- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Assessment of Metacaspase Activity in Phytoplankton
Published: Vol 9, Iss 16, Aug 20, 2019 DOI: 10.21769/BioProtoc.3341 Views: 5222
Reviewed by: Alba BlesaJörg ToepelDavid A. Cisneros
Original research article
The authors used this protocol in:

Feb 2019
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Programmed cell death (PCD) is an irreversible, genetically-controlled form of cell suicide in which an endogenous biochemical pathway leads to morphological changes and ultimately, cellular demise. PCD is accompanied by de-novo protein synthesis of a family of proteases-"caspases" that are often used as a diagnostic marker of PCD. Although phytoplankton do not contain true caspases, caspase-like activity (hypothetical proteins with analogous activity) has been traditionally used as a diagnostic marker of PCD in marine phytoplankton. Increased caspase-like proteolytic activity was demonstrated when synthetic fluorogenic activity substrates specific for caspases (with an Asp at the P1 position) were applied upon PCD induction. Metacaspases, cysteine proteases, share structural properties with those of caspases, yet they are highly specific for Arg and Lys cleavage site at the P1 position implying that caspase specific substrates are not indicative of metacaspase catalytic activity. This method specifically tests direct metacaspase activity in phytoplankton by the cleavage of the fluorogenic metacaspase substrate Ac-VRPR-AMC. Metacaspase activity was tested by the addition of a metacaspase specific peptide that is conjugated to the fluorescent reporter molecule. The cleavage of the peptide by the metacaspase releases the fluorochrome that, when excited by light, emits fluorescence. The level of metacaspase enzymatic activity in the cell lysate is directly proportional to the fluorescence signal detected. The use of specific standards in this test enables the quantification of the fluorescence results. This assay directly allows monitoring the metacaspase cleavage products and thereby tracing evidence for programmed cell death.
Keywords: Metacaspase activityBackground
PCD is an irreversible, genetically-controlled form of cell suicide which is a fundamental feature of prokaryotic and eukaryotic microbial life essential for the regulation of cellular and tissue homeostasis in metazoans (Aravind et al., 1999). In phytoplankton, PCD has been defined as a form of autocatalytic cell suicide in which an endogenous biochemical pathway leads to morphological changes and ultimately, cellular dissolution (Bidle and Falkowski, 2004). PCD has been demonstrated in diverse evolutionary lineages of prokaryotic and eukaryotic phytoplankton including cyanobacteria, coccolithophores, diatoms, and dinoflagellates (Vardi et al., 1999; Segovia et al., 2003; Berman-Frank et al., 2004; Bidle and Falkowski, 2004; Bidle and Bender, 2008). In phytoplankton, environmental stresses, such as cell age, nutrient stress, excessive salt concentrations or oxidative stress and viral attacks may initiate PCD (Berman-Frank et al., 2004; Bidle and Falkowski, 2004; Spungin et al., 2019). This process is accompanied by de novo protein synthesis of a family of proteases-the caspases, (cysteine aspartic proteases) cleaving specifically at Asp residues at the P1 position. Caspases are often used as a diagnostic marker of PCD. Classic caspases are unique in metazoans and in several viruses (Minina et al., 2017). Higher plants, unicellular protists, fungi, and bacteria lack true caspases but contain metacaspases (Uren et al., 2000), cysteine-proteases that share structural properties, specifically a histidine-cysteine catalytic dyad in the predicted active site (Tsiatsiani et al., 2011). Metacaspases are widespread among prokaryotic and eukaryotic phytoplankton genomes. It has been indicated that prokaryotic phytoplankton, as well as evolving eukaryotic lineages, not only express PCD that involves caspase-like activity, but also a core set of proteins that are orthologues of metazoan caspases (Bidle and Falkowski, 2004; Bidle, 2016; Koonin and Aravind 2002).
Although phytoplankton do not contain true caspases, upon PCD induction caspase proteolytic activity is demonstrated when synthetic fluorogenic activity substrates are applied with an Asp at the P1 position (Berman-Frank et al., 2004 and 2007; Bidle and Bender, 2008; Thamatrakoln et al., 2012; Bar-Zeev et al., 2013). Unlike caspases, metacaspases lack Asp specificity and cleave their peptide substrates after Arg or Lys in the P1 position (Tsiatsiani et al., 2011). This implies that caspase specific substrates are not indicative of metacaspase catalytic activity. Thus, it has been suggested to use substrates with Arg or Lys residues at the P1 position to detect metacaspase activities in cellular extracts (Tsiatsiani et al., 2011; Klemenčič et al., 2015). Recently developed substrates applied to investigate the activity of specific metacaspases (Tsiatsiani et al., 2011; Klemenčič et al., 2015) have enabled further examination into the roles of metacaspases in organisms possessing metacaspase genes.
The method we detail herewith specifically tests direct metacaspase activity in phytoplankton by examining the cleavage of the fluorogenic metacaspase substrate Ac-VRPR-AMC (Ac-Val-Arg-Pro-Arg-AMC). Metacaspase activity tests and method calibrations were performed specifically in the marine cyanobacteria Trichodesmium under conditions that induce PCD (e.g., iron starvation and oxidative stress) and in samples collected from the South West Pacific Ocean (Spungin et al., 2018; Spungin et al., 2019). In this method, Trichodesmium cell lysate was tested for protease activity by the addition of a metacaspase specific peptide that is conjugated to the fluorescent reporter molecule, for example, 7-amino-4-methyl coumarin (AMC). The cleavage of the peptide by the metacaspase releases the fluorochrome that, when excited by light at 380 nm wavelength, emits fluorescence at 460 nm. Enzymatic activity in the cell lysate is directly proportional to the fluorescence signal detected with a fluorimeter or a fluorescent microplate reader.
