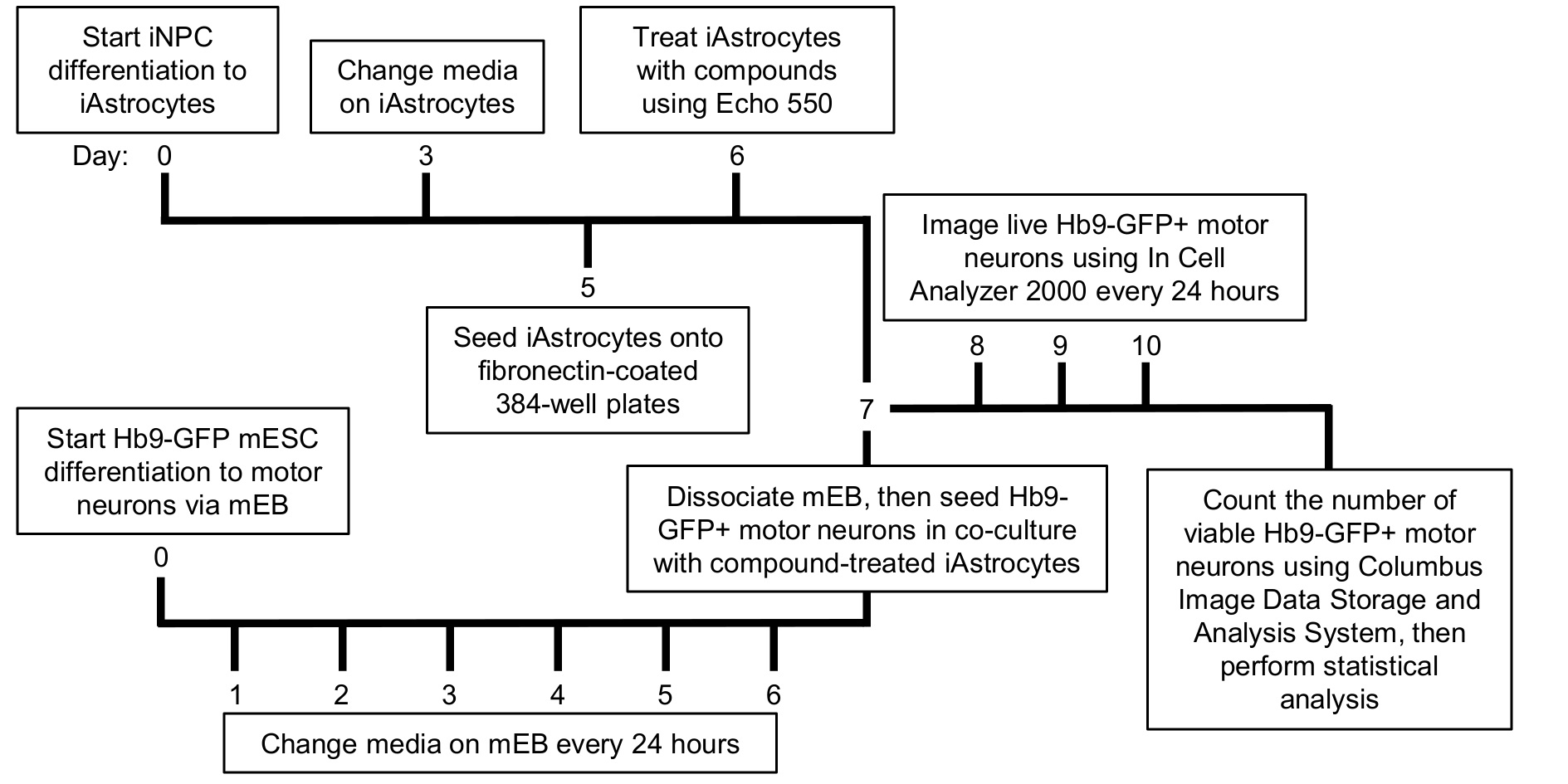
Figure 1. Timeline of the high-throughput human iAstrocyte–Hb9-GFP+ mouse motor neuron co-culture assay
Materials and Reagents
- Materials
- 10 cm tissue culture plates (Thermo Scientific, catalog number: 150350)
- 9 cm Petri dish (Scientific Laboratory Supplies, catalog number: PET2000)
- 384-well plates (Greiner-Bio, catalog number: 781091)
- 384-well PP source plates (Labcyte, catalog number: PP-0200)
- 5 ml Disposable Pipets (Fisher Scientific, catalog number: 13-676-10H)
- 10 ml Disposable Pipets (Fisher Scientific, catalog number: 13-676-10J)
- 25 ml Disposable Pipets (Fisher Scientific, catalog number: 13-678-11)
- 10 µl Pipet Tips (Fisher Scientific, catalog number: 02-707-441)
- 200 µl Pipet Tips (Fisher Scientific, catalog number: 02-707-422)
- 1,000 µl Pipet Tips (Fisher Scientific, catalog number: 02-707-402)
- 15 ml Falcon Tube (Greiner-Bio, catalog number: 188271)
- 50 ml Falcon Tube (Greiner-Bio, catalog number: 227261)
- 0.2 µm Syringe Filter (Sartorius, catalog number: 16534K)
- DMSO resistant foil lids (Brooks life sciences, catalog number: 4ti-0512)
- Steritop filter (Millipore, catalog number: S2GPT05RE)
- Sterile cell strainer, 40 µm mesh (Fisher Scientific, catalog number: 11587522)
- Sterile cell strainer, 70 µm mesh (Corning, catalog number: 431751)
- Reagents
- Primary Mouse Embryonic Fibroblasts (Sigma-Aldrich, catalog number: PMEF-CF)
- Pen-Strep (Lonza, catalog number: DE17-603E)
- 10x Trypsin (Lonza, catalog number: BE02-007E)
- 2-Mercaptoethanol (Sigma-Aldrich, catalog number: M3148)
- Accutase (Gibco, catalog number: 11599686)
- B-27 (Gibco, catalog number: 11530536)
- BDNF (Peprotec, catalog number: 450-02)
- CaCl (Sigma-Aldrich, catalog number: C1016)
- CNTF (Peprotec, catalog number: 450-13)
- D-(+)-Glucose (Sigma-Aldrich, catalog number: G7021)
- DMEM (Lonza, catalog number: 12-741F)
- DMEM/F-12, GlutaMAX (Gibco, catalog number: 11524436)
- DMSO, anhydrous (Sigma-Aldrich, catalog number: 276855)
- DNase I (Applichem Lifescience, catalog number: A3778)
- EDTA (Thermo Scientific, catalog number: 17892)
- ESC FBS (Gibco, catalog number: 11500526)
- FBS (Biosera, catalog number: FB-1090)
- FGF (Peprotec, catalogue number: 100-18B)
- GDNF (Peprotec, catalog number: 450-10)
- Ham’s F-12 Nutrient Mix (Gibco, catalog number: 15172529)
- Human Fibronectin (Sigma-Aldrich, catalog number: FC010-10MG)
- KCl (Sigma-Aldrich, catalog number: P5405)
- Knockout DMEM (Gibco, catalog number: 10389172)
- KnockOut Serum Replacement (Gibco, catalog number: 11520366)
- L-Cysteine (Sigma-Aldrich, catalog number: C7352)
- L-glutamine (Lonza, catalog number: BE-17-605E)
- MgSO4 (Sigma-Aldrich, catalog number: M2643)
- Mouse LIF (Sigma-Aldrich, catalog number: ESG1106)
- N-2 (Gibco, catalog number: 11520536)
- NaCl (Fisher Scientific, catalog number: 11904061)
- NaH2PO4 (Sigma-Aldrich, catalog number: S3139)
- NaHCO3 (Sigma-Aldrich, catalog number: S5761)
- Nitrogen (oxygen free) (BOC, catalog number: 44-Y)
- Non-Essential Amino Acids (Gibco, catalog number: 12084947)
- Papain (Sigma-Aldrich, catalog number: P4762)
- PBS Tablets (Thermo Scientific, catalog number: BR0014G)
- Retinoic Acid (Sigma-Aldrich, catalog number: R2625)
- SAG (Sigma-Aldrich, catalog number: 566660)
- Sterile 1x PBS (see Recipes)
- Sterile ultrapure water (see Recipes)
- Human iNPC Proliferation Media (see Recipes)
- Human iAstrocyte Differentiation Media (see Recipes)
- Mouse Embryonic Stem Cell (mESC) Proliferation Media (see Recipes)
- Mouse Embryonic Bodies (mEB) Differentiation Media (see Recipes)
- Mouse Motor Neuron (mMN) Media (see Recipes)
- EB Dissociation Buffer (see Recipes)
Equipment
- 4 °C fridge
- -20 °C freezer
- -80 °C freezer
- Autoclave
- Class II biosafety cabinet (NuAire, catalog number: NU-437-400E)
- CO2 Incubator (Sanyo, catalog number: MCO-19AIC)
- Echo 550 liquid handler (Labcyte, catalog number: Echo 550)
- Haemocytometer
- Harrier 15/80 benchtop centrifuge (MSE, catalog number: MSB.080.CX1.5)
- In Cell Analyzer 2000 (GE Healthcare, catalog number: 52-851714-001)
- Liquid nitrogen dewar
- Mechanical pipettes (P10, P50, P200, P1000)
- Multichannel mechanical pipettes (P10, P50)
- MultiPod Controller (Roylan Developments, catalog number: SPOD0012)
- Mechanical pipette gun
- PK120 centrifuge with T336 rota (ALC, catalog number: 11200030), and buckets for microplates (ALC, catalog number: 11210267)
- Polystyrene float for water bath
- StoragePod Enclosure (Roylan Developments, catalog number: SPOD0010)
- Water bath
- Sterile culture hood
Software
- Echo Liquid Handler Software (Labcyte)
- Echo Plate Reformat (Labcyte)
- In Cell Analyzer 2000 (GE Healthcare)
- Columbus Image Data Storage and Analysis System (PerkinElmer)
- Excel 2016 (Microsoft)
- GraphPad Prism 7.0 (GraphPad)
Procedure
Part I: Hb9-GFP Mouse Embryonic Stem Cell (mESC) Maintenance Protocol
- Prepare in advance of mESC maintenance protocol
Prepare Human iAstrocyte Differentiation Media, Mouse Embryonic Stem Cell (mESC) Proliferation Media, and sterile 1x PBS according to the recipes below in a sterile culture hood. - Defrost Primary Mouse Embryonic Fibroblasts (MEFs) the day before splitting or defrosting Hb9-GFP mESC in a sterile culture hood (unless stated otherwise)
- Place Human iAstrocyte Differentiation Media in a 37 °C water bath.
