- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Non-invasive Quantification of Cell Wall Porosity by Fluorescence Quenching Microscopy

Published: Vol 9, Iss 16, Aug 20, 2019 DOI: 10.21769/BioProtoc.3344 Views: 6172
Reviewed by: Samantha E. R. DundonVikash VermaAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Apr 2019
Advertisement




Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
All bacteria, fungi and plant cells are surrounded by a cell wall. This complex network of polysaccharides and glycoproteins provides mechanical support, defines cell shape, controls cell growth and influences the exchange of substances between the cell and its surroundings. Despite its name, the cell wall is a flexible, dynamic structure. However, due to the lack of non-invasive methods to probe the structure, relatively little is known about the synthesis and dynamic remodeling of cell walls. Here, we describe a non-invasive method that quantifies a key physiological parameter of cell walls, the porosity, i.e., the size of spaces between cell wall components. This method measures the porosity-dependent decrease of the plasma membrane-localized fluorescent dye FM4-64 in the presence of the extracellular quencher Trypan blue. This method is applied to bacteria, fungi and plant cell walls to detect dynamic changes of porosity in response to environmental cues.
Keywords: Cell wallBackground
Bacterial, fungal and plant cells are surrounded by a cell wall which has a multitude of different functions, including, defining size and shape, and controlling the exchange of substances within the environment. A key feature of cell walls is their flexibility. Long polysaccharides form a strong network and their structure is frequently adjusted, for example to allow for cell elongation, or to counter pathogen attack. In addition, cell wall porosity allows for molecular movement between the wall’s various components. Cell wall porosity is also a reliable indicator of digestibility and saccharification efficiency of cell wall material (Himmel et al., 2007; Ding et al., 2012; Tavares et al., 2015) and has been linked to anti-fungal drug uptake (Liu et al., 2019). Current methods for measuring cell wall porosity are based on relatively invasive methods, such as transmission electron microscopy (TEM) or field-emission scanning electron microscopy (FESEM) (Sugimoto et al., 2000; Xiao et al., 2016; Zheng et al., 2017), which alter the original cell wall structure and may lead to artifacts. Another method, cryo-FESEM, is less invasive but the resolution is limited to 20 nm (Derksen et al., 2011). Pore size distributions at sub-nanometer resolution can be obtained by gas adsorption, but this method also requires harsh sample pretreatment (Adani et al., 2011). Instead of determining pore sizes directly, other methods measure the capacity for molecular movement within the wall, which can be compatible with live-cell analysis. For example, the relative porosity of yeast cell walls assesses the measuring the effect of different-sized polycations on the leakage of UV-absorbing intracellular compounds (De Nobel et al., 1990). Similarly, the effect of fluorescent quenchers with different hydrodynamic radius on the autofluorescence of lignin can be measured to gain insight on differences in porosity between wood samples (Donaldson et al., 2015).
The method presented here makes use of the ability of quenchers to diffuse through the cell wall to determine relative porosity. In contrast to the approaches described above, this method works on living cells and can be applied to all accessible tissues and all organisms. It measures the fluorescence intensity of the dye FM4-64 located at the plasma membrane in the presence of extracellular quenchers. The quenching effect has been shown to depend on cell wall porosity (Liu et al., 2019). The method has been successfully used on bacteria, fungi and plants, for example to determine changes in wall porosity during drought-induced cell elongation in the model plant Arabidopsis (Liu et al., 2019).
The method described below uses Trypan blue as a quencher. In comparison with other quenchers, Trypan blue has a higher dynamic range (Liu et al., 2019), which results in more reliable results when analyzing cell walls with low porosity. Furthermore, Trypan blue is widely available, doesn’t require co-factors and is inexpensive. The protocol for yeast cells can be quickly adapted to any other microorganism, by choosing the right culture medium and growth conditions. Similarly, the protocols for Arabidopsis roots and Maize leaf samples can be adapted to other plant species. For root samples, however, growth on agar plates is recommended instead of cultivation on soil, because it can be difficult to clean all soil particles without damaging the root surface.