Materials and Reagents
- Pipette tips
- Polycarbonate filters (e.g., PC 25 mm 5 µm, Whatman 110613)
- Microcentrifuge tubes (e.g., Axygen, catalog number: 3110451)
- Ice
- 96-well plate-black (e.g., SPL, Cell Culture Plate, PS, 96-well, Black, catalog number: 30496)
- HEPES (Sigma-Aldrich, catalog number: H3375)
- Sodium chloride (e.g., Merck, catalog number: 105404)
- Sucrose (e.g., AMRESCO, catalog number: 0335)
- CHAPS (3-(3-cholamidopropyl)-dimethylammonio-1-propanesulfonate) (Sigma-Aldrich, catalog number: C9426)
- Dithiothreitol (DTT) (Bio-Rad, catalog number: 1610611)
- Dimethyl Sulfoxide (DMSO) (Sigma-Aldrich, catalog number: 154938)
- Fluorogenic metacaspase substrate, Ac-VRPR-AMC, Mw = 725.85
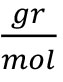 (BACHEM, catalog number: 4048494), store at -20 °C
(BACHEM, catalog number: 4048494), store at -20 °C - 7-amino-4-methyl coumarin (AMC) standard (Sigma-Aldrich, catalog number: A9891), Mw = 175.18 , store at -20 °C
- PierceTM BCA protein assay kit (Thermo, catalog number: 23225)
- Liquid nitrogen
- 5x Lauber buffer (see Recipe 1)
- 1 M AMC standard stock (see Recipe 2)
- 3 M Fluorogenic metacaspase substrate, Ac-VRPR-AMC (see Recipe 3)
- Working solution (Fluorogenic metacaspase aubstrate) (see Recipe 4)
Equipment
- Pipettes
- Ice bucket
- Forceps
- -20 °C freezer
- -80 °C freezer
- Ultra-cell disruptor (e.g., Sonic Dismembrator, Fisher Scientific, Waltham, MA)
- Microcentrifuge (e.g., Thermo Heraeus Pico 17 Microcentrifuge, Thermo Fisher, catalog number: 75002410)
- Vortex (e.g., Labnet, model: VX100 Vortex Mixer, catalog number: S-0100)
- Microplate reader equipped with fluorescence detection capabilities, kinetic reads and shake options (e.g., Synergy4 BioTek, Winooski, VT)
Procedure
- Preparation of cell extracts (for cell collection and volumes refer to Notes 1 and 2)
- Prepare 1x Lauber buffer (see Recipe 1).
- Add 1000 µl 1x Lauber buffer to the cells (Note 3).
- Place the tubes in a bucket of ice.
- Sonicate each sample for 30 s (short pulses). Repeat this step 4 times (each sample 4 x 30 s) (Note 4).
- Centrifuge at room temperature, maximum speed (14,000 x g) for 5 min.
- Transfer 500 µl of supernatant to a fresh tube containing 5 µl DTT (1 M) and place on ice for immediate assay or store at -80 °C for future use. The remaining sample in the tube can be stored at -80 °C for backup and for total protein concentration analysis.
- Preparation of Standards (Note 5)
Use Table 1 as a guide to prepare a set of standards by using two-fold dilutions.- Prepare a stock of 1 M AMC (see Recipe 2).
- Prepare 15 tubes.
- To each tube add the diluent (1x Lauber buffer).
- To tube 1 add 1 µl AMC standard stock (1 M) to a final concentration of 1,000 µM.
- Vortex the tube and remove 500 µl from tube 1 to tube 2 (2 fold dilution), thus in tube 2 the final concentration will be 500 µM, vortex the tube and remove 500 µl from tube 2 to tube 3, to a final concentration of 250 µM in tube 3. Repeat this to all tubes. Tube number 15 leave with 1x Lauber buffer only (0 µM).
Table 1. Preparation of diluted AMC standards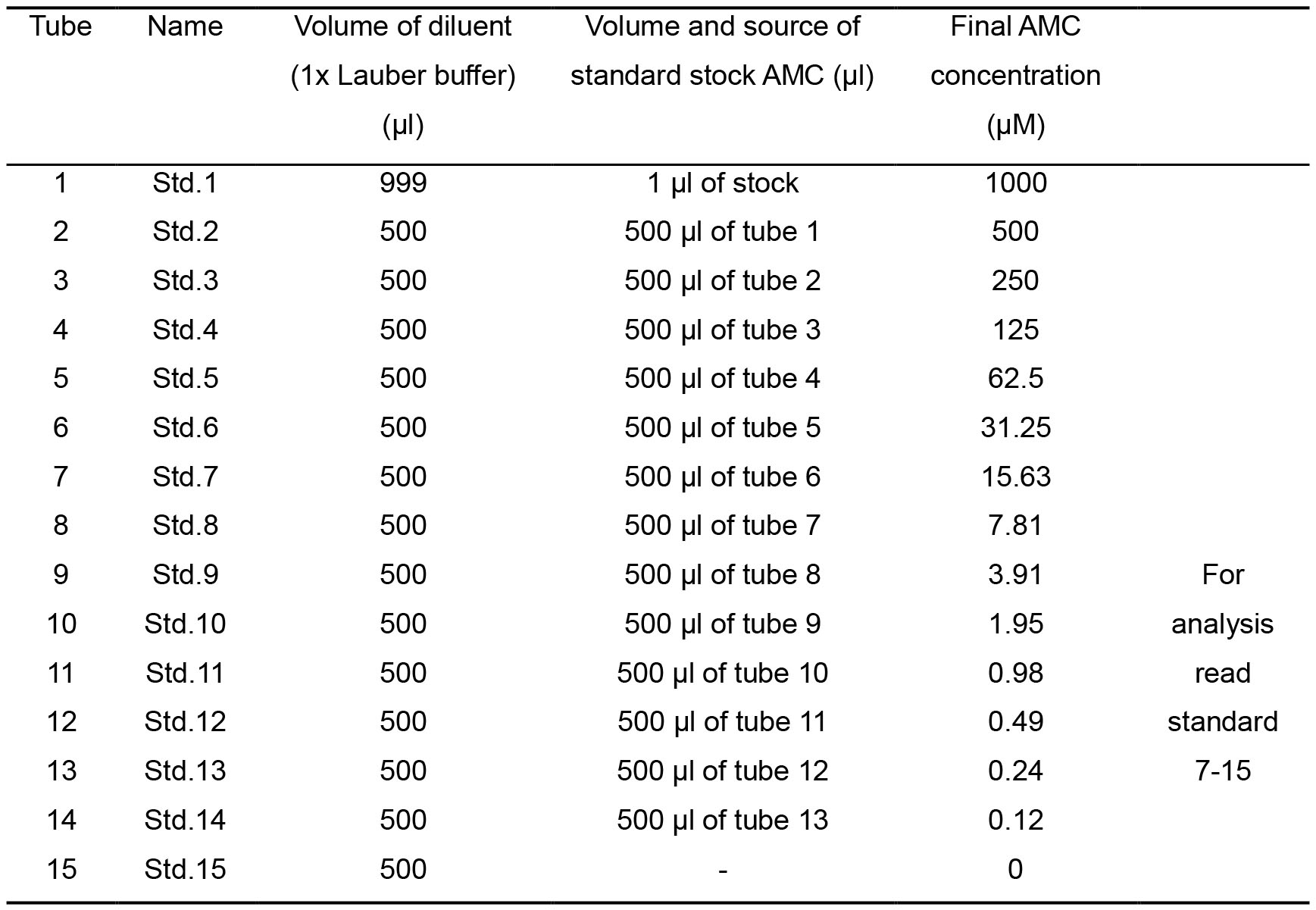
- Loading of the plate (Table 2)
- Additional controls that should be included in this assay are a) Sample blank (no substrate), b) Substrate blank (no cell lysate, substrate blanks will ensure the substrate is not spontaneously degraded over time) and c) DMSO control (as it is used as a solvent). The total reaction volume must be kept constant and therefore 1x Lauber buffer can be used to replace the volume normally occupied by either the cell lysate or the substrate reagent.
