- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Application of a Modified Smart-seq2 Sample Preparation Protocol for Rare Cell Full-length Single-cell mRNA Sequencing to Mouse Oocytes
Published: Vol 9, Iss 16, Aug 20, 2019 DOI: 10.21769/BioProtoc.3345 Views: 6980
Reviewed by: Gal HaimovichShyam SolankiVaibhav B. Shah
Original research article
The authors used this protocol in:

Feb 2019
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Endogenous retroviruses (ERV) are transposable retroelements that form ~10% of the murine genome and whose family members are differentially expressed throughout embryogenesis. However, precise regulation of ERV in germ cells remains unclear. To investigate ERV expression in oocytes, we adapted a single-cell mRNA-sequencing library preparation method to generate bulk sequencing libraries from growing oocytes in a time- and cost-efficient manner. Here, we present a modified Smart-seq2 protocol that yields full-length cDNA libraries from purified RNA obtained from low numbers of pooled immature or mature oocytes. Using this method, RNA-sequencing libraries can be generated from any rare or difficult-to-isolate populations for subsequent sequencing and retroelement expression analysis.
Keywords: OocyteBackground
Endogenous retroviruses (ERV) are retrovirus-like mobile DNA elements that form ~10% of the murine genome, and are expressed in both healthy tissues and disease states (Kazazian and Moran, 2017; Johnson, 2019). Expression of ERV families is tissue-specific (Faulkner et al., 2009), and many families of ERV are temporally and differentially regulated throughout embryogenesis (Peaston et al., 2004; Rowe and Trono, 2011). We observed that proviral ERV are globally dysregulated in C57BL/6N, but not C57BL/6J, mice, secondary to loss of two transcriptional repressors, suppressor of non-ecotropic ERV (SNERV)-1, and SNERV-2, that promote heterochromatinization at ERV loci (Treger et al., 2019). We observed that in the absence of SNERVs in C57BL/6N mice, ERV are upregulated in a variety of immune cells, as well as in murine embryonic fibroblasts and embryonic stem cells. To identify whether ERV dysregulation is similarly present in gametes, we needed a cost-effective method to successfully generate cDNA sequencing libraries from pooled samples of growing oocytes.
While primordial oocytes can now be efficiently isolated from fetal and neonatal ovaries by flow cytometry (Stewart et al., 2015), antral germinal vesicle (immature) and metaphase II-arrested (mature) oocytes require a technique employing microdissection of superovulated ovaries (Seli et al., 2005; Duselis and Vrana, 2007; Litscher and Wassarman, 2010; Stein and Schindler, 2011; Guzeloglu-Kayisli et al., 2012;). While superovulation increases the yield of immature and mature oocytes, only ~20-30 ovulated oocytes per mouse can be reliably obtained through this method (Litscher and Wassarman, 2010). Prior studies have generated mRNA sequencing libraries from ~700-1,500 oocytes ( Smallwood et al., 2011; Veselovska et al., 2015; Gahurova et al., 2017), whose isolation represents a significant investment of both time and money.
The transcriptomes of immature (GV) and mature (MII) oocytes have been successfully sequenced by single-cell RNA-seq (scRNA-sesq) technologies (Tang et al., 2010; Xue et al., 2013). scRNA-seq permits the identification of cell-to-cell heterogeneity in a sample by detecting high-abundance cellular transcripts (Liu and Trapnell, 2016; Picelli, 2016; Choi and Kim, 2019), and circumvents the need to isolate large numbers of oocytes for bulk sequencing library preparation. However, our aim was to sensitively detect even low-abundance retroelement transcripts in growing oocytes and to detect differential expression between substrains. For this goal, scRNA-seq was not a suitable technique. Additionally, a high percentage of repeat element sequencing reads map to multiple locations in the genome and transcriptome, and their mapping accuracy is improved by using longer sequencing read lengths and paired-end reads (Treangen and Salzberg, 2011). Importantly, while many scRNA-seq methods amplify only the 3′ end of polyadenylated transcripts, the Smart-seq2 method is a low-cost protocol that generates full-length cDNA from mRNA transcripts (Picelli et al., 2014; Haber et al., 2017; Ziegenhain et al., 2017) and could further improve mappability of repeat-associated reads. For these reasons, we developed a modified Smart-seq2 protocol to generate full-length cDNA libraries from low numbers of pooled oocytes for bulk sequencing to permit subsequent analysis of transposable element expression.
