- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Ex vivo Drosophila Wing Imaginal Disc Culture and Furin Inhibitor Assay

Published: Vol 9, Iss 16, Aug 20, 2019 DOI: 10.21769/BioProtoc.3336 Views: 5637
Reviewed by: Abhijit KaleSveta ChakrabartiAnonymous reviewer(s)
Original research article
The authors used this protocol in:

May 2019
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Furin is an evolutionarily conserved proprotein convertase (PC) family enzyme with a broad range of substrates that are essential for developmental, homeostatic, and disease pathways. Classical genetic approaches and in vitro biochemical or cell biological assays identified that precursor forms of most growth factor family proteins are processed by Furin. To quantitatively assess the potential role of Furin in cleaving and modulating intercellular dispersion of a Drosophila signaling protein, we developed a simple assay by combining genetics, ex vivo organ culture, pharmacological treatment, and imaging analyses. The protocol herein describes how to ex vivo culture Drosophila wing imaginal discs expressing a fluorescently tagged Drosophila Fibroblast Growth Factor (FGF, Branchless/Bnl) over a long period of time in the presence of Furin inhibitors and monitor the cleavage and intercellular dispersion of the truncated Bnl parts using microscopy. Although the assay described here is for assessing the effect of Furin inhibition on Bnl cleavage in the Drosophila larval wing imaginal disc, the principle and methodology can easily be adopted for any other signals, tissue systems, or organisms. This strategy and protocol provide an assay for examining Furin activity on a specific substrate by directly visualizing the spatiotemporal distribution of its truncated parts in an ex vivo-cultured organ.
Keywords: Proteolytic cleavageBackground
The range of intercellular dispersion and signaling activity of many morphogenetic signaling proteins depends on 'limited proteolysis' by proprotein convertases (PCs) (LeMosy, 2006; Künnapuu et al., 2009; Wharton and Serpe, 2013; Sohr et al., 2019). PCs are serine endoprotease family enzymes that are known to cleave various growth factors including Epidermal growth factor (EGF), the Transforming Growth Factor beta/Bone Morphogenetic Protein (TGF-β/BMP) family, several Fibroblast Growth Factor (FGF) family proteins, Platelet-Derived Growth Factor (PDGF), and Vascular Endothelial Growth Factor (VEGF), all of which play a critical role in development and diseases (Khatib and Geraldine, 2006; Tulin and Stathopoulos, 2010; Rousselet et al., 2011; Künnapuu et al., 2014; Constam, 2014; Anderson and Wharton, 2017; Sohr et al., 2019). Although biochemical and cell culture assays are sufficient to demonstrate a substrate cleavage by individual PC substrate-specificity, their functional characterization in vivo is largely dependent on classical genetic approaches. However, the presence of multiple PCs in different organisms and their functional redundancy can only provide limited information about an individual PC's function. There are nine PCs in vertebrates and three in Drosophila, all expressed broadly, that share a consensus cleavage site (-Arg-X-Lys/Arg-Arg-) in their substrates. Moreover, a broad range of cellular substrates as well as extracellular matrix proteins that can also influence a signaling or morphogenetic outcome makes the genetic results hard to interpret.