- Prepare working solution (WS) of substrate (1 mM) sufficient for all samples, standards and substrate blanks (see Recipe 4).
- Load the 96-well plate according to the content and amounts presented in Table 2 and table plan example presented in Figure 1.
- Cover the plate with a lid and go to the plate reader (Note 6).
Table 2. Description of the amounts and contents required for loading of the plate
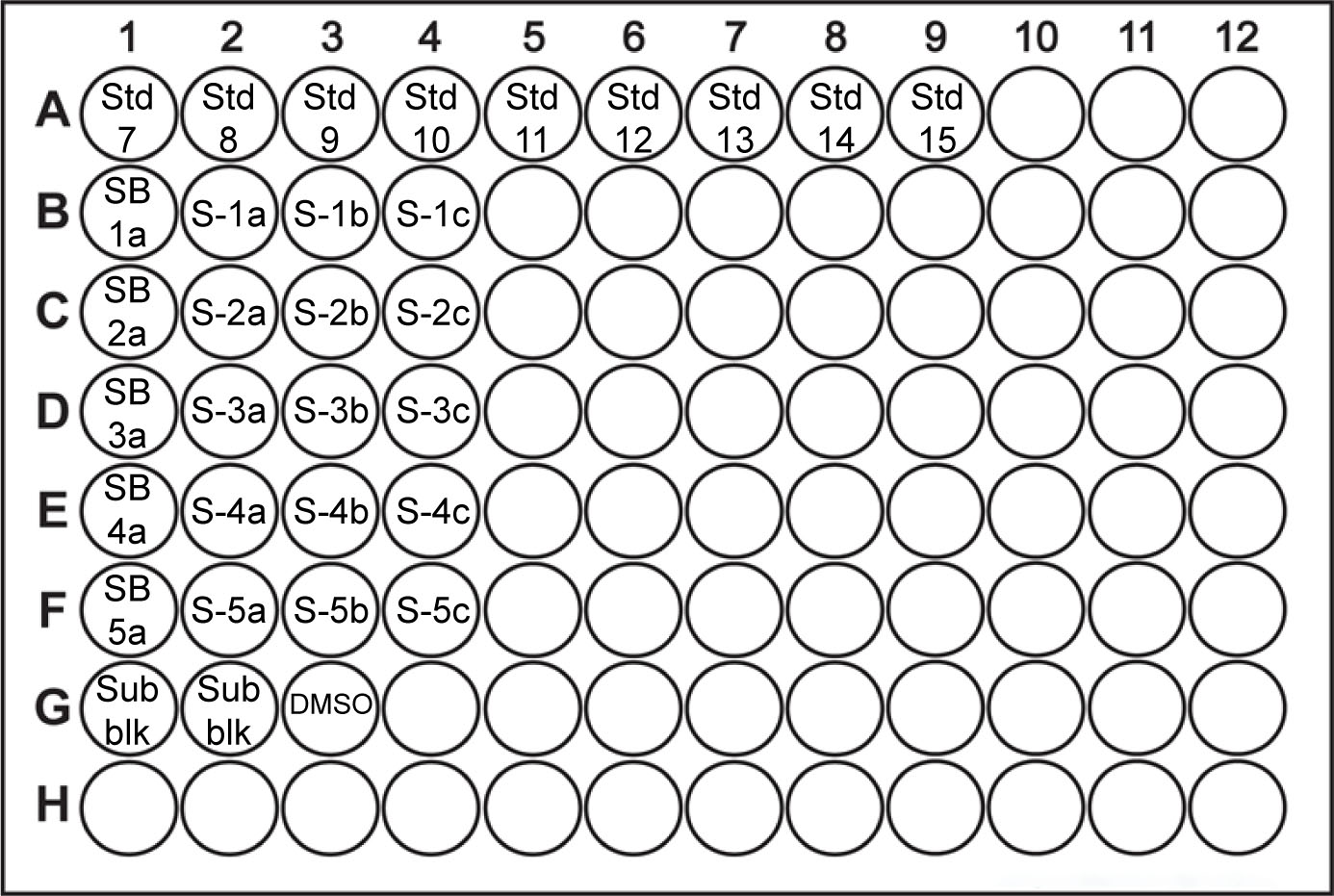
Figure 1. Example of 96-well plate samples, standards and control loading plan. Std- standard sample; SB- sample blank; S- sample; Sub blk- substrate blank; DMSO- Dimethyl Sulfoxide. - Plate reader
Substrate cleavage (release of AMC) is performed here by continuously monitoring the release over 2-3 h. Alternatively, end point analysis can be performed by measuring AMC after a defined time (30-40 min), but it is advisable to first monitor continuously to ensure that the time chosen is within the linear phase of the reactions.