Materials and Reagents
- Pipette tips
- Microscope cover slips, 50 x 20 mm and 20 x 20 mm (CITOTEST, P/N: 80340-0630, 80340-3610)
- Microscope slide (CITOTEST, P/N: 80320-2140)
- 15 ml round bottom tubes
- 10 cm glass culture dish
- 1.5 ml Centrifuge tube
- Tape
- Razor blades
- Filter paper
- Yeast (Saccharomyces cerevisiae, BY4742)
- Arabidopsis seedlings (Col-0)
- Maize (Zea mays, B73)
- FM4-64 (N-(3-Triethylammoniumpropyl)-4-(6-(4-(Diethylamino) Phenyl) Hexatrienyl) Pyridinium Dibromide) (Thermo Fisher Scientific, catalog number: T3166)
- DMSO (Dimethyl sulfoxide) (MP Biomedicals, catalog number: 196055)
- Trypan blue (Solarbio, catalog number: T8070)
- Murashige and Skoog (MS) salts (Phyto Technology Laboratories, catalog number: M519)
- Sucrose (Guangzhou Jinhuada Chemical Reagent, CAS: 57-50-1)
- Agar (Solarbio, catalog number: A8190)
- Tryptone (OXIOD, catalog number: LP0042)
- Yeast extract (OXIOD, catalog number: LP0021)
- NaCl (Guangzhou Jinhuada Chemical Reagent, CAS: 7647-14-5)
- Glucose (Guangzhou Jinhuada Chemical Reagent, CAS: 58367-01-4)
- Na2HPO4 (Guangzhou Jinhuada Chemical Reagent, CAS: 10039-32-4)
- KH2PO4 (Guangzhou Jinhuada Chemical Reagent, CAS: 7778-77-0)
- KCl (Guangzhou Jinhuada Chemical Reagent, CAS: 7447-40-7)
- KOH
- YDP medium (see Recipes)
- Half strength MS medium (see Recipes)
- PBS solution (pH 7.2-7.4) (see Recipes)
- FM4-64 stock solution (see Recipes)
- Trypan blue stock solution (see Recipes)
Equipment
- Forceps
- Scissors
- Staining jars
- Pipettes, 10, 100, 1000 μl (Thermo Scientific, models: PZ31631, PZ69795, QZ06709)
- Centrifuge (Sigma, model: 1-14)
- Analytical balance (Adam Equipment, model: PWC 124)
- Autoclave sterilizer (ZEALWAY, model: GR60DP)
- Water purification systems (Synergy, model: SYNSVHF00)
- Air clean bench (AIRTECH, model: SW-CJ-2FD)
- Shaker incubator (Nanrong, model: NRY-200)
- Plant growth chamber (NINGBO SOUTHEAST INSTRUMENT, model: GDN-1000D-4)
- Confocal fluorescence microscope (Leica, model: DMi8) with ThorLabs module (model: ThorLabs-CLS)
Note: The protocol has also been tested on fluorescence widefield microscope systems with sCMOS cameras. These can be used instead of a confocal microscope as long as a clearly focused section of plasma membrane can be visualized with high signal-to-noise ratio. - Computer (Dell, model: U2717D)
Software
- ThorImageLS 3.0 (Thorlabs)
- ImageJ 1.52n (National Institutes of Health. USA, http://imagej.nih.gov/ij) (Schindelin et al., 2012)
Procedure
- Yeast sample
- Culture yeast cells in 5 ml YPD medium (see Recipes) in round bottom tubes in a shaker incubator at 30 °C for 24 h or according to the requirements of a specific strain or a specific experiment.
- Transfer 500 μl of the culture into a centrifuge tube, centrifuge for 1 min at 4,000 x g and discard the supernatant.
- Resuspend the cells in 1 ml PBS (pH 7.2-7.4, see Recipes), centrifuge for 1 min at 4,000 x g and discard the supernatant.
- Resuspend the cells in 50 μl PBS-based FM4-64 solution (50 μM) and incubate for 3 min at room temperature in the dark.