Here we present a protocol that enables direct visualization of Furin-dependent signal cleavage by monitoring the spatiotemporal fates of the cleaved portions of a substrate in developing organs. The strategy and methodology was designed to examine Drosophila Bnl cleavage by Furin. The experiment takes advantage of the 3rd instar larval wing imaginal disc and an associated tracheal branch called the air sac primordium (ASP) (Sato and Kornberg, 2002). Bnl is expressed by a restricted group of wing disc cells and subsequently transported to the ASP to control its morphogenesis (Sato and Kornberg, 2002; Du et al., 2018). Bnl is cleaved by Furin-1 at the 164th amino acid residue prior to the interorgan transport of only its C-terminal truncated portion that contains the receptor binding site (Sohr et al., 2019). The truncated part of Bnl forms a characteristic long-range gradient in the ASP by binding to its receptor and traveling through actin-based filopodia or cytonemes extended by ASP cells (Du et al., 2018). To visually monitor the fates of the full-length Bnl and its truncated portions, we first generated a transgenic Drosophila line harboring a chimeric Bnl construct with an HA-tag at the 87th and a GFP tag at 432nd residue, flanking the cleavage site and conserved FGF domain. The construct was expressed in the wing disc Bnl source. Laval wing discs were dissected and ex vivo-cultured in the absence or presence of Furin protease inhibitors for variable amounts of time followed by α-HA immunostaining and fluorescence microscopic imaging of the discs. While in normal conditions only the C-terminal GFP-tagged fragment of Bnl was transported to the ASP, the addition of Furin inhibitors to the ex vivo culture resulted in uncleaved Bnl with both tags being transported to the recipient ASP cells (Figure 1). Since a broad range of signaling proteins involved in morphogenetic and disease pathways are modulated by Furin, the methodology described here can be a powerful tool to evaluate the role of Furin cleavage in various other signaling pathways. For instance, despite the wide-spread occurrence of signal cleavage modulating paracrine signaling activity, most full-length uncleaved forms of the signals were also found to activate receptors and are shown to be secreted when expressed in cultured cells (Künnapuu et al., 2009; Tulin and Stathopoulos, 2010; Sopory et al., 2010; Tokhunts et al., 2010; Constam, 2014). Therefore, a link between PC-dependent cleavage with the maturation and activity of the proteins remained obscure. The results from this assay together with other experiments showed that uncleaved Bnl is active and can be transported to the recipient cells to induce signaling. However, Bnl cleavage ensures its efficient polarized target-specific dispersion, thereby modulating its range and tissue-specific activity (Sohr et al., 2019). The ex vivo culture method with pharmacological Furin inhibition strategy is not limited to only the Drosophila wing imaginal disc and ASP tissue system. Similar assays can easily be applied to other Drosophila tissues, signaling proteins, or model systems.
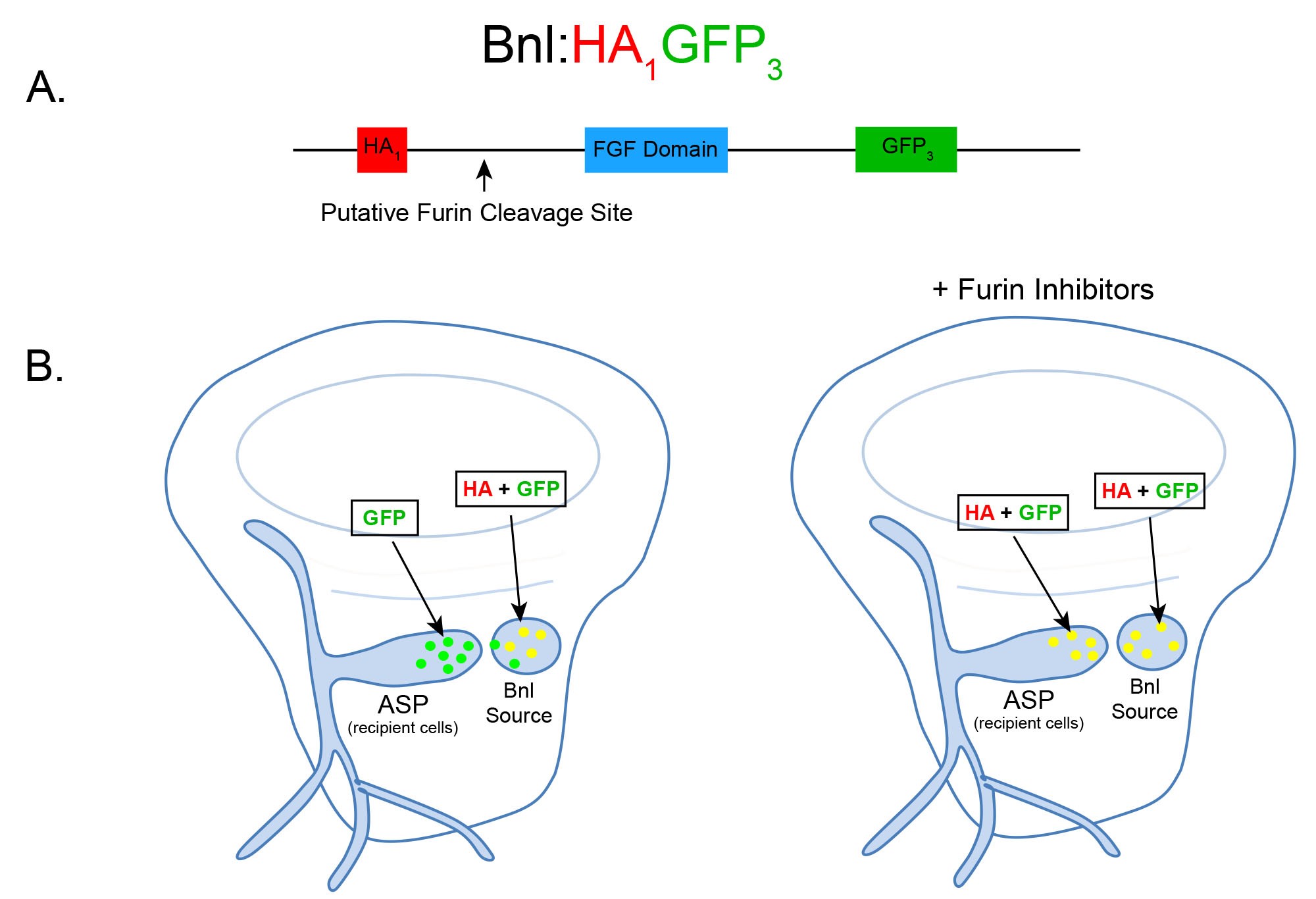