Read the plate with the next parameters:- Temperature: room temperature.
- Kinetics: 2-3 h with a read-every 10 min.
- Mix: all wells should be mixed (medium mix, 5 s) before each read.
- Ex: 380 nm, Em: 460 nm.
- Temperature: room temperature.
- Additional controls that should be included in this assay are a) Sample blank (no substrate), b) Substrate blank (no cell lysate, substrate blanks will ensure the substrate is not spontaneously degraded over time) and c) DMSO control (as it is used as a solvent). The total reaction volume must be kept constant and therefore 1x Lauber buffer can be used to replace the volume normally occupied by either the cell lysate or the substrate reagent.
- Calculations
- Standard curve calculation:
Prepare a graph of standard curve (Fluorescence unit [FU] against standard concentration) and calculate the standard curve equation. See Table 3, and Figure 2 for example.
Table 3. Example of fluorescence units (FU) obtained for the different AMC standard concentrations. Only standard tubes 7-15 are read (Note 5).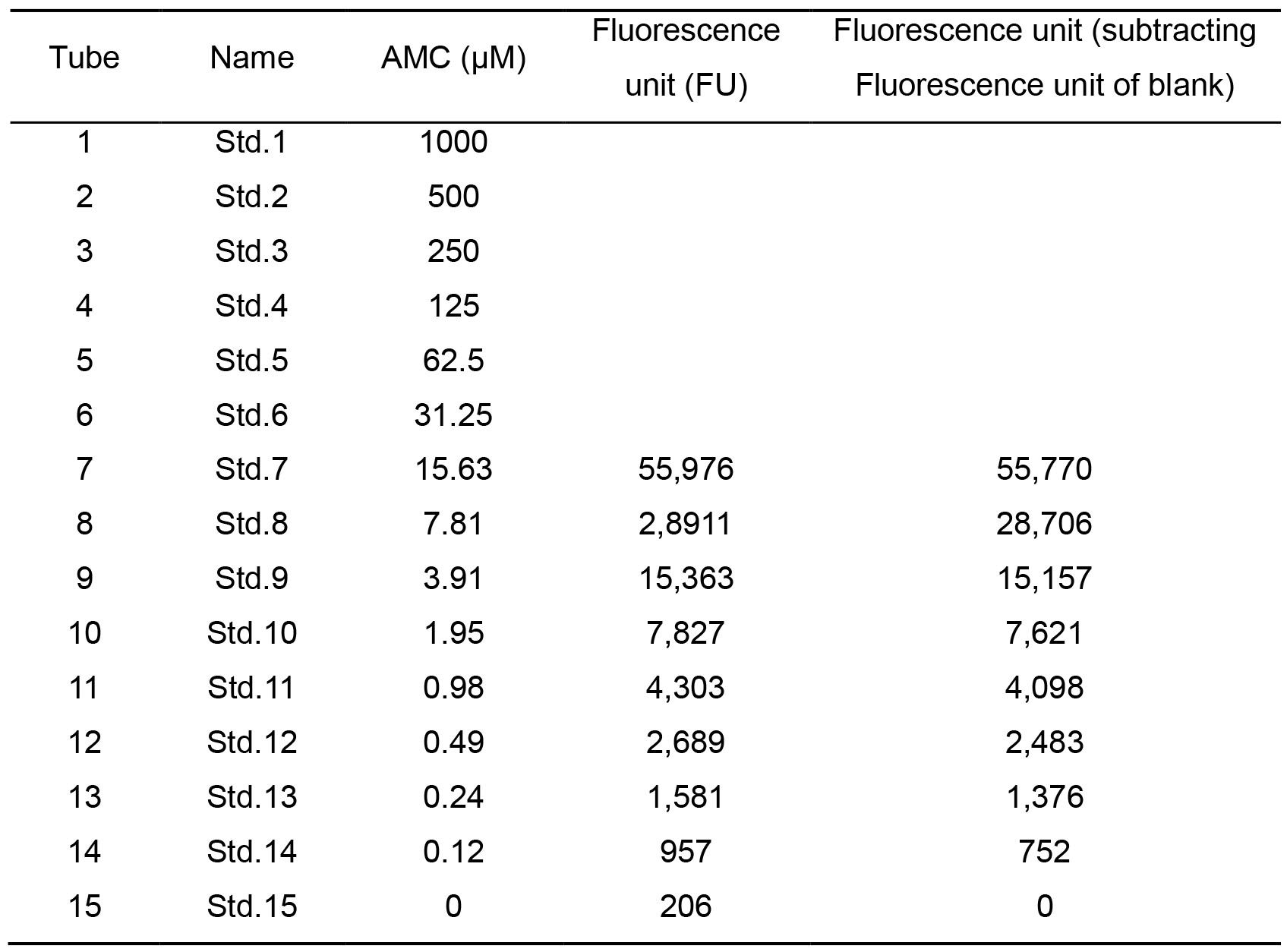
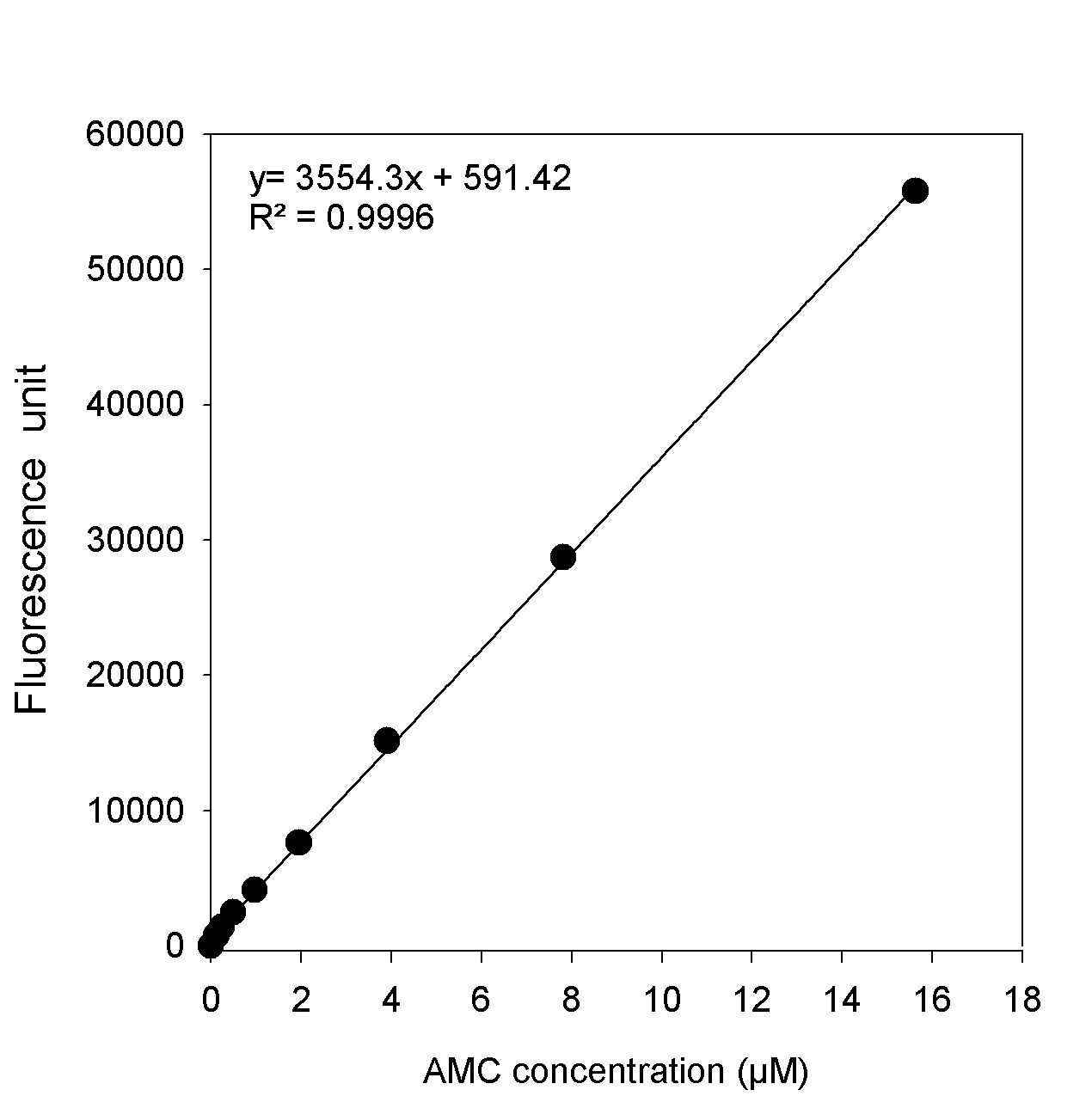
Figure 2. Example of AMC standard curve obtained from the fluorescence reads against AMC concentrations (µM) - Sample calculations:
- If background controls (sample blanks or substrate blanks) give a substantial reading, these values should be subtracted from the experimental results.Subtract the sample blanks read from the sample results reads.
- Using the standard curve equation insert the sample fluorescence values obtained in the standard equation, to obtain AMC concentrations (Table 4).
- Plot the data on a graph (AMC concentration against time) (Figure 3).
- For each sample calculate the slope, obtained from the linear time points. This is an indicator for change in concentration over time (µM AMC min-1).
- Multiply the slope (µM min-1) by the filtration volume and divide by the resuspension volume (1x Lauber buffer added) (Table 5).
- Protein normalization: it is suggested to normalize each kinetic rate to its total protein amount. This will also allow comparing between the different samples. Protein concentration can be determined from the remaining sample (see Step A6). The protein content of the cell lysate can be estimated using a protein determination assay that is compatible with detergents present in the Lauber Buffer where no DTT is added (e.g., PierceTM BCA protein assay kit).
- Final units for metacaspase activity rate (cleavage of substrate rate) are µM AMC min-1. If results are normalized to mg L-1 protein, final units are µmol AMC mg protein-1 min-1 (For unit equation refer to Note 7, unit equation 1).
- Average the 3 results obtained from the 3 technical replicates (Table 5).