This Smart-seq2-based protocol provides a cost-effective method to generate libraries for bulk mRNA-sequencing of oocytes, which results in data suitable for downstream analysis of population-level differences in retroelement expression. This protocol can be further modified to generate sequencing libraries from any rare or difficult-to-isolate population, with broad applications ranging from analysis of transduced primary cells with low transduction efficiency to investigation of transcriptional differences in scarce tissue-resident immune cell populations.
Materials and Reagents
- 30 gauge needles (BD, catalog number: 305128)
- 100 mm Petri dish (Falcon, catalog number: 351029)
- 1.5 ml autoclaved sterile microcentrifuge tubes (USA Scientific, catalog number: 1615-5500)
- SHARP Precision barrier tips (Denville, catalog numbers: P1120, P1121, P1122, P1126)
- Twintec PCR 96-well PCR plates, colorless (Eppendorf, catalog number: 951020401)
- Microseal B PCR Plate Sealing Film (Bio-Rad, catalog number: MSB1001)
- Qubit Assay tubes (ThermoFisher, catalog number: Q32856)
- 200 μl pipette tip (Fisherbrand, catalog number: 02-681-147 or equivalent)
- Pregnant mare serum gonadotropin (Biovendor, catalog number: RP1782721000)
- Human chorionic gonadotropin (Sigma, catalog number: CG10-10VL)
- M2 medium (Millipore, catalog number: MR-015P-5F)
- Hyaluronidase (Sigma, catalog number: H4272)
- Milrinone (Sigma, catalog number: M4659)
- RNase Away (Ambion, catalog number: 10328011)
- β-mercaptoethanol (Sigma, catalog number: M6250)
- RLT buffer (Qiagen, catalog number: 79216)
- Dry ice
- RNeasy Micro Kit (Qiagen, catalog number: 74004)
- Ultrapure water (AmericanBio, catalog number: AB02128)
- RNAClean XP beads (Agencourt, catalog number: A63987)
- 80% ethanol
- 10 mM dNTPs (Thermo Fisher, catalog number: R1092)
- RNase Inhibitor (Takara, catalog number: 2313A)
- Maxima RNase H-minus RT 5x Buffer (Thermo Fisher, catalog number: EP0751)
- Maxima RNase H-minus RT (Thermo Fisher, catalog number: EP0751)
- Betaine (Sigma, catalog number: 61962)
- 1 M MgCl2 (Sigma, catalog number: M1028)
- Kapa HiFi HotStart ReadyMix (Kapa Biosystems, catalog number: KK2601)
- AMPure XP beads (Agencourt, catalog number: A63882)
- 70% ethanol
- TE buffer (AmericanBio, catalog number: AB14033)
- Qubit dsDNA HS Assay Kit (Thermo Fisher, catalog number: Q32851)
- High Sensitivity DNA Kit (Agilent, catalog number: 5067-4626)
- Illumina’s Nextera XT Kit
- Oligos
- 3′ RT primer
5′-AAGCAGTGGTATCAACGCAGAGTACT30VN-3′ (Sigma)
Note: T30VN corresponds to 30 T’s followed by a “V” (A, C, or G), followed by an “N” (any base). - Template Switching Oligo
5′-AAGCAGTGGTATCAACGCAGAGTACATrGrG+G-3′ (Exiqon)
Note: “rG” stands for riboguanosine, while “+G” indicates that the last deoxyguanosine is modified to be a locked nucleic acid. - ISPCR
5′-AAGCAGTGGTATCAACGCAGAGT-3′ (Sigma)
Note: All oligos should be ordered as desalted and diluted to 100 μM using 0.1x TE and stored at -20 °C prior to use.
- 3′ RT primer
- Master Mix 1 (see Recipes)
- Master Mix 2 (see Recipes)
- Master Mix 3 (see Recipes)
Equipment
- Dissecting scissors (Fine Science Tools, catalog number: 91460-11 or equivalent)
- Curved tip forces (Fine Science Tools, catalog number: 11273-20 or equivalent)
- Watchmaker’s forceps (Fine Science Tools, catalog number: 11251-20 of equivalent)
- Dissecting microscope (Laxco, LMS-S2103-LS200 or equivalent)
- 1.0 ml pipette controller (Hamilton, catalog number: 0010PC)
- Aspirator tube assemblies for calibrated microcapillary pipettes (Sigma, catalog number: A5177-5EA)
- Capillaries without filament (World Precision Instruments, catalog number: 1B100-6)
- Vortex machine (Scientific Industries, catalog number: 00-SI-0236)
- Swinging bucket centrifuge (Eppendorf, model: 5810R)
- 96-well magnet plate (Alpaqua Magnum, model: FLX A000400 or equivalent)
- 96-well heated thermal cycler (Bio-Rad, model: S1000 or equivalent)
- Qubit Fluorometer (Invitrogen, model: Q33226 or equivalent)
- Agilent 2100 Bioanalyzer System (Agilent, G2939BA, G2953CA)
Procedure
Part I: Oocyte isolation
This protocol should yield ~80-120 mature oocytes and ~200-300 immature oocytes per pooled sample (from four mice).