Figure 1. Schematic and expected dispersion patterns of Bnl:HA1GFP3. A. Schematic of the chimeric Bnl:HA1GFP3 protein, where an HA and GFP tag are located upstream and downstream of a Furin cleavage site, respectively. Bnl is cleaved at 164th amino acid and the HA and GFP tags are located at the 87th and 432nd residues, respectively. B. Cartoons of wing discs expressing Bnl:HA1GFP3 (yellow: GFP + α-HA red immunostaining) in the Bnl source. In ex vivo culture conditions without Furin inhibitors (left wing disc), Bnl is cleaved and only the GFP-tagged fragment of Bnl is transported to the ASP recipient cells. In ex vivo conditions with Furin inhibitors included, the amount of full length Bnl:HA1GFP3 (yellow) puncta in the ASP gradually increases with longer incubation time (right wing disc).
Materials and Reagents
- Drosophila rearing
- Fly Vials (Genesee Scientific, catalog number: 32-117)
- Vial Plugs (Genesee Scientific, catalog number: 49-101)
- Transgenic Drosophila lines: bnl:Gal4/TM6 and UAS-bnl:HA1GFP3 (Sohr et al., 2019)
- Standard Fly Nutrient Medium (Genesee Scientific, catalog number: 66-114)
- Propionic Acid (Sigma, catalog number: 402907)
- Late third instar larval dissection and washing
- Small paint brush (VWR, catalog number: 149-2130)
- Pipette tips (VWR, catalog number: 83007-376)
- Sterile Eppendorf tubes (Genesee Scientific, catalog number: 24-282)
- Pyrex spot plate (Fisher Scientific, catalog number: 13748B)
- 35 mm Plastic Petri dishes (Falcon, catalog number: 351008)
- Filter unit to sterilize WM1 media (VWR, catalog number: 89220-694)
- 100% ethanol (Pharmco, catalog number: 111000200)
- Deionized water
- Schneider’s medium (Sigma, catalog number: S0146)
- Insulin solution from bovine pancreas (Sigma, catalog number: I0516)
- Fly extract (DGRC, catalog number: 1645670)
- Penicillin/Streptomycin solution (Corning, catalog number: 30-002-CI)
- 70% ethanol
- WM1 media (Zartman et al., 2013) (see Recipes)
- Ex vivo culturing of dissected larvae
- Endotoxin-free water (Sigma, catalog number: 210-7)
- Furin inhibitor I (EMD Millipore, catalog number: 344930)
- Furin inhibitor II (EMD Millipore, catalog number: 344931)
- Phosphate buffered saline (PBS) (Hardy Diagnostics, catalog number: U137)
- Furin inhibitor cocktail (see Recipes)
- Antibody staining of larval carcasses
- 10 ml Syringe (Becton Dickinson, catalog number: 301029)
- Sterile syringe filter, 0.2 μm (VWR, catalog number: 28145-501)
- 16% formaldehyde (Ted Pella, Inc., catalog number: 18505)
- Triton X-100 (Sigma, catalog number: T8787)
- Normal goat serum powder (Jackson ImmunoResearch, catalog number: 005-000-121)
- α-HA antibody, 3F10 (Roche, catalog number: 11867423001)
- α-Discs Large antibody, 4F3 (DSHB, catalog number: AB_528203)
- Alexa Fluor 647 Goat anti-Rat IgG (Invitrogen, catalog number: A21247)
- Alexa Fluor 555 Goat anti-Mouse IgG (Invitrogen, catalog number: A21422)
- 4% formaldehyde (see Recipes)
- 0.1% PBS-T (see Recipes)
- 5% Normal goat serum in PBS-T (see Recipes)
- Mounting and imaging wing imaginal discs
- Microscope slides (Fisher Scientific, catalog number: 12-550-18)
- Coverslips (Corning, catalog number: 2845-22)
- Vectashield mounting medium (Vector, catalog number: H-1000)
- Nail polish (Electron Microscopy Sciences, catalog number: 72180)
Equipment
- Drosophila rearing
- Stereomicroscope (e.g., Standard Olympus SZ61 or Nikon SMZ745)
- Drosophila workstation with CO2 pad (Flowbuddy Complete; Genesee Scientific, catalog number: 59-122WC)
- Incubator (25 °C, Peltier Refrigerated incubator)
- Late third instar larval dissection and washing
- Dumont forceps #5 (Fine Science Tools, catalog number: 11252-00)
- Stereomicroscope (as described in section A)
- Pipette (1,000 μl)
- Scissors (VWR, catalog number: 89259-936)