Table 4. Example of FU obtained for sample 1 (3 technical sample replicates 1a, 1b and 1c) in 10 min time intervals with a total reaction time of 110 min and values obtained after inserting the FU in the standard curve equation (µM AMC).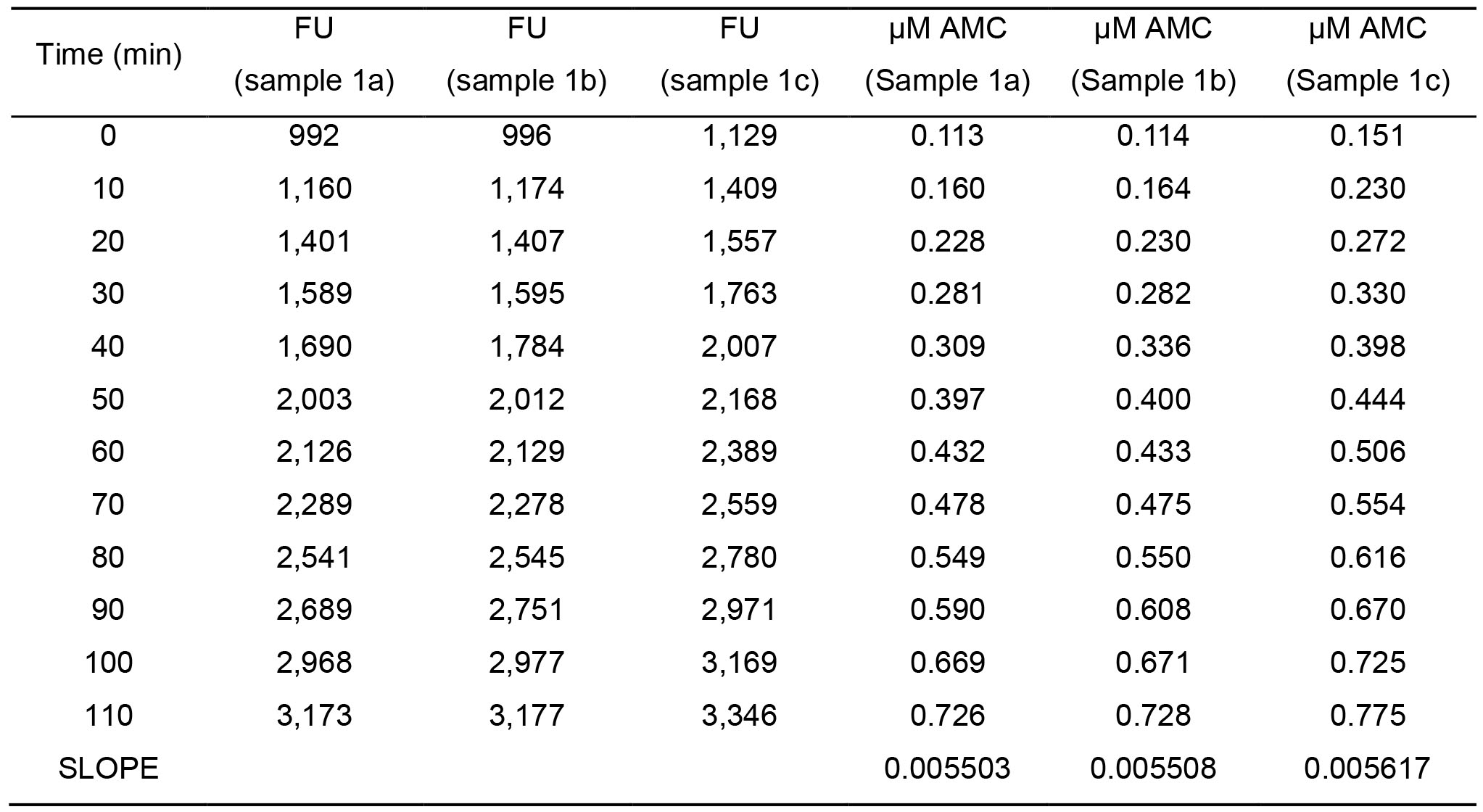
Table 5. Data calculation example, after the slope is obtained. The slope (µM AMC min-1) is multiplied by the filtration volume (ml) and divided by resuspension volume (ml) for normalization. Normalization to biomass (e.g., protein) is advised. Technical repeats are then averaged, to a final result of metacaspase activity, representing the rate of AMC release min-1 mg protein-1.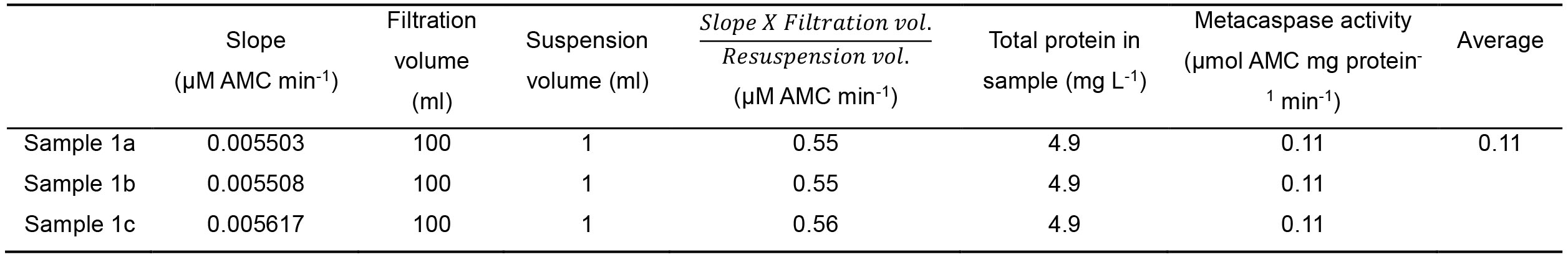
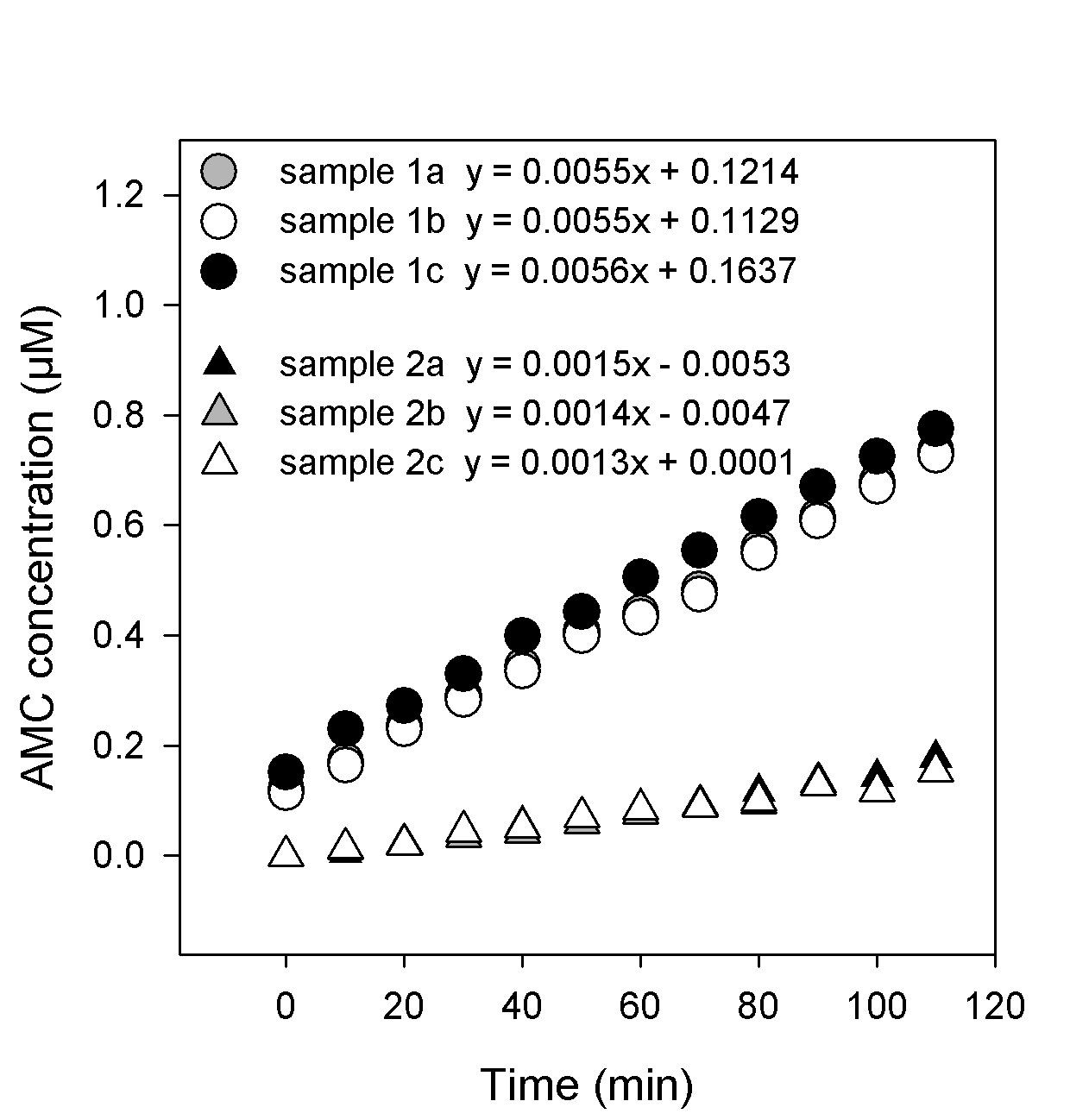
Figure 3. Example of curves obtained from sample 1, and sample 2, representing the change in AMC concentration (µM) over time. The 3 curves represent 3 technical reads of each sample (1a, 1b and 1c or 2a, 2b and 2c).
- Standard curve calculation:
Notes
- Collection of cells by filtration: Filtration process is necessary only when no other way (e.g., centrifuge) of cell isolation is suitable. For filtration process follow the next steps:
- Filter cells on a polycarbonate filter (For Trichodesmium 5-10 µM pore size).
Filtration volume depends on the source of the samples and concertation. For dense cultures of Trichodesmium (~1-5 µg ml-1 total protein) 100-200 ml are filtered. For natural marine samples, where concentrations are low (~0.02 µg ml-1 total protein) 2,000-4,000 ml should be filtered. Filtration volume may vary between different cells; a specific test should be done to verify filtration volumes. - Insert the filter in a microcentrifuge tube.
- Quick freeze the filter in liquid nitrogen. Use forceps and immerse the tube in the liquid nitrogen for ~15 s.
- Shake the tube or gently vortex until cells are released from the filter, then take the filter out of the tube.
- Proceed to the sonication step (Step A4).
For further information regarding, sample collection of Trichodesmium during our experiments, refer to Spungin et al., 2016, 2018 and 2019.
- Filter cells on a polycarbonate filter (For Trichodesmium 5-10 µM pore size).
- After freezing the sample in liquid nitrogen, samples can be stored in -80 °C until further analysis. For long term storage it is advisable to proceed to the next steps, of buffer addition and cell extraction and then store the samples at -80 °C.
- It is advised to study different cell contractions to ensure that no saturation effects occur during the assay. The amount of suspension volume can be changed according to cell concentration, thus 1,000 µl is suggested, but if low cell concentrations are detected, reduce the suspension volume.