Note: Prior to beginning this protocol, assemble the Hamilton pipette-based micropipette oocyte handling device as described in Xing (2010). Videos that detail the following procedures for oviduct removal and dissection can be found in the Journal of Visualized Experiments (Duselis and Vrana, 2007; Stein and Schindler, 2011).
- RLT buffer preparation
- Clean pipettes and workspace thoroughly with RNase Away to avoid RNase contamination.
- Using PCR-rated filter tips, add 10 μl of β-mercaptoethanol to 1 ml of RLT buffer in a 1.5 ml Eppendorf tube and pipette to mix well.
Note: 350 μl of the RLT buffer containing β-mercaptoethanol will be needed per RNA sample. Increase volumes proportionally to obtain the desired final volume. RLT buffer containing β-mercaptoethanol can be stored for up to 1 month at room temperature. - Transfer 350 μl of the RLT buffer containing β-mercaptoethanol to each of two 1.5 ml Eppendorf tubes.
- Mice superovulation and tissue harvesting
- Mature oocyte generation and oviduct isolation
- Inject four 3-week-old female mice intraperitoneally with 5 IU of pregnant mare serum gonadotropin (PMSG) to induce oocyte growth.
- Forty-eight hours later, inject these same mice intraperitoneally with 5 IU of human chorionic gonadotropin (hCG) to induce oocyte ovulation.
- Euthanize the mice by cervical dislocation fourteen hours after hCG injection.
- Use sterilized fine forceps and fine scissors to remove the oviducts with attached uterine horn from each mouse (total of 8). Place tissues into a droplet of pre-warmed room temperature M2 media on an untreated 100 mm Petri dish.
- Transfer one oviduct-uterine horn sample at a time to a new 300 μl drop of pre-warmed M2 media on a new untreated 100 mm Petri dish.
- Under a dissecting microscope, use two pairs of watchmaker’s forceps to tear the ampulla of the oviduct and release the cumulus-oocyte complexes into the surrounding media.
- Add 300 μl of 0.6 mg/ml hyaluronidase to the drop of media (final concentration of 0.3 mg/ml).
- Immature oocyte generation and ovary isolation
- Inject four 3-week-old female mice intraperitoneally with 5 IU of pregnant mare serum gonadotropin to induce superovulation.
- Euthanize the mice by cervical dislocation forty-four hours after PMSG injection.
- Use sterilized fine forceps and fine scissors to remove the ovaries from each mouse (total of 8). Place tissues into a droplet of pre-warmed room temperature M2 media on an untreated 100 mm Petri dish.
- Transfer one ovary at a time to a new 300 μl drop of pre-warmed M2 media supplemented with 10 μm milrinone on a new 100 mm Petri dish. Milrinone helps to ensure metaphase arrest of the oocytes (Stein and Schindler, 2011).
- Add 300 μl of 0.6 mg/ml hyaluronidase to the drop of media (final concentration of 0.3 mg/ml).
- Mature oocyte generation and oviduct isolation
- Oocyte isolation
Note: During the processing of immature oocytes, use M2 media supplemented with 10 μm milrinone.- Under a dissecting microscope, puncture and gently dissociate the tissue with two 30-gauge needles to aid the release of oocytes into the surrounding media.
- Detach adherent cumulus cells from the oocytes by gently and repeatedly pipetting the cumulus-oocyte complexes using the micropipette.
- Once denuded of cumulus cells, wash oocytes three times by transferring the oocytes into new droplets of pre-warmed M2 media on the untreated 100 mm Petri dish using the micropipette.
- Count the total number of oocytes as they are drawn up into the micropipette. Aspirate the minimal amount of media possible (less than 6 μl), as large volumes will dilute the RLT buffer.