- Prepare fibronectin-coated tissue culture plates.
- Prepare 18 ml of 2.5 µg/ml Fibronectin Coating Solution per 10 cm plate by adding the 1 mg/ml Human Fibronectin to room temperature PBS in a 1:400 ratio.
- Mix the 2.5 µg/ml Fibronectin Coating Solution and transfer 6 ml to each of 3 x 10 cm tissue culture plates using a 10 ml pipet.
- Incubate the plates with the 2.5 µg/ml Fibronectin Coating Solution at room temperature for a minimum of 5 min.
- Defrost and seed MEFs into Human iAstrocyte Differentiation Media.
- Take one vial of MEFs from the liquid nitrogen dewar, and defrost the vial in a 37 °C water bath using a float.
- Remove Fibronectin Coating Solution from 10 cm tissue culture plates using a 10 ml pipet, and add 10 ml warm Human iAstrocyte Differentiation Media to each 10 cm tissue culture plate.
- Transfer 1 ml of the defrosted MEFs from the vial to 2 ml warm Human iAstrocyte Differentiation Media in a 15 ml Falcon using a P1000 (1 ml) mechanical pipette, mix and then transfer 1 ml of this MEF mixture to each 10 cm tissue culture plate.
- Rock the plates containing the MEFs back and forward, then side to side to mix.
- Incubate the plates in a 37 °C/5% CO2 incubator overnight, ready to add defrosted mESC or split mESC the following day.
- Defrost or split Hb9-GFP mESC in a sterile culture hood (unless stated otherwise)
- Place Mouse Embryonic Stem Cell (mESC) Proliferation Media and 1x Trypsin solution in a 37 °C water bath.
- Change the media on the 10 cm plates containing MEFs.
- Prepare 45 ml of mESC Proliferation Media + LIF by adding 6 µl LIF to 45 ml warm mESC Proliferation Media in a 50 ml Falcon tube using a P10 (10 µl) mechanical pipette.
- Remove Human iAstrocyte Differentiation Media from MEFs using a 10 ml pipet.
- Add 15 ml warm mESC Media + LIF to each 10 cm plate containing MEFs using a 25 ml pipet.
- Incubate the plates of MEFs in a 37 °C/5% CO2 for a minimum of 30 min, or until mESC are ready to plate.
- Defrost and seed mESC onto MEF feeder plates.
- Take one vial of mESC from the liquid nitrogen dewar, and defrost the vial in a 37 °C water bath using a float.
- Transfer 1 ml of the defrosted MEFs from the vial to 2 ml warm mESC Proliferation Media in a 15 ml Falcon using a P1000 (1 ml) mechanical pipette, mix and then transfer 1 ml of this mESC mixture to each 10 cm plate of MEFs.
- Rock the plates containing the mESC back and forward, then side to side to mix.
- Incubate the mESC plates in a 37 °C/5% CO2 incubator.
- Alternatively to Step C3, the mESC may already be in culture, and can be split (instead of being defrosted) onto the MEFs (according to the ‘split the mESC’ section below).
- Change the media on mESC every day in a sterile culture hood (unless stated otherwise)
- Place mESC Proliferation Media in a 37 °C water bath.
- Change the media on the 10 cm plates containing mESC.
- Prepare 45 ml of mESC Media + LIF by adding 6 µl LIF to 45 ml warm mESC Proliferation Media in a 50 ml Falcon tube using a P10 (10 µl) mechanical pipette.
- Remove most of the media from mESCs plates using a 25 ml pipet, leaving a few ml to cover the mESC.
- Add 15 ml warm mESC Media + LIF to each mESC plate using a 25 ml pipet.
- Incubate the mESC plates in a 37 °C/5% CO2 for 24 h.
- Repeat steps 1 and 2a-2d every day.
- Split the mESC every 3-4 days in a sterile culture hood
- Place mESC Proliferation Media and 1x Trypsin solution in a 37 °C water bath.
- Split Hb9-GFP mESC into mEB Differentiation Media in a sterile culture hood (unless stated otherwise).
- Remove all mESC Proliferation Medium from mESC plates using a 25 ml pipet, and wash the 10 cm plate of cells in 8 ml room temperature PBS using a 10 ml pipet.
- Remove all the PBS wash using a 10 ml pipet, and add 1 ml 1x trypsin to each plate of mESC using a P1000 (1 ml) mechanical pipette.
- Incubate the mESC with trypsin in a 37 °C incubator for 5 min.
- Incubate longer if necessary-ideally mESC come off in suspension with some MEFs as well.
- Add 10 ml mESC Proliferation Media to each plate using a 10 ml pipet.
- For each plate of mESC, inspire the mESC and media using a 10 ml pipet, and release the suspension back into the 10 cm tissue culture plate whilst pressing the pipet nozzle to the bottom of the plate and applying pressure. Repeat this step 10 times. This process helps to break up the mESC colonies into single cells.
- Transfer the contents of each 10 cm plate into separate, clean 10 cm tissue culture plates using a 10 ml pipet, and incubate in a 37 °C/5% CO2 incubator for 30 min.
- For each plate, inspire the mESC suspension and wash the plate a few times with the mESC suspension using a 10 ml pipet, and then transfer all mESC suspension to a 50 ml Falcon tube. Combine up to three 10 cm plates of mESC suspension in one 50 ml Falcon tube.
- On the bench, centrifuge the mESC suspension at 200 x g for 4 min at room temperature.
- Remove the supernatant using a 25 ml pipet, and then add 4 ml mESC Proliferation Media for each 10 cm plate of mESC that was transferred to the Falcon tube.
- Re-suspend the mESC pellet using a 10 ml pipet.
- Place a sterile 70 µm cell strainer in a 50 ml Falcon tube, and then transfer the mESC suspension through the strainer using a 10 ml pipet.
- Rinse the strainer with 1 ml mESC Proliferation Media.
- Add 150-200 µl of the mESC suspension to each 10 cm plate of MEF in mESC Media + LIF using a P200 (200 µl) mechanical pipette.
- Rock the plates containing the mESC back and forward, then side to side to mix.