- Take at least 10-15 s break between each sonication per sample as the sonication process heats the samples. Let the sample rest on ice between each sonication round.
- As metacaspase activity rate may vary in different organisms or under different cell concentrations, the range of the standard curve should suit the range of the activity rate (the amount of AMC substrate released via metacaspase activity in the sample). Thus, it is important to specifically test the suitable standard range. For analyses we use standard in tubes 7-15 (concentration ranging between 0 to 15.6 µM. However, a test should be done to ensure the appropriate range).
- Reaction occurs instantaneously at room temperature so fluorescence should be read right after the loading of the plate.
- Final units for metacaspase activity rate (cleavage of substrate rate) are µM AMC min-1. If results are normalized to mg L-1 protein, final units are µmol AMC mg protein-1 min-1 (Unit equation 1).
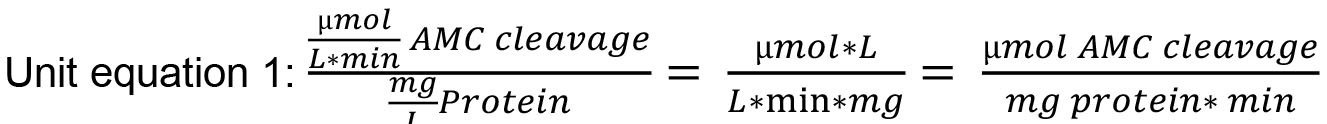
Recipes
- 1.5x Lauber buffer
50 mM HEPES (5.96 g in 100 ml DDW)
100 mM NaCl (2.92 g in 100 ml DDW)
10% Sucrose (50 g in 100 ml DDW)
0.1% CHAPS (0.5 g in 100 ml DDW)
10 mM DTT (10 μl of 1 M DTT in 1 ml Lauber buffer, will yield a final concentration of 10 mM DTT)- for metacaspase activity assay prepare 5x Lauber buffer with no DTT (DTT will be added during assay procedure, see Step A6)
For preparing 1x Lauber buffer: Dilute 5x Lauber buffer 1:4 with DDW. - 1 M AMC standard stock
7-amino-4-methylcoumarin (AMC) standard (Sigma)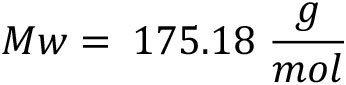
Prepare stock of 1 M:
For 1 M AMC stock, add 0.175 mg to 1 ml DMSO
Divide to aliquots and store at -20 °C - 3 M Fluorogenic metacaspase substrate, Ac-VRPR-AMC (BACHEM)
Amount in tube: 1 mg= 0.001 g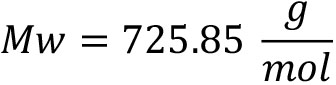
Prepare stock of 3 mM:
For 3 mM substrate stock, add 460 µl DMSO to stock
Following reconstitution, aliquot and freeze (-20 °C). Stock solutions are stable for up to 6 months at -20 °C - Working solution (Fluorogenic metacaspase aubstrate)
For assay we use working solution of 1 mM, thus dilute the amount needed for the assay: 1:2 with 1x Lauber buffer
Acknowledgments
This work was supported by the United States-Israel Binational Science Foundation (BSF) grant 2008048 to IBF and KDB, a collaborative grant from MOST Israel and the High Council for Science and Technology (HCST)-France to IBF, and GIF funding No. 1133/2011 to IBF. This protocol was adapted and modified from our previous studies (Spungin et al., 2018 and 2019).