Note: Keep FM4-64 solution in the dark and stock solution on ice. Samples that have been labeled with FM4-64 should be imaged within 10 min, as the dye will eventually be endocytosed. Endocytosis of FM4-64 is indicated by internal signal, typically occuring in the form of small spheres. - Add 1 ml PBS solution and centrifuge for 1 min at 4,000 x g, discard the supernatant. Repeat this step one time.
- Transfer 2 μl stained cells onto a microscope slide and add 10 μl PBS, mix by stirring with the pipet tip and place a coverslip.
Note: If there are issues with cell movement during imaging, yeast cells can be immobilized on slides coated with polylysine or an agarose pad. - Mount the slide on the microscope and locate cells using bright field mode with a 100x objective.
Note: Especially with samples of microorganisms, it is important to minimize exposure to laser light, which will cause bleaching. Therefore, it is recommended to adjust focus in bright field mode before switching to confocal mode and immediately acquire an image. - Switch to confocal acquisition with 488 nm or 514 nm laser and a 582/75 nm bandpass emission filter to detect FM4-64 emission (see Notes). Pinhole diameter should be below two airy discs (about 100 μm).
- Set imaging parameters (laser power, detector gain or exposure time) to get a bright signal without overexposure.
- Acquire images. Typically, one image of 3-6 different areas per sample can be taken.
- Prepare additional samples following Steps A2 to A8, but instead of adding PBS to the slide in Step A6, add PBS-based Trypan blue solution, each time with a different concentration (1, 5, 10, 50 and 100 μM). Acquire images of each sample without changing any imaging parameters.
- Arabidopsis thaliana seedling root sample
- Grow Arabidopsis seedlings in half-strength MS medium on plates (see Recipes) at 22 °C with 16 h light/8 h dark for 7 d (Figure 1).
- Put a whole seedling into a tube containing 20 μM PBS-based FM4-64 solution, incubate for 3 min and wash with PBS solution in a staining jar.
- Add 50 μl PBS onto a microscope slide, arrange the seedling on the slide and add cover slip onto the root part of the seedling (Figure 1).
- Find the target region, for example the elongation zone, using a 40x objective and bright filed mode.
- Switch to confocal acquisition with 488 nm or 514 nm laser and a 582/75 nm bandpass emission filter to detect FM4-64 emission (see Notes). Pinhole diameter should be below two airy discs (about 100 μm).
- Set imaging parameters (laser power, detector gain or exposure time) to give a bright signal without overexposure.
- Acquire images. Typically, one image of 1-3 different areas per sample can be taken.
- Prepare additional samples following Steps B2 to B5, but instead of adding PBS to the slide in Step B3, add PBS-based Trypan blue solution, each time with a different concentration (1, 5, 10, 50 and 100 μM). Acquire images of each sample without changing any imaging parameters.

Figure 1. Preparation of Arabidopsis root samples. A. Seven-day-old seedlings grown on plates. B. Seedling with root submerged in FM4-64 solution. C. Seedling washed in PBS buffer in a staining jar. D. Seedling mounted on a slide with root covered by the cover slip.
- Maize leaf sample
- Put two layers of tape along the long edge of a large cover slip, leaving a groove in the middle (Figure 2). This facilitates the efficient exchange of quencher solution without moving the sample.
Note: Although this operation is more complicated than the ones described for the other samples, it can lead to higher data reliability when the effect of all quencher concentrations was measured on the same cell. The whole protocol should be finished within 10 to 15 min to prevent FM4-64 endocytosis from influencing the results. - Make a cut into a maize leaf using a razor blade and start peeling the lower epidermis using forceps. Cut a small piece of leaf together with the peeled epidermis (Figure 2).
Note: Separating the maize leaf epidermis from the rest of the leaf helps to provide more reliable results through minimization of chlorophyll fluorescence, whose spectrum overlaps with that of FM4-64. - Stain sample with 50 μM PBS-based FM4-64 solution in a centrifuge tube for 3 min, wash with PBS buffer in a staining jar (Figure 2).