- Incubate the mESC plates in a 37 °C/5% CO2 incubator.
Part II: Human iNPC Maintenance Protocol
- Prepare in advance of Human iNPC maintenance protocol
Prepare Human iNPC Proliferation Media and sterile 1x PBS according to the recipes below in a sterile culture hood. - Split iNPC
- Place Human iNPC Proliferation Media and 1x Accutase solution in a 37 °C water bath.
- Prepare fibronectin-coated tissue culture plates for iNPC maintenance.
- Prepare 6 ml of 5 µg/ml Fibronectin Coating Solution per 10 cm tissue culture plate by adding the 1 mg/ml Human Fibronectin to room temperature PBS in a 1:200 ratio.\
- Mix the 5 µg/ml Fibronectin Coating Solution and transfer 6 ml to each 10 cm tissue culture plate using a 10 ml pipette.
- Incubate the plates with the 5 µg/ml Fibronectin Coating Solution at room temperature for a minimum of 5 min.
- Split iNPC into Human iNPC Proliferation Media in a sterile culture hood (unless stated otherwise).
- Remove all Human iNPC Proliferation Media from iNPC using a 10 ml pipet, and wash the cells in 5 ml room temperature PBS using a 10 ml pipet.
- Remove all the PBS wash using a 10 ml pipet, and add 1 ml Accutase to the iNPC using a P1000 (1 ml) mechanical pipette.
- Incubate the iNPC with Accutase in a 37 °C incubator for 4 min.
- Gently tap the iNPC plate to dislodge the cells from the plate completely.
- Add 5 ml room temperature PBS to the lifted iNPC, mix and transfer to a 15 ml Falcon tube using a 10 ml pipet.
- On the bench, centrifuge the iNPCs at 200 x g for 4 min at room temperature.
- Pour off the supernatant, and re-suspend the iNPC pellet in an appropriate amount of Human iNPC Proliferation Media.
- Remove the Fibronectin Coating Solution from the 10 cm tissue culture plates using a 10 ml pipet.
- Add 12 ml warm Human iNPC Proliferation Media to the 10 cm tissue culture plates coated with 5 µg/ml Fibronectin Coating Solution using a 10 ml pipet.
- Add an appropriate volume of the re-suspended iNPC solution to the Human iNPC Proliferation Media in the 10 cm tissue culture plates.
- Incubate the iNPC proliferation plates in a 37 °C/5% CO2 incubator for 2-4 days, or until the iNPCs reach 80-90% confluency, and then split the iNPCs again as described above.
Part III: Human iAstrocyte-mouse Hb9-GFP+ Motor Neuron Co-culture Protocol
- Prepare in advance of co-culture protocol
- Prepare Human iNPC Proliferation Media, Human iAstrocyte Differentiation Media, Mouse Embryonic Stem Cell (mESC) Proliferation Media, Mouse Embryonic Bodies (mEB) Differentiation Media, sterile 1x PBS, and sterile ultrapure water according to the recipes below in a sterile culture hood.
- Prepare compound library in 100% anhydrous DMSO on 384-well PP source plates, with DMSO resistant foil lid. Store in the StoragePod Enclosure and maintain dry nitrogen atmosphere using the MultiPod Controller.
- Write compound transfer protocol using the Echo liquid handler software. Briefly, set the source plate to ‘384PP_DMSO’, set the destination plate to ‘Griener_384PS_781096’, and use a custom mapping mode. Set the protocol to transfer 40 nl of compound in DMSO from the source plate to each well on the destination plate. Given there will be 40 µl media in each well on the destination plate, the final DMSO concentration will be 0.1% (v/v). Set the protocol to deliver 6 technical replicate wells for each test compound, and 12-16 technical replicate wells for the positive control and 12-16 technical replicate wells for the negative control (DMSO only). We discovered a compound in house that increased Hb9-GFP+ mMN survival in the co-culture assay, which we will refer to as compound A, and used that as our positive control. For a comprehensive dose response of select test compounds, we recommend testing a 7-point dose response at every half-log, i.e., 0.01, 0.03, 0.1, 0.3, 1, 3, and 10 µM.
- Write an ‘Hb9-GFP MN’ protocol for imaging the Hb9-GFP+ motor neurons on the In Cell Analyzer 2000 software. Briefly, set the following criteria: objective = Nikon 10x; number of fields = 4 per well; wavelength = FITC (Ex490 nm/Em525 nm); exposure = 0.5 s; focus = laser autofocus at 10% power; deconvolution = enhanced ratio method, 5 cycles; plate temperature = 37 °C. Note that other high-throughput fluorescence microscopes and software could be used instead of the In Cell Analyzer 2000 and In Cell Analyzer 2000 software respectively, however, the imaging protocols would need adapting accordingly.
- Differentiate Human iNPC into Human iAstrocytes (Day 0)
- Place Human iNPC Proliferation Media, Human iAstrocyte Differentiation Media, Mouse Embryonic Bodies (mEB) Differentiation Media, Mouse Embryonic Stem Cell (mESC) Proliferation Media, 1x Trypsin solution and 1x Accutase solution in a 37 °C water bath.
- Prepare fibronectin-coated tissue culture plates for iAstrocyte differentiation.
- Prepare 6 ml of 2.5 µg/ml Fibronectin Coating Solution per 10 cm plate by adding the 1 mg/ml Human Fibronectin to room temperature PBS in a 1:400 ratio.
- Mix the 2.5 µg/ml Fibronectin Coating Solution and transfer 6 ml to each 10 cm tissue culture plate using a 10 ml pipet.
- Incubate the plates with the 2.5 µg/ml Fibronectin Coating Solution at room temperature for a minimum of 5 min.
- Prepare fibronectin-coated tissue culture plates for iNPC maintenance (see Human iNPC Maintenance Protocol above).
- Split iNPC into Human iAstrocyte Differentiation Media or Human iNPC Proliferation Media in a sterile culture hood (unless stated otherwise).
- Remove all Human iNPC Proliferation Media from iNPC using a 10 ml pipet, and wash the cells in 5 ml room temperature PBS using a 10 ml pipet.
- Remove all the PBS wash using a 10 ml pipet, and add 1 ml Accutase to the iNPC using a P1000 (1 ml) mechanical pipette.
- Incubate the iNPC with Accutase in a 37 °C incubator for 4 min.
- Gently tap the iNPC plate to dislodge the cells from the plate completely.
- Add 5 ml room temperature PBS to the lifted iNPC, mix and transfer to a 15 ml Falcon tube using a 10 ml pipet.
- On the bench, centrifuge the iNPCs at 200 x g for 4 min at room temperature.
- Pour off the supernatant, and re-suspend the iNPC pellet in an appropriate amount of Human iNPC Proliferation Media.
- Remove the Fibronectin Coating Solution from the 10 cm tissue culture plates using a 10 ml pipet.
- Add 12 ml warm Human iAstrocyte Differentiation Media to the 10 cm tissue culture plates coated with 2.5 µg/ml Fibronectin Coating Solution using a 10 ml pipet.
- Add 12 ml warm Human iNPC Proliferation Media to the 10 cm tissue culture plates coated with 5 µg/ml Fibronectin Coating Solution using a 10 ml pipet.
- Add an appropriate volume of the re-suspended iNPC solution to the Human iAstrocyte Differentiation Media in the 10 cm tissue culture plates.
- Add an appropriate volume of the re-suspended iNPC solution to the Human iNPC Proliferation Media in the 10 cm tissue culture plates.
- Incubate the iAstrocyte differentiation plates in a 37 °C/5% CO2 incubator for 3 days.
- Incubate the iNPC proliferation plates in a 37 °C/5% CO2 incubator for 2-4 days, or until the iNPCs reach 80-90% confluency, and then split the iNPCs as described above.
- Differentiate mESC into mEB (Day 0)
- Change the media on the 10 cm plates containing MEFs (as described in the Hb9-GFP mESC Maintenance Protocol above).
- Split the Hb9 mESC into mEB Differentiation Media in a sterile culture hood (unless stated otherwise).
- Split the mESC (as described in the Hb9-GFP mESC Maintenance Protocol above).
- Add 18 ml warm mEB Differentiation media to a 9 cm Petri dish, then add 1 ml of the mESC suspension using a P1000 (1 ml) mechanical pipette to start mESC differentiation into motor neurons via mEB. Each 10 cm plate of mEB will yield approximately 4 x 106 Hb9-GFP mouse motor neurons after 7 days of differentiation.