- Transfer the counted immature or mature oocytes into the corresponding Eppendorf tube containing previously prepared RLT buffer.
- Vortex the tube at high speed for 45 s to homogenize the sample. Place tube on dry ice until it can be stored at -80 °C.
Part II: Oocyte RNA library preparation
- RNA isolation
- Thaw RLT samples on ice.
- Isolate RNA from the thawed samples using the RNeasy Micro Kit, according to the manufacturer’s instructions.
- Elute RNA in 14 μl of ultra-pure water and store at -80 °C until ready to proceed with library preparation.
- cDNA generation
Note: The heated lid should be set to 105 °C for all thermal cycler programs. Preheat the PCR machine before running each program.- Allow RNAClean XP beads to equilibrate to room temperature for 30 min before use in Step B4.
- Prepare Master Mix 1 and place on ice until ready to be used.
- Transfer 10 μl of each RNA sample to a corresponding well of a 96-well skirted PCR plate.
- Add 22 μl (2.2x) RNAClean XP beads to each sample and pipette gently to mix. Allow to incubate for 10 min at room temperature. The RNA is now bound to the beads.
- Place the 96-well plate containing beads and RNA on a 96-well magnet plate and incubate for 5 min. The magnetic beads will aggregate along the sidewalls of the wells.
- Use a multichannel pipette to carefully remove the supernatant without disturbing the beads.
- Wash the beads by gently pipetting 100 μl of 80% ethanol over the beads. Let stand for 1 min and then remove the supernatant without disturbing the beads.
- Repeat Step B7 for a total of two ethanol washes.
- Allow the beads to air dry on the magnet for 5 min.
- Remove the 96-well plate from the magnet stand and resuspend each well of dried beads in 8 μl of Master Mix 1.
- Seal the plate with a Microseal B film, quick-spin the plate in a swinging bucket centrifuge to remove liquid from the sidewalls, and incubate the plate for 3 min at 72 °C. This step denatures the RNA.
- Immediately place the sealed 96-well plate on ice for 1 min. This step minimizes RNA secondary structure formation.
- Prepare Master Mix 2 and place on ice until ready to be used.
- Add 14 μl of Master Mix 2 to each well for a total volume of 22 μl and gently pipette to mix.
- Seal the plate with a Microseal B film and quick-spin the plate in a swinging bucket centrifuge to remove liquid from the sidewalls. Place the plate in a preheated thermal cycler with the heated lid set to 105 °C and run PCR Program 1 to perform reverse transcription of the RNA.
PCR Program 1:
50 °C for 90 min
85 °C for 5 min - Prepare Master Mix 3 and place on ice until ready to be used.
- Following reverse transcription, quick-spin the plate in a swinging bucket centrifuge to remove liquid from the sidewalls and remove the adhesive seal.
- Add 28 μl of Master Mix 3 to each well for a total volume of 50 μl and pipette gently to mix.
- Seal the plate with a Microseal B film and quick-spin the plate in a swinging bucket centrifuge to remove liquid from the sidewalls. Place the plate in a preheated thermal cycler with the heated lid set to 105 °C and run PCR Program 2 to amplify cDNA.
Note: Because of the difficulty of isolating oocytes in large numbers, coupled with the high cost of library preparation and sequencing, we chose a PCR amplification cycle number in Step B19 based upon Qubit results we obtained in Step B31 using equivalent input numbers of splenocytes. Ideally, the PCR amplification cycle number will be determined empirically using test samples of the same cell type and input cell number being sequenced. An ideal final concentration of cDNA is ~0.5 ng/μl, which permits further dilution of the cDNA sample to 0.2 ng/μl.
PCR Program 2:
98 °C for 3 min
Then 12 cycles of:
98 °C for 15 s
67 °C for 20 s
72 °C for 6 min
Followed by:
72 °C for 5 min - Allow AMPure XP beads to equilibrate to room temperature for 30 min before use in Step B22.
- Quick-spin the plate in a swinging bucket centrifuge to remove liquid from the sidewalls and remove the adhesive seal.\
- Add 35 μl (0.7x) AMPure XP beads to each sample and pipette gently to mix. Allow to incubate for 5 min at room temperature. The cDNA is now bound to the beads.
- Place the 96-well plate on a 96-well magnet plate and incubate for 6 min.
- Use a multichannel pipette to carefully remove the supernatant without disturbing the beads.