- Add 100 μl PBS to the groove of the large cover slip and arrange the maize leaf sample in it. Cover the sample with a square cover slip (Figure 2).
- Find the target region using a 40x objective in bright field mode.
- Switch to confocal acquisition with 488 nm or 514 nm laser and a 582/75 nm bandpass emission filter to detect FM4-64 emission (see Notes). Pinhole diameter should be below two airy discs (about 100 μm).
- Set imaging parameters (laser power, detector gain or exposure time) to give a bright signal without overexposure.
- Acquire one image of the target region.
- Without removing the sample from the microscope, use filter paper to drain the buffer and immediately add PBS-based Trypan blue solution, starting with the lowest concentration (1 μM).
- Aquire one image and change to the next higher concentration as described in the step above. Repeat until images at all concentrations (1, 5, 10, 50, 100 μM) have been acquired.

Figure 2. Preparation of maize leaf samples. A. Maize seedling that was used here as sample material. B. A stretch of epidermis (arrow) is peeled from the leaf using forceps and cut out together with a small piece of leaf. C. Sample submerged in FM4-64 solution. D. Sample washed in PBS in a staining jar. E. Sample mounted on a large coverslip on which a central groove was created by taping the edges. F. Sample after application of a small coverslip. G. Change of quencher solution without removal of the sample from the microscope.
Data analysis
- Export all image data in a format compatible with ImageJ (e.g., tif) from the imaging system and import into ImageJ.