- Incubate the plates of mEB in a 37 °C/5% CO2 for 24 h.
- Change media on mEBs (Days 1-6)
- Place mEB Differentiation Media in a 37 °C water bath.
- Change media on EBs.
- Transfer mEB and media from 9 cm Petri dish to a 50 ml Falcon using a 25 ml pipet.
- Incubate the Falcon tubes for 10 min in a Falcon rack in the hood, allowing the mEB to sink to the bottom of the Falcon tube.
- Remove the media in the Falcon from the top downwards, leaving the final 5 ml at the bottom of the Falcon tube that contains the soft pellet of mEBs.
- Add 15 ml fresh mEB Differentiation Media to each Falcon tube.
- On Days 2-6 only, add 9 µl 4 mM Retinoic Acid (in ethanol) and 18 µl 1 mM SAG to each Falcon tube of mEBs using a P20 (20 µl) mechanical pipette.
- Mix the mEB and media using a 25 ml pipet, and then transfer to a fresh 9 cm Petri dish.
- Incubate the plates of mEBs in a 37 °C/5% CO2 incubator for 24 h.
- Repeat steps 1 and 2a-2g every day until day of EB dissociation.
- Change media on iAstrocytes (Day 3)
- Place Human iAstrocyte Differentiation Media in a 37 °C water bath.
- Remove all Human iAstrocyte Differentiation Media from iAstrocytes using a 10 ml pipet.
- Add 12 ml fresh Human iAstrocyte Differentiation Media to each 10 cm tissue culture plate of iAstrocytes.
- Incubate the plates in a 37 °C/5% CO2 incubator for 2 days.
- Seed iAstrocytes onto 384-well plates (Day 5)
- Place Human iAstrocyte Differentiation Media and 1x Accutase solution in a 37 °C water bath.
- Prepare fibronectin-coated 384-well plates.
- Prepare 2 ml of 2.5 µg/ml Fibronectin Coating Solution per 384-well plate by adding the 1 mg/ml Human Fibronectin to room temperature PBS in a 1:400 ratio.
- Mix the 2.5 µg/ml Fibronectin Coating Solution and transfer it to a 9 cm Petri dish to act as a reservoir.
- Transfer 5 µl of the Fibronectin Coating Solution to each well of the 384-well plate excluding the outermost wells using a P10 (10 µl) multichannel mechanical pipette.
- Transfer 40 µl of PBS to each of the outermost wells on the 384-well plate to act as a firewall using a P50 (50 µl) multichannel mechanical pipette.
- Incubate the plates with the 2.5 µg/ml Fibronectin Coating Solution at room temperature for a minimum of 5 min.
- Lift iAstrocytes and seed 2,000 iAstrocytes/well onto 384-well plates in a sterile culture hood (unless stated otherwise).
- Remove all Human iAstrocyte Differentiation Media from iAstrocytes using a 10 ml pipet, and wash the cells in 5 ml room temperature PBS using a 10 ml pipet.
- Remove all the PBS wash using a 10 ml pipet, and add 1 ml Accutase to the iAstrocytes using a P1000 (1 ml) mechanical pipette.
- Incubate the iAstrocytes with Accutase in a 37 °C incubator for 4 min.
- Gently tap the iAstrocyte plate to dislodge the cells from the plate completely.
- Add 5 ml room temperature PBS to the lifted iAstrocyte, mix and transfer to a 15 ml Falcon tube using a 10 ml pipet.
- On the bench, centrifuge the iAstrocytes at 200 x g for 4 min at room temperature.
- Pour off the supernatant, flick the bottom of the Falcon tube to gently vortex the iAstrocyte pellet, and re-suspend the iAstrocytes in an appropriate amount of Human iAstrocyte Differentiation Media.
- Prepare a Haemocytometer, and transfer 10 µl of the iAstrocyte suspension onto the counting grid.
- Count the number of iAstrocytes, and calculate the cells/mL in suspension.
- Dilute the iAstrocytes to 5.7 x 104 iAstrocytes/ml in Human iAstrocyte Differentiation Media, and transfer the iAstrocyte suspension to a 9 cm Petri dish to act as a reservoir.
- Transfer 35 µl of the 5.7 x 104 iAstrocytes/ml suspension to each well of the 384-well plate–excluding the outermost wells–using a P50 (50 µl) multichannel mechanical pipette.
- Centrifuge the 384-well plates at 400 x g for 60 s using a PK120 centrifuge with T336 rota and buckets for microplates to collect media and cells to base of wells.
- Incubate the 384-well plates in a 37 °C/5% CO2 incubator for 24 h to allow iAstrocytes to adhere to the plates.
- Treat iAstrocytes with compounds (Day 6)
- De-pressurise the StoragePod Enclosure using the MultiPod Controller, unlock and take the 384-well PP source plate out of the StoragePod Enclosure.
- Calibrate and focus the Echo 550 liquid handler.
- Centrifuge the 384-well PP source plate at 1,200 x g for 120 s using a PK120 centrifuge with T336 rota and buckets for microplates to de-gas the compounds in DMSO.
- Survey the 384-well PP source plate containing the compounds in DMSO. There should be adequate volume to deliver compounds from the source plate to the destination plate using the desired compound transfer protocol (described in the ‘prepare in advance of co-culture protocol’ section above). In addition, the compounds should have low water content-below 70% DMSO is unacceptable and drugs should be refreshed on the source plate.
- Transfer compounds in DMSO from 384-well PP source plate to 384-well plate’s destination plate containing iAstrocytes using the compound transfer protocol using the Echo 550 liquid handler. The final concentration of DMSO should not exceed 0.5% (v/v) in the iAstrocyte media and should be consistent in all wells in any given experiment. Include DMSO only wells as negative controls.
- Centrifuge the 384-well plates at 400 x g for 60 s using a PK120 centrifuge with T336 rota and buckets for microplates to collect media and cells to base of wells.
- Incubate the 384-well plates in a 37 °C/5% CO2 incubator for 24 h.
- Put the DMSO resistant foil lid back on the 384-well PP source plate and return to the StoragePod Enclosure, lock and then re-pressurise the StoragePod Enclosure with nitrogen using the MultiPod Controller.
- Dissociate mEBs and seed mouse GFP+ motor neurons in co-culture with compound-treated iAstrocytes (Day 7)
- Prepare EB Dissociation Buffer according to the recipe below in a sterile tissue culture hood.
- Prepare an appropriate volume of Mouse Motor Neuron (mMN) Media according to the recipe below in a sterile tissue culture hood.
- Place mMN Media, EB Dissociation Buffer, and FBS in a 37 °C water bath.
- Dissociate mEBs
- Transfer the mEBs and media from one or two mEB plates into a 50 ml Falcon tube.
- On the bench, centrifuge the mEBs at 200 x g for 2 min at room temperature.
- Remove the supernatant using a 25 ml pipet, and re-suspend the mEB in 10 ml PBS.
- On the bench, centrifuge the mEBs at 200 x g for 2 min at room temperature.
- Remove the PBS wash using a 10 ml pipet.
- Add 4.75 ml warm EB Dissociation Buffer and 100 µl 200 U/ml Papain to each Falcon of mEBs.
- Gently pipette up and down ten times using a P1000 (1 ml) mechanical pipette.
- Incubate the Falcon tubes in a 37 °C water bath for 3 min.
- Remove the tube and gently shake, then return to the 37 °C water bath for 2 min.
- Gently pipette the mEB solution up and down ten times using a P1000 (1 ml) mechanical pipette.
- Repeat steps h-j up to 3 more times, or until the solution is cloudy with no large mEB clumps.
- Depending on the activity of the papain, more 200 U/ml Papain may need to be added to the Falcons to completely dissociate the mEBs.
- Equally, avoid excessive papain treatment because the cells will lyse.
- On the bench, centrifuge the mEBs at 300 x g for 5 min at room temperature.
- For each 50 ml Falcon tube, prepare 2.7 ml EB Dissociation Buffer and add 300 µl FBS and 150 µl 0.5 mg/ml DNase I using a P200 (200 µl) mechanical pipette, and mix the FBS/DNAse I solution using a 10 ml pipet.
- Remove supernatant from the dissociated mEBs using a 10 ml pipet, and add 3 ml of the FBS/DNAse I solution using a 10 ml pipet.
- Gently pipette the mEB solution up and down five times using a P1000 (1 ml) mechanical pipette.