Competing interests
The authors declare no competing financial interests.
References
- Aravind, L., Dixit, V. M. and Koonin, E. V. (1999). The domains of death: evolution of the apoptosis machinery. Trends Biochem Sci 24(2): 47-53.
- Bar-Zeev, E., Avishay, I., Bidle, K. D. and Berman-Frank, I. (2013). Programmed cell death in the marine cyanobacterium Trichodesmium mediates carbon and nitrogen export. ISME J 7(12): 2340-2348.
- Berman-Frank, I., Bidle, K. D., Haramaty, L. and Falkowski, P. G. (2004). The demise of the marine cyanobacterium, Trichodesmium spp., via an autocatalyzed cell death pathway. Limnol Oceanogr 49(4): 997-1005.
- Berman-Frank, I., Rosenberg, G., Levitan, O., Haramaty, L. and Mari, X. (2007). Coupling between autocatalytic cell death and transparent exopolymeric particle production in the marine cyanobacterium Trichodesmium. Environ Microbiol 9(6): 1415-1422.
- Bidle, K. D. (2016). Programmed cell death in unicellular phytoplankton. Curr Biol 26(13): R594-R607.
- Bidle, K. D. and Bender, S. J. (2008). Iron starvation and culture age activate metacaspases and programmed cell death in the marine diatom Thalassiosira pseudonana. Eukaryot Cell 7(2): 223-236.
- Bidle, K. D. and Falkowski, P. G. (2004). Cell death in planktonic, photosynthetic microorganisms. Nat Rev Microbiol 2(8): 643-655.
- Klemenčič, M., Novinec, M. and Dolinar, M. (2015). Orthocaspases are proteolytically active prokaryotic caspase homologues: the case of Microcystis aeruginosa. Mol Microbiol 98(1): 142-150.
- Koonin, E. V. and Aravind, L. (2002). Origin and evolution of eukaryotic apoptosis: the bacterial connection. Cell Death Differ 9(4): 394-404.
- Minina, E. A., Coll, N. S., Tuominen, H. and Bozhkov, P. V. (2017). Metacaspases versus caspases in development and cell fate regulation. Cell Death Differ 24(8): 1314-1325.
- Segovia, M., Haramaty, L., Berges, J. A. and Falkowski, P. G. (2003). Cell death in the unicellular chlorophyte Dunaliella tertiolecta. A hypothesis on the evolution of apoptosis in higher plants and metazoans. Plant Physiol 132(1): 99-105.
- Spungin, D., Belkin, N., Foster, R.A., Stenegren, M., Caputo, A., Pujo-Pay, M., Leblond, N., Dupouy, C., Bonnet, S. and Berman-Frank, I. (2018). Programmed cell death in diazotrophs and the fate of organic matter in the western tropical South Pacific Ocean during the OUTPACE cruise. Biogeosciences 15: 3893-3908.
- Spungin, D., Bidle, K. D. and Berman-Frank, I. (2019). Metacaspase involvement in programmed cell death of the marine cyanobacterium Trichodesmium. Environ Microbiol 21(2): 667-681.
- Spungin, D., Pfreundt, U., Berthelot, H., Bonnet, S., AlRoumi, D., Natale, F., Hess, W. R., Bidle K. D. and Berman-Frank, I. (2016). Mechanisms of Trichodesmium demise within the New Caledonian lagoon during the VAHINE mesocosm experiment. Biogeosciences 13: 4187-4203.
- Thamatrakoln, K., Korenovska, O., Niheu, A. K. and Bidle, K. D. (2012). Whole-genome expression analysis reveals a role for death-related genes in stress acclimation of the diatom Thalassiosira pseudonana. Environ Microbiol 14(1): 67-81.
- Tsiatsiani, L., Van Breusegem, F., Gallois, P., Zavialov, A., Lam, E. and Bozhkov, P. V. (2011). Metacaspases. Cell Death Differ 18(8): 1279-1288.
- Uren, A. G., O'Rourke, K., Aravind, L. A., Pisabarro, M. T., Seshagiri, S., Koonin, E. V. and Dixit, V. M. (2000). Identification of paracaspases and metacaspases: two ancient families of caspase-like proteins, one of which plays a key role in MALT lymphoma. Mol Cell 6(4): 961-967.
- Vardi, A., Berman-Frank, I., Rozenberg, T., Hadas, O., Kaplan, A. and Levine, A. (1999). Programmed cell death of the dinoflagellate Peridinium gatunense is mediated by CO2 limitation and oxidative stress. Curr Biol 9(18): 1061-1064.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Spungin, D. and Berman-Frank, I. (2019). Assessment of Metacaspase Activity in Phytoplankton. Bio-protocol 9(16): e3341. DOI: 10.21769/BioProtoc.3341.
Category
Microbiology > Microbial biochemistry > Protein > Activity
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.