- Eliminate background signal by checking the background signal intensity in the space between cells (in case of yeast or bacterial cells) or the center of a cell (in case of larger cells), adjusting the black level to this value and pressing “apply” (Figure 3).
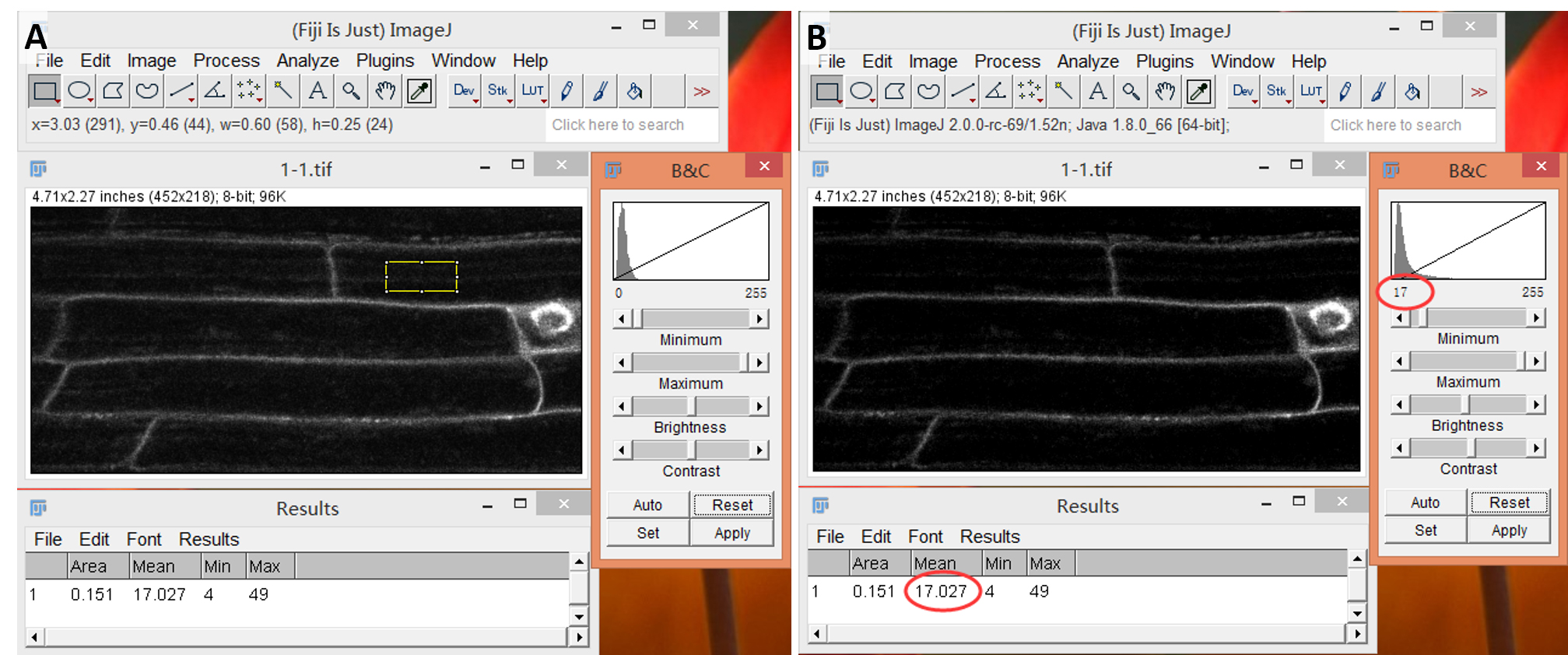
Figure 3. Background subtraction through adjustment of black level. A. Arabidopsis root sample with region of interest drawn at an area not containing the plasma membrane. B. The mean value of this area is used as black level and intensity values for the image redistributed by pressing “Apply”. - Draw a rectangular region of interest (ROI) covering the cell periphery of a cell (Figure 4) and measure mean grey value by pressing “Ctrl + M”. Do not change the size of the ROI between different images of one sample type. If the same region should be measured on several images, e.g., for the maize leaf sample, add the ROI to the ROI manager.
- Measure at least 50 ROIs for each type of sample and each quencher concentration. In yeast samples define one ROI per cell. In plant samples, several ROIs can be defined on the different walls of the same cell.
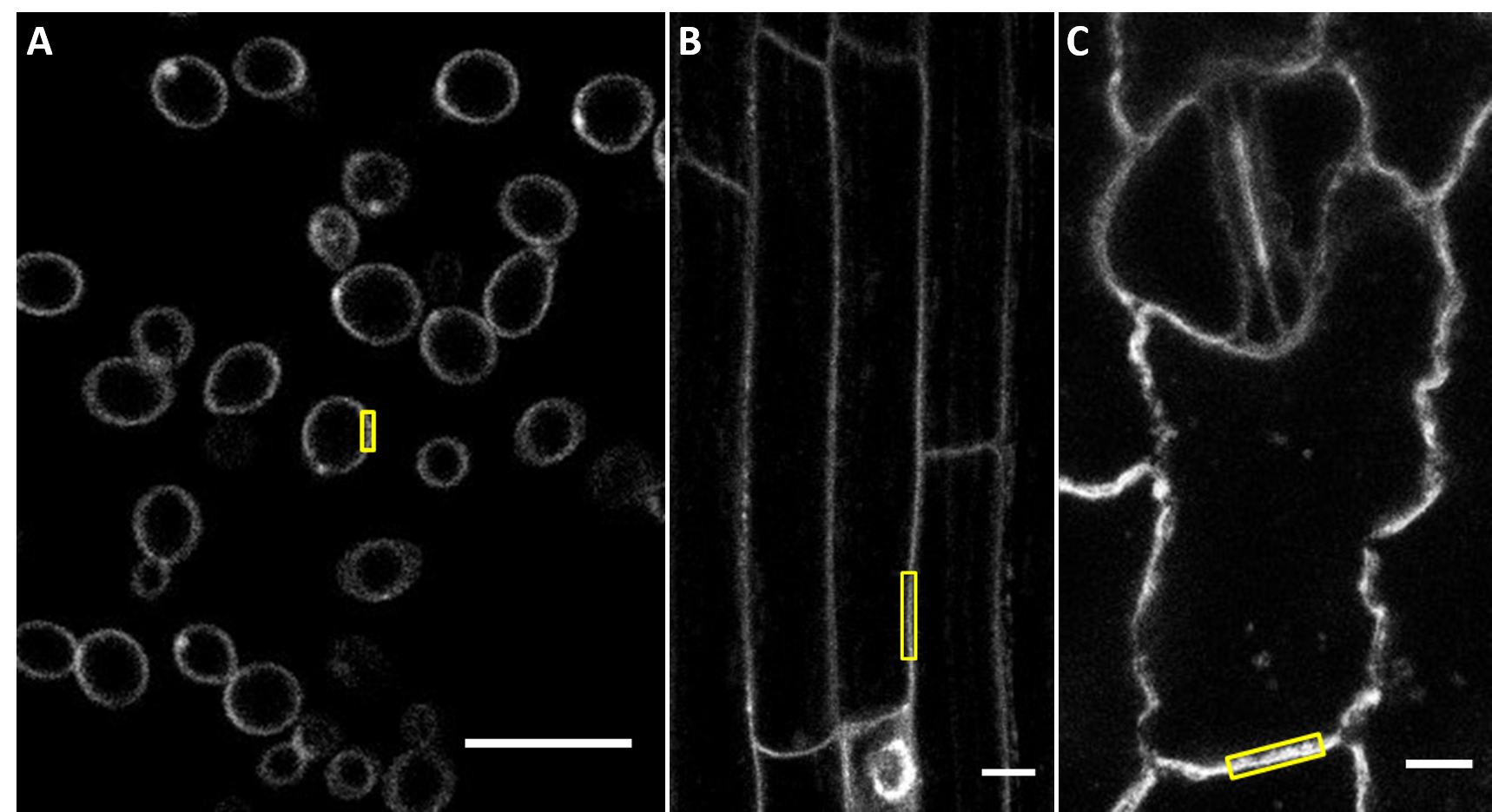
Figure 4. Exemplary regions of interest for the measurement of signal intensity. A. Yeast cells. B. Arabidopsis root cells. C. Maize leaf cells. Scale bars, 10 μm. - Export measurement data to spreadsheet software and calculate averages.
- Calculate quenching efficiency according to Lehrer (1971) with:

where, F0 is the fluorescence intensity of the sample in the absence of Trypan blue in the buffer; F is the fluorescence intensity of the sample in the presence of Trypan blue at the concentration of [Q];