- Add 5 ml FBS very slowly to the bottom of the 50 ml Falcon containing the dissociated mEBs using a 10 ml pipet. This creates a cushion, and an interface between the mEB solution (upper fraction) and the FBS (lower fraction) should be clearly visible.
- On the bench, centrifuge the mEBs at 100 x g for 6 min at room temperature, to pellet the dissociated cells from the mEBs whilst leaving debris in the upper supernatant fraction.
- Remove the supernatant using a 10 ml pipet, and then gently re-suspend the cells in an appropriate volume of mMN Media.
- Gently pipette the mMN solution up and down five times using a P1000 (1 ml) mechanical pipette.
- Place a sterile 40 µm cell strainer in a 50 ml Falcon tube, and then transfer the mMN suspension through the strainer using a 5 ml pipet.
- Rinse the strainer with 1 ml mMN Media using a P1000 (1 ml) mechanical pipette.
- Prepare a Haemocytometer, and transfer 10 µl of the mMN suspension onto the counting grid.
- Count the bright round cells, exclude the darker rough-edged cells and potential debris (Figure 2), and then calculate the neuronal cells/ml in suspension.
- Dilute the mMN to 2.5 x 106 neuronal cells/ml in mMN Media, and transfer the mMN suspension to a 9 cm Petri dish to act as a reservoir.
- Transfer 10 µl of the 2.5 x 106 neuronal cells/ml suspension to each well of the 384-well plate containing the compound-treated iAstrocytes-excluding the outermost wells-using a P50 (50 µl) multichannel mechanical pipette.
- Centrifuge the 384-well plates at 400 x g for 60 s using a PK120 centrifuge with T336 rota and buckets for microplates to collect media and cells to base of wells.
- Incubate the 384-well Human iAstrocyte-Mouse Motor Neuron co-culture plates in a 37 °C/5% CO2 incubator for 24 h.
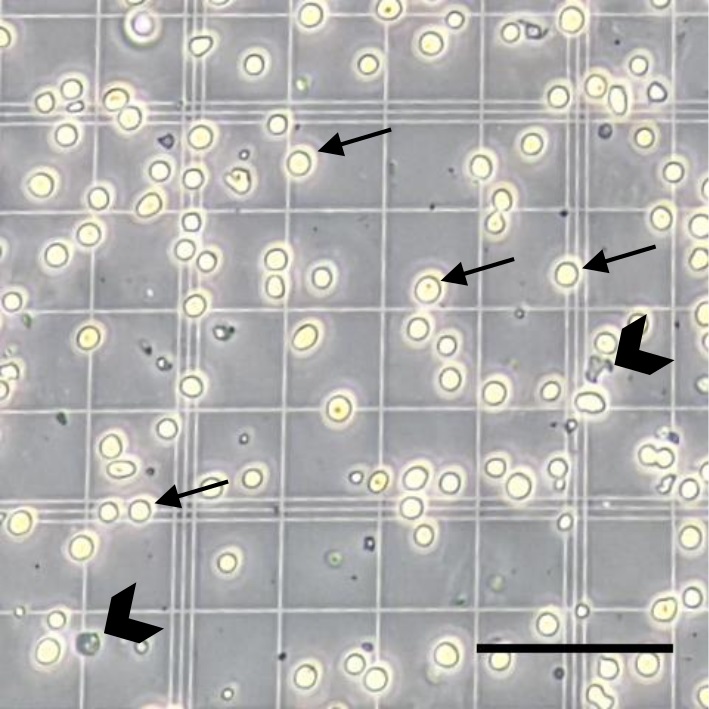
Figure 2. Counting cells derived from the Mouse Embryonic Bodies using a haemocytometer. The bright and round cells yielded from the Mouse Embryonic Bodies (mEBs) are included in the neuronal cell count (the four small arrows indicate examples). The darker rough-edged cells and potential debris are excluded from the neuronal cell count (the two large chevrons indicate examples). Scale bar = 100 µm.
- Add media to co-culture plates (Day 8)
- Prepare an appropriate volume of Mouse Motor Neuron (mMN) Media according to the recipe below in a sterile tissue culture hood.
- Place mMN Media in a 37 °C water bath.
- Add 15 µl warm mMN Media to each well of the 384-well plate containing the iAstrocyte-MN co-culture-excluding the outermost wells using a P50 (50 µl) multichannel mechanical pipette.
- Image the Hb9-GFP+ MN on a high throughput microscope (Days 8 and 10)
- Turn on the illuminator and pre-heat the In Cell Analyzer 2000 to 37 °C.
- Once the illuminator is ready, run the ‘Hb9-GFP MN’ protocol (described in the ‘prepare in advance of co-culture protocol’ section above) to image Hb9-GFP+ motor neurons in the co-culture plates using an In Cell Analyzer 2000 (Figure 3). For downstream analysis, the Hb9-GFP+ motor neurons imaged at Days 8 and 10 of the co-culture protocol are simply referred to as Day 1 and 3 respectively.
- Incubate the co-culture plates in a 37 °C/5% CO2 incubator.
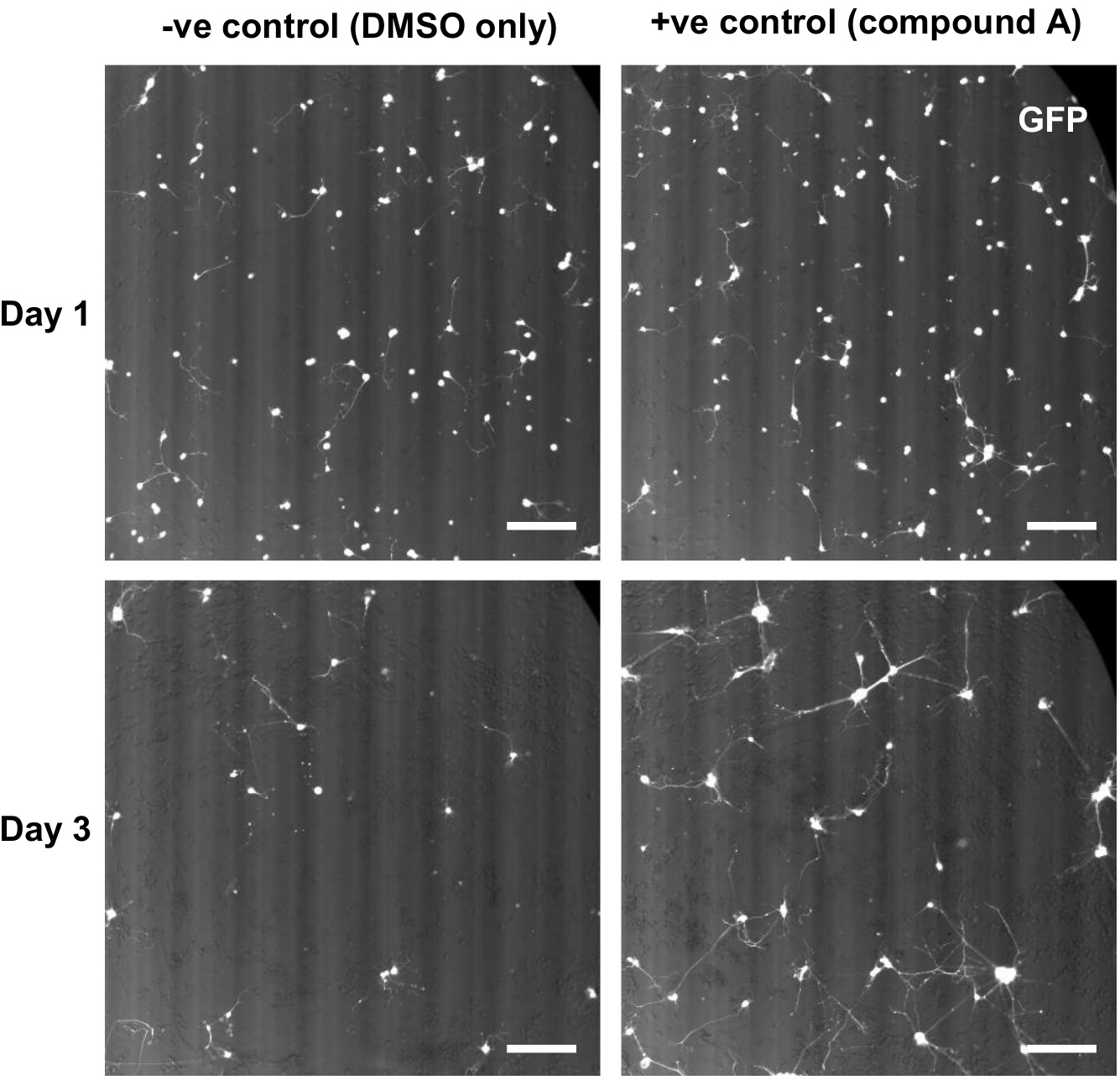
Figure 3. Example images of human iAstrocyte-Hb9-GFP+ mMN co-cultures. Representative images of human iAstrocytes and Hb9-GFP+ mMN in co-culture at 1 and 3 days post-mMN seeding, imaged on an In Cell Analyzer 2000 using the GFP channel. iAstrocytes were treated with either DMSO only (-ve control), or compound A (+ve control) 24 h prior to mMN seeding in co-culture. After 3 days of co-culture, there is greater mMN survival in the wells treated with compound A compared to wells treated with DMSO only. Scale bars = 200 µm.