- Wash the beads by gently pipetting 100 μl of 70% ethanol over the beads. Let stand for 1 min and then remove the supernatant without disturbing the beads.
- Repeat Step B25 for a total of two ethanol washes.
- Allow the beads to air dry on the magnet for 5 min.
- Remove the 96-well plate from the magnet stand and resuspend each well of dried beads in 40 μl of 1x TE Buffer to elute the cDNA from the beads for 5 min.
- Place the 96-well plate on a 96-well magnet plate and incubate for 5 min. The magnetic beads will aggregate along the sidewalls of the wells. The cDNA is contained in the supernatant.
- Use a multichannel pipette to carefully transfer 36 μl of supernatant from each well into a new well of the same 96-well plate (one well per pooled sample).
- Quantify the cDNA concentration of each sample using a Qubit dsDNA HS kit, according to the manufacturer’s instructions.
- The cDNA quality can be analyzed using a 2100 Bioanalyzer with the high sensitivity DNA kit, according to the manufacturer’s instructions. The average size of the PCR-amplified cDNA library should be 1.5-2 kb, as described alongside representative Bioanalyzer traces (Picelli et al., 2014).
- Normalize the cDNA concentration of each well to 0.2 ng/μl using 1x TE Buffer.
- Proceed to RNA-seq library generation using Illumina’s Nextera XT Kit with 1 ng of cDNA as input.
Data analysis
cDNA was generated from total RNA isolated from immature and mature C57LB/6J oocytes as described in the above protocol. Sequencing libraries were then constructed using tagmentation with Illumina’s Nextera XT kit and paired-end 75bp reads were sequenced on the NextSeq500. Sequencing reads were mapped to 680 LTR families using the RepEnrich (Criscione et al., 2014) alignment strategy and are plotted below. Detailed methods for data analysis have been previously published (Treger et al., 2019).
We observe that expression of 197 of the 680 LTR families is significantly increased or decreased more than two-fold in mature oocytes when compared to immature oocytes (Figure 1). The 191 of these 197 LTR families are downregulated in mature oocytes, and the expression of only 6 LTR families–all of which correspond to virus-like 30 (VL30) elements or VL30-associated LTRs–is increased in mature oocytes (Figure 1). VL30 elements are proviral-like retroelements that lack envelope-coding regions and do not encode intact open reading frames (Markopoulos et al., 2016). VL30 expression is induced by both epidermal growth factor and steroid hormones