K is the quenching efficiency of this sample system, which could be derived from slope of the concentration [Q] (x axis) plotted against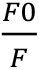 (y axis).
(y axis).
A higher quenching efficiency indicates better access of the quencher to the plasma membrane, which can serve as a good indicator of cell wall porosity (Liu et al., 2019). A detailed discussion of the relationship between quenching efficiency and cell wall structure can be found in Liu et al., (2019). The variation of results obtained from the same sample depends on the sample type (Figure 5).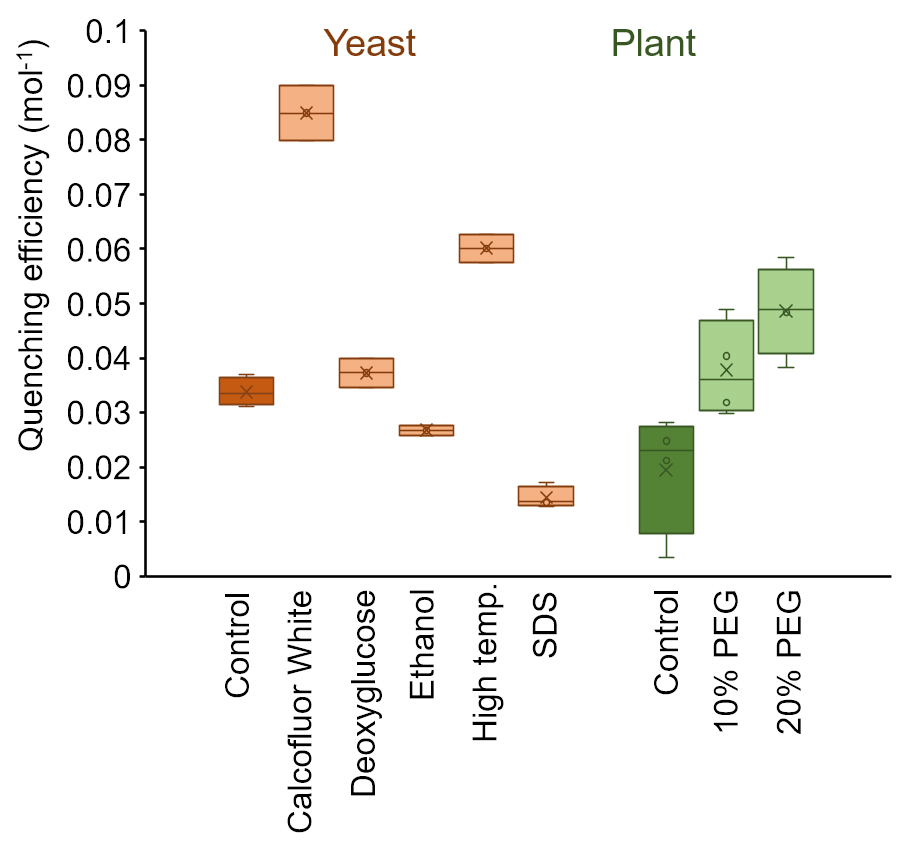
Figure 5. Exemplary quenching efficiency data measured on yeast and plant cells. Samples were exposed to chemical treatment or change in growth conditions for 12 h (yeast) or 24 h (plant). Variation within one treatment group was generally higher for plant samples than for yeast samples.
Notes
The emission of FM4-64 shows broad peaks at around 640 nm and 750 nm. Trypan blue was shown to emit fluorescence when bound to certain cell wall components between 640 and 680 nm (Liesche et al., 2015). To avoid overlap with the potential Trypan blue emission, it is recommended to detect FM4-64 around 580 nm or with a longpass filter beyond 700 nm.
Recipes
Note: Prepare all media using ultrapure water (drawn from a purification system set to a resistivity of 18 MΩ cm at 25 °C).
- YDP medium
2% (w/v) Tryptone
1% (w/v) Yeast extract
2% (w/v) glucose
Note: The mixture of tryptone and yeast extract, as well as the glucose solution should be sterilized at 121 °C for 20 min and adjusted to the final concentration after cooling. - Half strength MS medium