Data analysis
- Motor neuron viability assessment
- Import the .XCDE/.tif files captured on the In Cell Analyzer 2000 to the Columbus Image Data Storage and Analysis System.
- Write an analysis protocol on the Columbus Image Data Storage and Analysis System that can identify and count GFP+ mMN. The analysis protocol is made up of several ‘building blocks’ or steps in the Image Analysis section on the Columbus Image Data Storage and Analysis System, which are detailed and described below:
- Input Image. Here, the images are loaded into the analysis protocol (Figure 4A). Set stack processing as ‘individual planes’ and set no ‘flat field correction’ and no ‘quick tune’.
- Find Nuclei. Identify the GFP+ cell bodies of the motor neurons (Figure 4B). Use method ‘M’ on the GFP channel. Method ‘M’ is a robust method provided on the Columbus Image Data Storage and Analysis System used to detect round objects, termed ‘nuclei’, from a range of different types of images.
- Calculate Morphology Properties. This step allows the calculation of standard morphological properties such as area or roundness of the GFP+ cell bodies or ‘nuclei’ in the images. Use the ‘standard method’ in this step, which allows the images to be filtered based on cell area size.
- Select Population (1). Select the population of GFP+ cell bodies that are greater than 120 pixels2, and also have a width to length ratio of greater than 0.2 (Figure 4C). This filters out small GFP+ items such as debris.
- Calculate Intensity Properties. This step allows the calculation of standard intensity properties, such as the average pixel intensity of the GFP+ cell bodies or ‘nuclei’ in the images. Use the ‘standard method’ option in the Columbus Image Data Storage and Analysis System in this step.
- Select Population (2). Select the population of GFP+ cell bodies that have an average pixel intensity greater than 2,000 (arbitrary units) (Figure 4D). This filters out dim GFP+ items such as debris.
- Find Neurites. This step identifies GFP+ neurites that are growing from the GFP+ cell bodies (defined by steps b-f) (Figure 4E). Use the ‘CSIRO Neurite Analysis 2’ analysis method, which is done by an algorithm developed by the Australian CSIRO research institute. The exact parameters used in this step may require some slight modification for different experimental repeats.
- Select Population (3). Select the population of GFP+ cell bodies (defined by steps b-f) that have at least one neurite (defined in step g) connected (Figure 4F). This population is the number of viable GFP+ mMN. GFP+ cell bodies that do not have an axon attached are dead, and therefore filtered out of the viable GFP+ mMN count.
- Define Results. This step performs statistical analysis on multiple parameters including the number of GFP+ cell bodies, the number of GFP+ cell bodies with at least one neurite (i.e., viable GFP+ mMN), and other morphological and intensity properties from the images.
- Count the number of viable GFP+ mMN in the images from Days 1 and 3 using the analysis protocol described in step 2 using the batch analysis section of the Columbus Image Data Storage and Analysis System.
- Export result file to Excel 2016.
- Plot the number of viable GFP+ mMN at Day 3 in GraphPad 7.0.
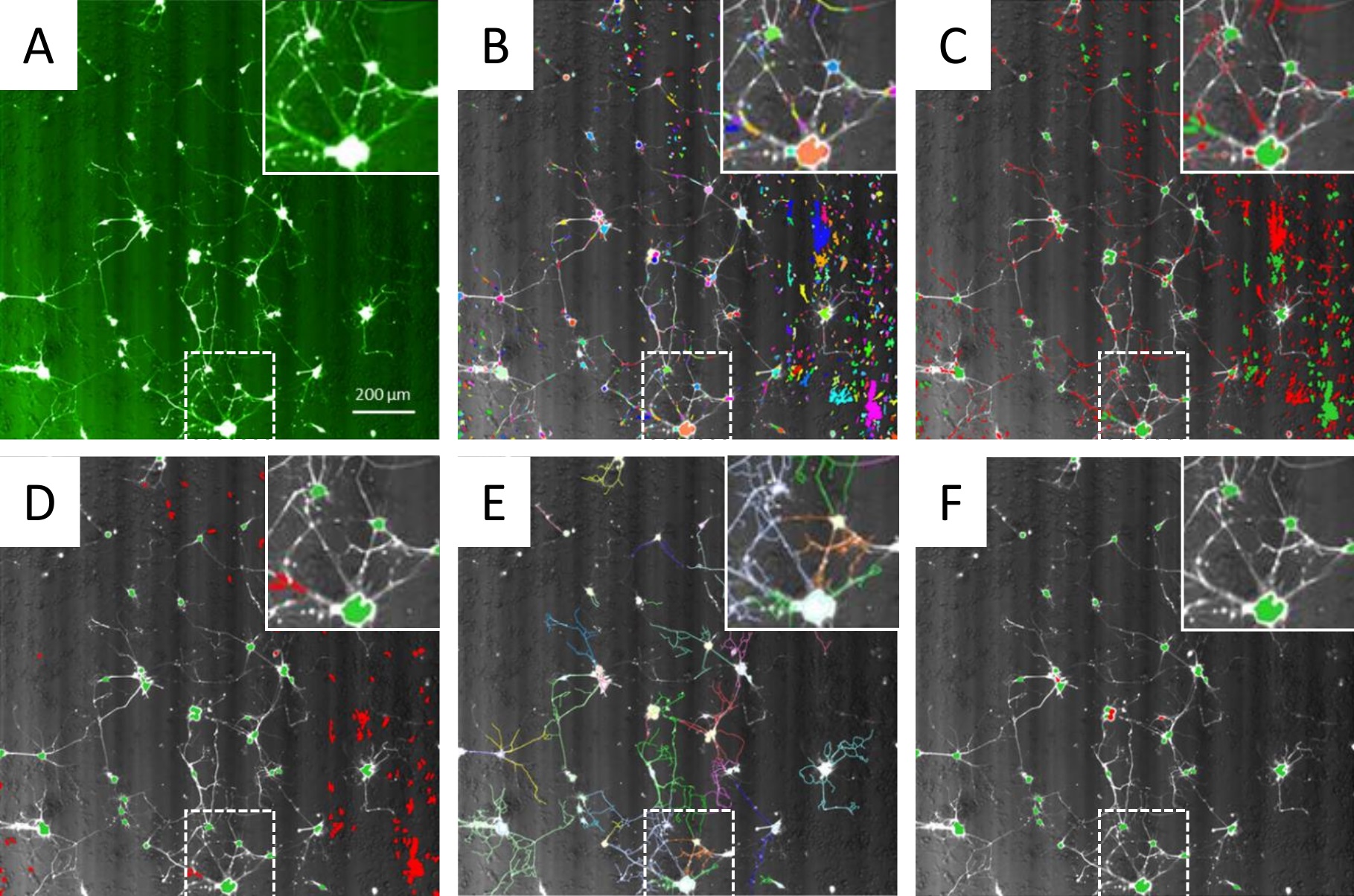
Figure 4. Analysis protocol on the Columbus Image Data Storage and Analysis System to identify and count GFP+ mMN. A. The images from the GFP channel are loaded into the Columbus Image Analysis software. Scale bar = 200 µm. B. Round GFP+ items are identified and then highlighted in multiple different colors to distinguish the separate items. These Round GFP+ items include the mMN cell bodies. C. GFP+ items that are greater than 120 pixels2 in size are selected (green), whilst GFP+ items that are smaller than 120 pixels2 are excluded (red). D. GFP+ items that have an average intensity greater than 2,000 are selected (green), whilst GFP+ items that have an average pixel intensity of less than 2,000 are excluded (red). E. GFP+ neurites that are growing from the GFP+ cell bodies are identified and then highlighted in multiple different colors to distinguish the separate neurites. F. The number of GFP+ cell bodies with at least one neurite attached (green) are counted, whilst GFP+ cell bodies with no neurites are excluded (red). The area highlighted with the dashed white box within each image is magnified 2x in the box inset at the top right of each image.