such as estradiol and progesterone in vitro (Courtney et al., 1982; Tzavaras et al., 2003). Transcription of VL30-type LTRs may be directly induced by growth factors and/or hormones as the germinal vesicle oocytes mature and arrest at metaphase II. Alternatively, these LTR elements may be coordinately regulated by the transcriptional activation of nearby cellular genes that are activated during oocyte maturation.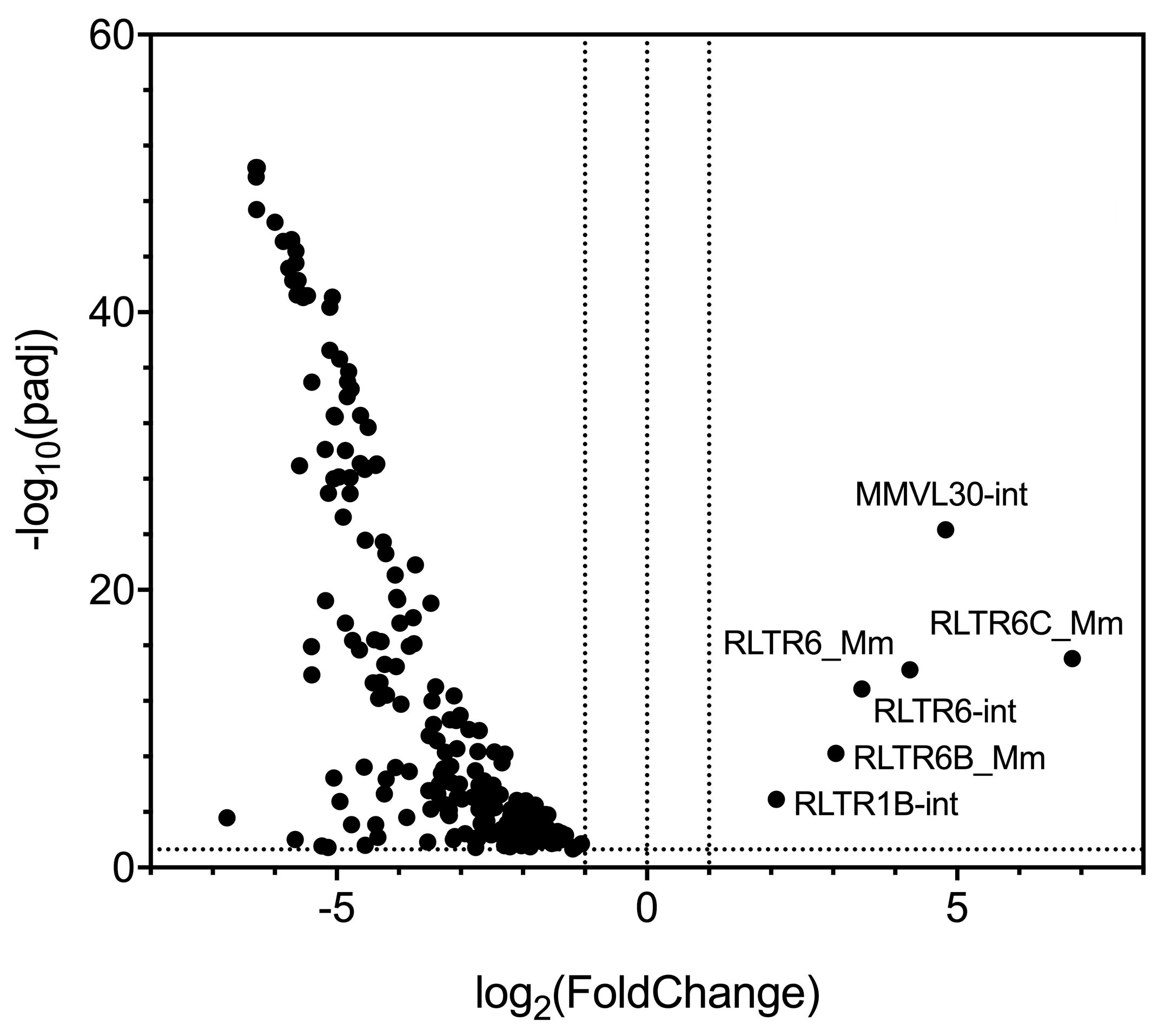
Figure 1. While VL30 LTR family members are upregulated, the expression of other LTR elements are globally decreased in mature oocytes. Volcano plot of significantly differentially expressed long-terminal repeat (LTR) families from mRNA sequencing comparing immature and mature oocytes. The log2 fold-change in expression is graphed against the negative log10 of the adjusted P-value calculated by DESeq2 (Love et al., 2014). Positive fold-change values indicate LTR families whose expression is increased in mature oocytes, while negative fold-change values indicate LTR families whose expression is decreased in mature oocytes. mRNA-sequencing libraries were generated using the modified Smart-seq2-based protocol presented in this paper.
Recipes
- Master Mix 1
2.5 μM 3′ RT primer
2.5 mM dNTPs
1 unit/μl RNase inhibitor
For making 8 μl (per well) Master Mix 1:
0.2 μl of 100 μM 3′ RT primer
2 μl of 10 mM dNTPs
0.2 μl of 40 U/μl RNase inhibitor
5.6 μl of Ultrapure water - Master Mix 2
1.4x Maxima RNase H-minus RT Buffer
1.4 M Betaine, 12.9 mM MgCl2
1.4 μM Template Switching Oligo
1.4 units/μl RNAase inhibitor
2.9 units/μl Maxima RNase H-minus RT
For making 14 μl (per well) Master Mix 2:
4 μl of 5x Maxima RNase H-minus RT Buffer
4 μl of 5 M Betaine
0.18 μl of 1 M MgCl2
0.2 μl of 100 μM Template Switching Oligo
0.5 μl of 40 U/μl RNase inhibitor
0.2 μl of 200 U/μl Maxima RNase H-minus RT
4.92 μl of Ultrapure water - Master Mix 3
0.4 μM ISPCR primer
1.8x Kapa HiFi HotStart ReadyMix
For making 28 μl (per well) Master Mix 2:
25 μl of 2x Kapa HiFi HotStart ReadyMix
0.1 μl of 100 μM ISPCR primer
2.9 μl of Ultrapure water
Acknowledgments
This work was supported in part by Howard Hughes Medical Institute (to A.I.) and by NIH award R01 AI054359 and R01 AI127429 (to A.I.). R.T. was supported by NIH training grant (5-T32-GM00720540) and F30 (5-F30-AI129265-02). An abbreviated version of this protocol was originally published in “The Lupus Susceptibility Locus Sgp3 Encodes the Suppressor of Endogenous Retrovirus Expression SNERV” (Treger et al., 2019).