0.22% (w/v) Murashige and Skoog (MS) salts
1% (w/v) sucrose
Adjust the pH to 5.8 with 2 M KOH
0.8% (w/v) agar
Sterilize at 121 °C for 20 min, pour 25 ml medium to each glass culture dish - PBS solution (pH 7.2-7.4)
NaCl 130 mM
KCl 2.6 mM
Na2HPO4 7 mM
KH2PO4 1.2 mM - FM4-64 stock solution
Prepare 2 mM FM4-64 stock solution in DMSO
Store in aliquots of 100 µl at -20 °C in the dark - Trypan blue stock solution
Prepare 10 mM Trypan blue stock solution in PBS, keep at 4 °C in the dark
Note: Weigh with a balance placed inside a fume hood or under suction and wear a mask when handling the solid. Trypan blue is suspected of causing cancer, especially in the powdered form as it can easily be inhaled.
Acknowledgments
This protocol was adapted from our previous work (Liu et al., 2019). The work was supported by the National Natural Science Foundation China (project grant number 31700313), the Science Foundation of Shaanxi Province (100 Talent Program), and Villum Foundation (project grant number 022868).
Competing interests
The authors declare that no competing interests exist.
References
- Adani, F., Papa, G., Schievano, A., Cardinale, G., D'Imporzano, G. and Tambone, F. (2011). Nanoscale structure of the cell wall protecting cellulose from enzyme attack. Environ Sci Technol 45(3): 1107-1113.
- De Nobel, J. G., Klis, F. M., Munnik, T., Priem, J. and van den Ende, H. (1990). An assay of relative cell wall porosity in Saccharomyces cerevisiae, Kluyveromyces lactis and Schizosaccharomyces pombe. Yeast (Chichester, England) 6(6): 483-490.