- Statistical analysis
The exact statistical analysis required depends on the experimental set-up. Typically, One-way ANOVA with Dunnett’s post-hoc test (comparing each condition to the DMSO control) is performed when screening multiple compounds at a single dose, or multiple doses of a single compound in one iAstrocyte cell line. Statistical analysis is performed in Graphpad 7.0 (Figure 5).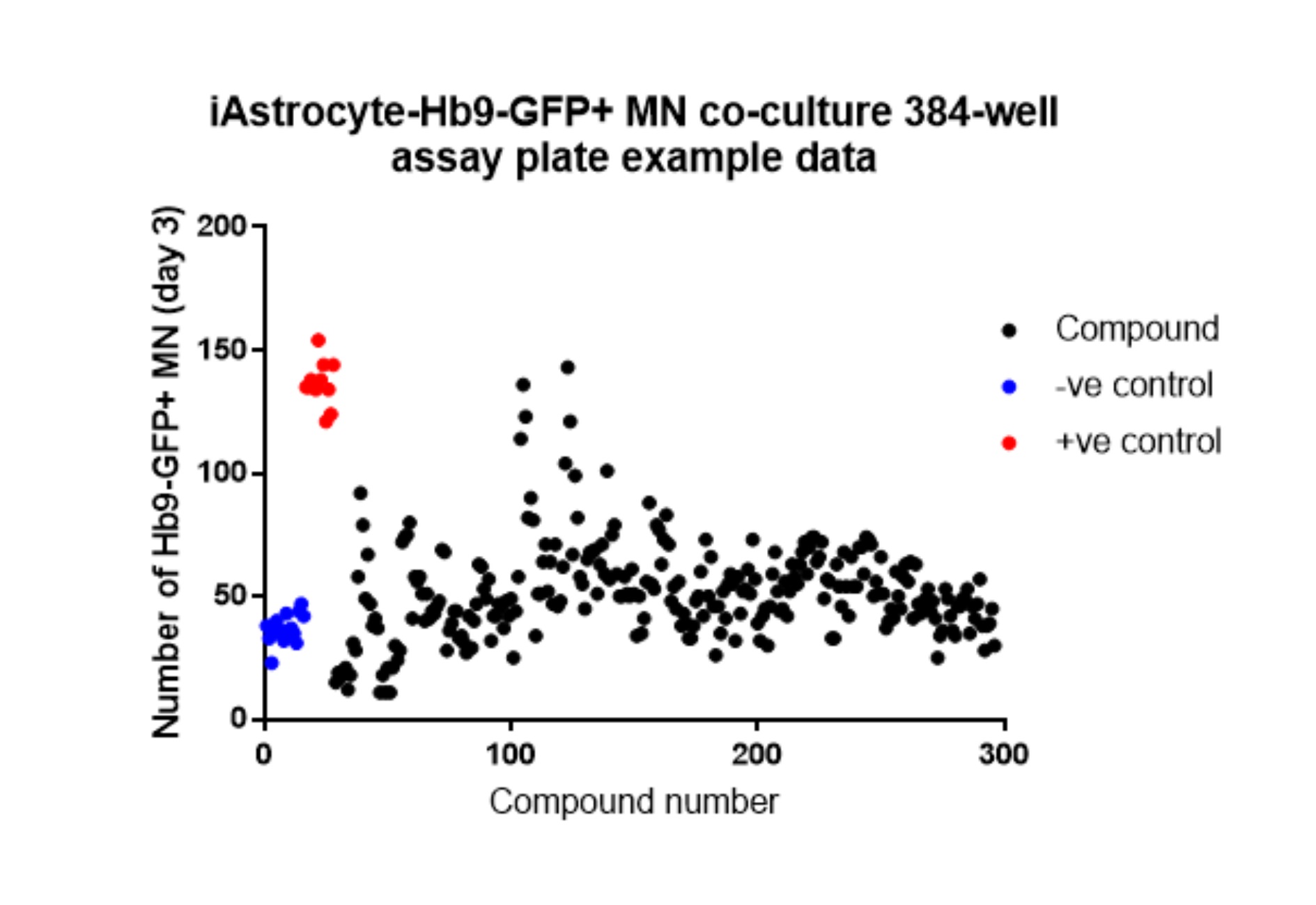
Figure 5. Example Hb9-GFP+ mMN counts at day 3 from one 384-well plate of the iAstrocyte-Hb9-GFP+ mMN co-culture assay. –ve control = DMSO only, +ve control = compound A.
Notes
- Note the lot number of serum used because this will affect the co-culture results. We recommend serum with the same lot number be used for any given batch of work, for example, screening a library of compounds.
- The mESC should appear as large bright colonies with clearly defined edges when viewed on a light microscope. If the colonies become darker grey, or have rough or poorly defined edges, the mESC have differentiated and will produce a lower yield of Hb9-GFP+ MNs after differentiation.
- The mESC are grown in culture until passage 18, and then replaced with younger passage stocks. This is because when the mESCs reach a higher passage, the yield of Hb9-GFP+ MN they can produce becomes lower.
- On Day 7 of the co-culture protocol, observe the mEBs using a green fluorescence microscope. If the mEB have differentiated well, there should be a high percentage of GFP+ cells with a high level of GFP expression. If the mEB are dim, or there is a low level of GFP expression, the MN yield will be low, so it is advisable not to use the mEB in co-culture.
- When performing high-throughput drug screening experiments using the iAstrocyte-Hb9-GFP+ MN co-culture assay, we recommend using a minimum of 12 technical replicates (separate wells on the 384-well plate) for both negative controls (DMSO only) and positive controls (compound A identified in house). For experimental conditions, 1 technical replicate is sufficient, but we recommend using 6 technical replicates. Additionally, in secondary validation experiments, we strongly recommend using 6 technical replicates for each experimental condition (and a minimum of 12 technical replicates for the positive and negative controls). A minimum of 3 experimental repeats is also recommended for both high-throughput screens and secondary validation experiments.
- When coating the 10 cm tissue culture plates with either the 2.5 or 5.0 µg/ml Fibronectin Coating Solution, there is no optimal length of time for the incubation. However, we recommend a minimum of 5 min at room temperature, or alternatively, overnight at 4 °C.
Recipes
- Sterile1x PBS
Add 5 PBS tablets to 500 ml Milli-Q ultrapure water and autoclave at 121 °C for 15 min
Allow the PBS to cool to room temperature before using - Sterile ultrapure water
Autoclave 500 ml Milli-Q ultrapure water at 121 °C for 15 min
Allow the water to cool to room temperature before using - Human iNPC Proliferation Media
DMEM/F-12, GlutaMAX 500 ml N-2 5 ml B-27 5 ml 4 mg/ml FGF 5 µl - Human iAstrocyte Differentiation Media
DMEM 500 ml N-2 1 ml FBS 50 ml Pen-Strep 5 ml - Mouse Embryonic Stem Cell (mESC) Proliferation Media
KnockOut DMEM 400 ml ESC FBS 75 ml L-glutamine 5 ml Non-Essential Amino Acids 5 ml 2-Mercaptoethanol 3.6 µl - Mouse Embryonic Bodies (mEB) Differentiation Media
Filter using a steritop filterKnockOut DMEM 222.5 ml Ham’s F-12 Nutrient Mix 222.5 ml KnockOut Serum Replacement 50 ml L-glutamine 2.5 ml 30% (w/v) filtered glucose 2.5 ml N-2 5 ml 2-Mercaptoethanol 4 µl - Mouse Motor Neuron (mMN) Media
mEB Differentiation Media 40 ml 200 µg/ml BDNF 4 µl 200 µg/ml CNTF 4 µl 200 µg/ml GDNF 4 µl - EB Dissociation Buffer
Filter using a 0.2 µm Syringe FilterSterile Milli-Q ultrapure water 31.472 ml 1 M NaCl 4.64 ml 1 M KCl 0.216 ml 1 M NaHCO3 1.04 ml 0.1 M NaH2PO4 0.4 ml 0.1 M CaCl2 0.6 ml 0.1 M MgSO4 0.4 ml 30% (w/v) filtered glucose 1 ml 0.5 M EDTA 0.04 ml 25 mg/ml L-Cysteine 0.192 ml
Acknowledgments
We would like to thank all the ALS patients who have kindly donated samples for this study. Hb9-GFP mESC were a kind gift from Prof. Thomas Jessell. The iNPC reprogramming and iAstrocyte differentiation protocols were developed by Dr Kathrin Meyer and Dr Laura Ferraiuolo in Prof. Brian Kaspar’s laboratory. This work was funded by a Motor Neuron Disease Association Senior Fellowship award to S.P.A. (grant number 956-799, MNDA-registered charity number 294354). L.F. and M.J.S. are funded by BenevolentAI. L.F. is funded by the Academy of Medical Sciences (SBF002\1142).