Competing interests
The authors declare no competing interests.
Ethics
Mice were housed in SPF conditions and care was provided in accordance with Yale University IACUC guidelines (protocol #10365).
References
- Choi, Y. H. and Kim, J. K. (2019). Dissecting cellular heterogeneity using single-cell RNA sequencing. Mol Cells 42(3): 189-199.
- Courtney, M. G., Schmidt, L. J. and Getz, M. J. (1982). Organization and expression of endogenous virus-like (VL30) DNA sequences in nontransformed and chemically transformed mouse embryo cells in culture. Cancer Res 42(2): 569-576.
- Criscione, S. W., Zhang, Y., Thompson, W., Sedivy, J. M. and Neretti, N. (2014). Transcriptional landscape of repetitive elements in normal and cancer human cells. BMC Genomics 15: 583-583.
- Duselis, A. R. and Vrana, P. B. (2007). Retrieval of mouse oocytes. JoVE (3): 185-185.
- Faulkner, G. J., Kimura, Y., Daub, C. O., Wani, S., Plessy, C., Irvine, K. M., Schroder, K., Cloonan, N., Steptoe, A. L., Lassmann, T., Waki, K., Hornig, N., Arakawa, T., Takahashi, H., Kawai, J., Forrest, A. R., Suzuki, H., Hayashizaki, Y., Hume, D. A., Orlando, V., Grimmond, S. M. and Carninci, P. (2009). The regulated retrotransposon transcriptome of mammalian cells. Nat Genet 41(5): 563-571.
- Gahurova, L., Tomizawa, S.-I., Smallwood, S. A., Stewart-Morgan, K. R., Saadeh, H., Kim, J., Andrews, S. R., Chen, T. and Kelsey, G. (2017). Transcription and chromatin determinants of de novo DNA methylation timing in oocytes. Epigenetics Chromatin 10: 25-25.
- Guzeloglu-Kayisli, O., Lalioti, M. D., Aydiner, F., Sasson, I., Ilbay, O., Sakkas, D., Lowther, K. M., Mehlmann, L. M. and Seli, E. (2012). Embryonic poly(A)-binding protein (EPAB) is required for oocyte maturation and female fertility in mice. Biochemical J 446(1): 47-58.
- Haber, A. L., Biton, M., Rogel, N., Herbst, R. H., Shekhar, K., Smillie, C., Burgin, G., Delorey, T. M., Howitt, M. R., Katz, Y., Tirosh, I., Beyaz, S., Dionne, D., Zhang, M., Raychowdhury, R., Garrett, W. S., Rozenblatt-Rosen, O., Shi, H. N., Yilmaz, O., Xavier, R. J. and Regev, A. (2017). A single-cell survey of the small intestinal epithelium. Nature 551(7680): 333-339.
- Johnson, W. E. (2019). Origins and evolutionary consequences of ancient endogenous retroviruses. Nat Rev Microbiol 17(6): 355-370.
- Kazazian, H. H., Jr. and Moran, J. V. (2017). Mobile DNA in health and disease. N Engl J Med 377(4): 361-370.
- Litscher, E. S. and Wassarman, P. M. (2010). Chapter 5: Isolation and manipulation of mouse gametes and embryos. In: Methods in Enzymology. Wassarman, P. M. and Soriano, P. M. (Eds.). Academic Press. 73-84.
- Liu, S. and Trapnell, C. (2016). Single-cell transcriptome sequencing: recent advances and remaining challenges. F1000Res 5.
- Love, M. I., Huber, W. and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15(12): 550.
- Markopoulos, G., Noutsopoulos, D., Mantziou, S., Gerogiannis, D., Thrasyvoulou, S., Vartholomatos, G., Kolettas, E. and Tzavaras, T. (2016). Genomic analysis of mouse VL30 retrotransposons. Mob DNA 7: 10.
- Peaston, A. E., Evsikov, A. V., Graber, J. H., de Vries, W. N., Holbrook, A. E., Solter, D. and Knowles, B. B. (2004). Retrotransposons regulate host genes in mouse oocytes and preimplantation embryos. Dev Cell 7(4): 597-606.