- Derksen, J., Janssen, G. J., Wolters-Arts, M., Lichtscheidl, I., Adlassnig, W., Ovecka, M., Doris, F. and Steer, M. (2011). Wall architecture with high porosity is established at the tip and maintained in growing pollen tubes of Nicotiana tabacum. Plant J 68(3): 495-506.
- Ding, S. Y., Liu, Y. S., Zeng, Y., Himmel, M. E., Baker, J. O. and Bayer, E. A. (2012). How does plant cell wall nanoscale architecture correlate with enzymatic digestibility? Science 338(6110): 1055-1060.
- Donaldson, L. A., Kroese, H. W., Hill, S. J. and Franich, R. A. (2015). Detection of wood cell wall porosity using small carbohydrate molecules and confocal fluorescence microscopy. J Microsc 259(3): 228-236.
- Himmel, M. E., Ding, S. Y., Johnson, D. K., Adney, W. S., Nimlos, M. R., Brady, J. W. and Foust, T. D. (2007). Biomass recalcitrance: engineering plants and enzymes for biofuels production. Science 315(5813): 804-807.
- Liesche, J., Marek, M., Günther Pomorski, T. (2015). Cell wall staining with Trypan Blue enables quantitative analysis of morphological changes in yeast cells. Front Microbiol 6:107.
- Lehrer, S. S. (1971). Solute perturbation of protein fluorescence. The quenching of the tryptophyl fluorescence of model compounds and of lysozyme by iodide ion. Biochemistry 10(17): 3254-3263.
- Liu, X., Li, J., Zhao, H., Liu, B., Günther-Pomorski, T., Chen, S. and Liesche, J. (2019). Novel tool to quantify cell wall porosity relates wall structure to cell growth and drug uptake. J Cell Biol 218(4): 1408-1421.
- Schindelin, J., Arganda-Carreras, I., Frise, E., Kaynig, V., Longair, M., Pietzsch, T., Preibisch, S., Rueden, C., Saalfeld, S., Schmid, B., Tinevez, J. Y., White, D. J., Hartenstein, V., Eliceiri, K., Tomancak, P. and Cardona, A. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9(7): 676-682.
- Sugimoto, K., Williamson, R. E. and Wasteneys, G. O. (2000). New techniques enable comparative analysis of microtubule orientation, wall texture, and growth rate in intact roots of Arabidopsis. Plant Physiol 124(4): 1493-1506.
- Tavares, E.Q., De Souza, A.P. and Buckeridge, M.S. (2015). How endogenous plant cell-wall degradation mechanisms can help achieve higher efficiency in saccharification of biomass. J Exp Bot 66(14): 4133-4143.
- Xiao, C., Zhang, T., Zheng, Y., Cosgrove, D. J. and Anderson, C. T. (2016). Xyloglucan deficiency disrupts microtubule stability and cellulose biosynthesis in Arabidopsis, altering cell growth and morphogenesis. Plant Physiol 170(1): 234-249.
- Zheng, Y., Cosgrove, D. J. and Ning, G. (2017). High-resolution field emission scanning electron microscopy (FESEM) imaging of cellulose microfibril organization in plant primary cell walls. Microsc Microanal 23(5): 1048-1054.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Liu, X., Pomorski, T. G. and Liesche, J. (2019). Non-invasive Quantification of Cell Wall Porosity by Fluorescence Quenching Microscopy. Bio-protocol 9(16): e3344. DOI: 10.21769/BioProtoc.3344.
- Liu, X., Li, J., Zhao, H., Liu, B., Günther-Pomorski, T., Chen, S. and Liesche, J. (2019). Novel tool to quantify cell wall porosity relates wall structure to cell growth and drug uptake. J Cell Biol 218(4): 1408-1421.
Category
Microbiology > Microbial cell biology > Cell imaging
Plant Science > Plant cell biology > Cell wall
Cell Biology > Cell structure > Cell surface
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.