Competing interests
M.J.S. and L.F. are funded by the biotech company BenevolentAI.
Ethics
Informed consent was obtained from all human subjects before skin sample collection (Study number STH16573, Research Ethics Committee reference 12/YH/0330). All applicable international, national, and/or institutional guidelines for the care and use of animals were followed.
References
- Allen, S. P., Hall, B., Castelli, L. M., Francis, L., Woof, R., Siskos, A. P., Kouloura, E., Gray, E., Thompson, A. G., Talbot, K., Higginbottom, A., Myszczynska, M., Allen, C. F., Stopford, M. J., Hemingway, J., Bauer, C. S., Webster, C. P., De Vos, K. J., Turner, M. R., Keun, H. C., Hautbergue, G. M., Ferraiuolo, L. and Shaw, P. J. (2019). Astrocyte adenosine deaminase loss increases motor neuron toxicity in amyotrophic lateral sclerosis. Brain 142(3): 586-605.
- Ferraiuolo, L., Higginbottom, A., Heath, P. R., Barber, S., Greenald, D., Kirby, J. and Shaw, P. J. (2011a). Dysregulation of astrocyte-motoneuron cross-talk in mutant superoxide dismutase 1-related amyotrophic lateral sclerosis. Brain 134(Pt 9): 2627-2641.
- Ferraiuolo, L., Kirby, J., Grierson, A. J., Sendtner, M. and Shaw, P. J. (2011b). Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat Rev Neurol 7(11): 616-630.
- Ferraiuolo, L., Meyer, K., Sherwood, T. W., Vick, J., Likhite, S., Frakes, A., Miranda, C. J., Braun, L., Heath, P. R., Pineda, R., Beattie, C. E., Shaw, P. J., Askwith, C. C., McTigue, D. and Kaspar, B. K. (2016). Oligodendrocytes contribute to motor neuron death in ALS via SOD1-dependent mechanism. Proc Natl Acad Sci U S A 113(42): E6496-E6505.
- Frakes, A. E., Braun, L., Ferraiuolo, L., Guttridge, D. C. and Kaspar, B. K. (2017). Additive amelioration of ALS by co-targeting independent pathogenic mechanisms. Ann Clin Transl Neurol 4(2): 76-86.
- Haidet-Phillips, A. M., Hester, M. E., Miranda, C. J., Meyer, K., Braun, L., Frakes, A., Song, S., Likhite, S., Murtha, M. J., Foust, K. D., Rao, M., Eagle, A., Kammesheidt, A., Christensen, A., Mendell, J. R., Burghes, A. H. and Kaspar, B. K. (2011). Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat Biotechnol 29(9): 824-828.
- Hautbergue, G. M., Castelli, L. M., Ferraiuolo, L., Sanchez-Martinez, A., Cooper-Knock, J., Higginbottom, A., Lin, Y. H., Bauer, C. S., Dodd, J. E., Myszczynska, M. A., Alam, S. M., Garneret, P., Chandran, J. S., Karyka, E., Stopford, M. J., Smith, E. F., Kirby, J., Meyer, K., Kaspar, B. K., Isaacs, A. M., El-Khamisy, S. F., De Vos, K. J., Ning, K., Azzouz, M., Whitworth, A. J. and Shaw, P. J. (2017). SRSF1-dependent nuclear export inhibition of C9ORF72 repeat transcripts prevents neurodegeneration and associated motor deficits. Nat Commun 8: 16063.
- Kim, J., Efe, J. A., Zhu, S., Talantova, M., Yuan, X., Wang, S., Lipton, S. A., Zhang, K. and Ding, S. (2011). Direct reprogramming of mouse fibroblasts to neural progenitors. Proc Natl Acad Sci U S A 108(19): 7838-7843.
- Lin, C. L., Bristol, L. A., Jin, L., Dykes-Hoberg, M., Crawford, T., Clawson, L. and Rothstein, J. D. (1998). Aberrant RNA processing in a neurodegenerative disease: the cause for absent EAAT2, a glutamate transporter, in amyotrophic lateral sclerosis. Neuron 20(3): 589-602.
- McGown, A. and Stopford, M. J. (2018). High-throughput drug screens for amyotrophic lateral sclerosis drug discovery. Expert Opin Drug Discov 13(11): 1015-1025.
- Mertens, J., Paquola, A. C. M., Ku, M., Hatch, E., Bohnke, L., Ladjevardi, S., McGrath, S., Campbell, B., Lee, H., Herdy, J. R., Goncalves, J. T., Toda, T., Kim, Y., Winkler, J., Yao, J., Hetzer, M. W. and Gage, F. H. (2015). Directly reprogrammed human neurons retain aging-associated transcriptomic signatures and reveal age-related nucleocytoplasmic defects. Cell Stem Cell 17(6): 705-718.
- Meyer, K., Ferraiuolo, L., Miranda, C. J., Likhite, S., McElroy, S., Renusch, S., Ditsworth, D., Lagier-Tourenne, C., Smith, R. A., Ravits, J., Burghes, A. H., Shaw, P. J., Cleveland, D. W., Kolb, S. J. and Kaspar, B. K. (2014). Direct conversion of patient fibroblasts demonstrates non-cell autonomous toxicity of astrocytes to motor neurons in familial and sporadic ALS. Proc Natl Acad Sci U S A 111(2): 829-832.
- Myszczynska, M. and Ferraiuolo, L. (2016). New in vitro models to study amyotrophic lateral sclerosis. Brain Pathol 26(2): 258-265.
- Pellerin, L. and Magistretti, P. J. (1994). Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc Natl Acad Sci U S A 91(22): 10625-10629.
- Re, D.B., Le Verche, V., Yu, C., Amoroso, M.W., Politi, K.A., Phani, S., Ikiz, B., Hoffmann, L., Koolen, M., Nagata, T., Papadimitriou, D., Nagy, P., Mitsumoto, H., Kariya, S., Wichterle, H., Henderson, C.E. and Przedborski, S. (2014). Necroptosis drives motor neuron death in models of both sporadic and familial ALS. Neuron 81(5): 1001-1008.
- Takahashi, K. and Yamanaka, S. (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126(4): 663-676.
- Vandoorne, T., De Bock, K. and Van Den Bosch, L. (2018). Energy metabolism in ALS: an underappreciated opportunity? Acta Neuropathologica 135(4): 489-509
- Varcianna, A., Myszczynska, M. A., Castelli, L. M., O'Neill, B., Kim, Y., Talbot, J., Nyberg, S., Nyamali, I., Heath, P. R., Stopford, M. J., Hautbergue, G. M. and Ferraiuolo, L. (2019). Micro-RNAs secreted through astrocyte-derived extracellular vesicles cause neuronal network degeneration in C9orf72 ALS. EBioMedicine 40: 626-635.
- Wichterle, H., Lieberam, I., Porter, J. A. and Jessell, T. M. (2002). Directed differentiation of embryonic stem cells into motor neurons. Cell 110(3): 385-397.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Stopford, M. J., Allen, S. P. and Ferraiuolo, L. (2019). A High-throughput and Pathophysiologically Relevant Astrocyte-motor Neuron Co-culture Assay for Amyotrophic Lateral Sclerosis Therapeutic Discovery. Bio-protocol 9(17): e3353. DOI: 10.21769/BioProtoc.3353.
Category
Neuroscience > Basic technology > High-throughput screening
Cell Biology > Cell-based analysis > Fluorescent Co-culture
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.