- Picelli, S. (2017). Single-cell RNA-sequencing: The future of genome biology is now. RNA Biol 14(5): 637-650.
- Picelli, S., Faridani, O. R., Bjorklund, A. K., Winberg, G., Sagasser, S. and Sandberg, R. (2014). Full-length RNA-seq from single cells using Smart-seq2. Nat Protoc 9(1): 171-181.
- Rowe, H. M. and Trono, D. (2011). Dynamic control of endogenous retroviruses during development. Virology 411(2): 273-287.
- Seli, E., Lalioti, M.D., Flaherty, S.M., Sakkas, D., Terzi, N., and Steitz, J.A. (2005). An embryonic poly(A)-binding protein (ePAB) is expressed in mouse oocytes and early preimplantation embryos. PNAS 102, 367-372.
- Smallwood, S. A., Tomizawa, S., Krueger, F., Ruf, N., Carli, N., Segonds-Pichon, A., Sato, S., Hata, K., Andrews, S. R. and Kelsey, G. (2011). Dynamic CpG island methylation landscape in oocytes and preimplantation embryos. Nat Genet 43(8): 811-814.
- Stein, P. and Schindler, K. (2011). Mouse oocyte microinjection, maturation and ploidy assessment. J Vis Exp (53). pii: 2851. doi: 10.3791/2851.
- Stewart, K. R., Veselovska, L., Kim, J., Huang, J., Saadeh, H., Tomizawa, S., Smallwood, S. A., Chen, T. and Kelsey, G. (2015). Dynamic changes in histone modifications precede de novo DNA methylation in oocytes. Genes Dev 29(23): 2449-2462.
- Tang, F., Barbacioru, C., Nordman, E., Li, B., Xu, N., Bashkirov, V. I., Lao, K. and Surani, M. A. (2010). RNA-Seq analysis to capture the transcriptome landscape of a single cell. Nat Protoc 5(3): 516-535.
- Treangen, T. J. and Salzberg, S. L. (2011). Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat Rev Genet 13(1): 36-46.
- Treger, R. S., Pope, S. D., Kong, Y., Tokuyama, M., Taura, M. and Iwasaki, A. (2019). The lupus susceptibility locus Sgp3 encodes the suppressor of endogenous retrovirus expression SNERV. Immunity 50(2): 334-347 e339.
- Tzavaras, T., Eftaxia, S., Tavoulari, S., Hatzi, P. and Angelidis, C. (2003). Factors influencing the expression of endogenous reverse transcriptases and viral-like 30 elements in mouse NIH3T3 cells. Int J Oncol 23(4): 1237-1243.
- Veselovska, L., Smallwood, S. A., Saadeh, H., Stewart, K. R., Krueger, F., Maupetit-Mehouas, S., Arnaud, P., Tomizawa, S., Andrews, S. and Kelsey, G. (2015). Deep sequencing and de novo assembly of the mouse oocyte transcriptome define the contribution of transcription to the DNA methylation landscape. Genome Biol 16: 209.
- Xing, X. (2010). An embryo handling device with a hamilton pipette (International Society for Transgenic Technologies at https://www.transtechsociety.org/) (Membership user ID and password are required for access.)
- Xue, Z., Huang, K., Cai, C., Cai, L., Jiang, C. Y., Feng, Y., Liu, Z., Zeng, Q., Cheng, L., Sun, Y. E., Liu, J. Y., Horvath, S. and Fan, G. (2013). Genetic programs in human and mouse early embryos revealed by single-cell RNA sequencing. Nature 500(7464): 593-597.
- Ziegenhain, C., Vieth, B., Parekh, S., Reinius, B., Guillaumet-Adkins, A., Smets, M., Leonhardt, H., Heyn, H., Hellmann, I. and Enard, W. (2017). Comparative analysis of single-cell RNA sequencing methods. Mol Cell 65(4): 631-643 e634.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Treger, R. S., Pope, S. D., Xing, X. and Iwasaki, A. (2019). Application of a Modified Smart-seq2 Sample Preparation Protocol for Rare Cell Full-length Single-cell mRNA Sequencing to Mouse Oocytes. Bio-protocol 9(16): e3345. DOI: 10.21769/BioProtoc.3345.
Category
Molecular Biology > RNA > RNA sequencing
Developmental Biology > Cell growth and fate > Oocyte
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.