- Ex vivo culturing of dissected larvae
- Sterile 25 °C incubator (Thermo Scientific, model: Heracell150i)
- Sterile biosafety cabinet for cell culture (Labconco, model: 302481101)
- Antibody staining of larval carcasses
Nutating mixer (VWR, model: 82007-002) - Mounting and imaging wing imaginal discs
- Forceps
- Laser scanning confocal microscope (e.g., Leica SP5X) or spinning disc confocal microscope (e.g., Nikon)
Software
- Imaging Software: IQ3 (Andor)
- ImageJ/Fiji
- Adobe Photoshop, Adobe Illustrator
- Microsoft Excel
Procedure
- Drosophila rearing/crossing
- Use the CO2 pad to anesthetize and collect ~20 female virgin bnl-Gal4 flies as well as ~10 male UAS-bnl:HA1GFP3 flies.
- Place the flies together in a vial with fresh fly food to set up the cross: bnl-Gal4 × UAS-bnl:HA1GFP3.
- Keep the crossed flies in a 25 °C incubator meant for rearing Drosophila.
- Transfer the crossed flies to a new vial containing fresh food every other day, but keep all of the older vials where the larvae for ex vivo culturing will be collected from.
- After egg laying, mid-late third instar larvae will be ready for dissection after 5-6 days.
- Preparation of late 3rd instar Drosophila larvae for dissection
- Dip a paint brush into deionized water and use it to remove late 3rd instar larvae that are wandering on sides of the fly vial.
- Place ~10 larvae into a spot plate well containing 1x PBS.
- Gently swirl them around with the paint brush to remove any large food debris that may be stuck on the surface of the larvae.
- With forceps, gently transfer these larvae to a separate spot plate well containing 70% ethanol.
- Swirl the larvae around gently and ensure that they are submerged. Incubate for 1-2 min to surface sterilize them.
- Remove the larvae from ethanol with forceps and place them into a spot plate well containing fresh 1x PBS (different well from Step B2).
- Rinse the larvae twice with 1x PBS in a separate spot plate well to completely remove the ethanol.
- 3rd Instar larva dissection
- Remove one washed larva from the group and place in a plastic Petri dish containing WM1 media (see Recipes). The larva should be completely submerged in the media.
- Using forceps and the dissection microscope, remove and discard the bottom 1/3 of the posterior portion of the larva. The anterior portion will contain all imaginal discs aside from the genital discs (Figure 2).
- Invert the anterior portion of the larva by using forceps to push the mouth hooks (tip of anterior half) through the interior of the larva towards the posterior end, resulting in the imaginal discs and other interior tissues being externalized.
Note: A video showing the initial larval dissection can be found at Youtube–Larval Brain Dissection in Drosophila Melanogaster (published by WilliamsCollege) - Completely remove the gut from where it attaches to the mouth hooks without disturbing the imaginal discs to minimize bacterial contamination during the ex vivo culture.
- Place this inverted anterior portion of the larva with gut removed into a sterile Eppendorf tube containing 1 ml WM1 media.
- Repeat Steps C1-C5 until all washed larvae are dissected. To avoid contamination, wash and dissect only 10-15 larvae per batch.
- Repeat Procedures B and C until enough dissected larvae are obtained for the experiments required.
Note: The total number of larvae dissected is determined by the number of time points and treatment groups that are being used (see Step E6 below).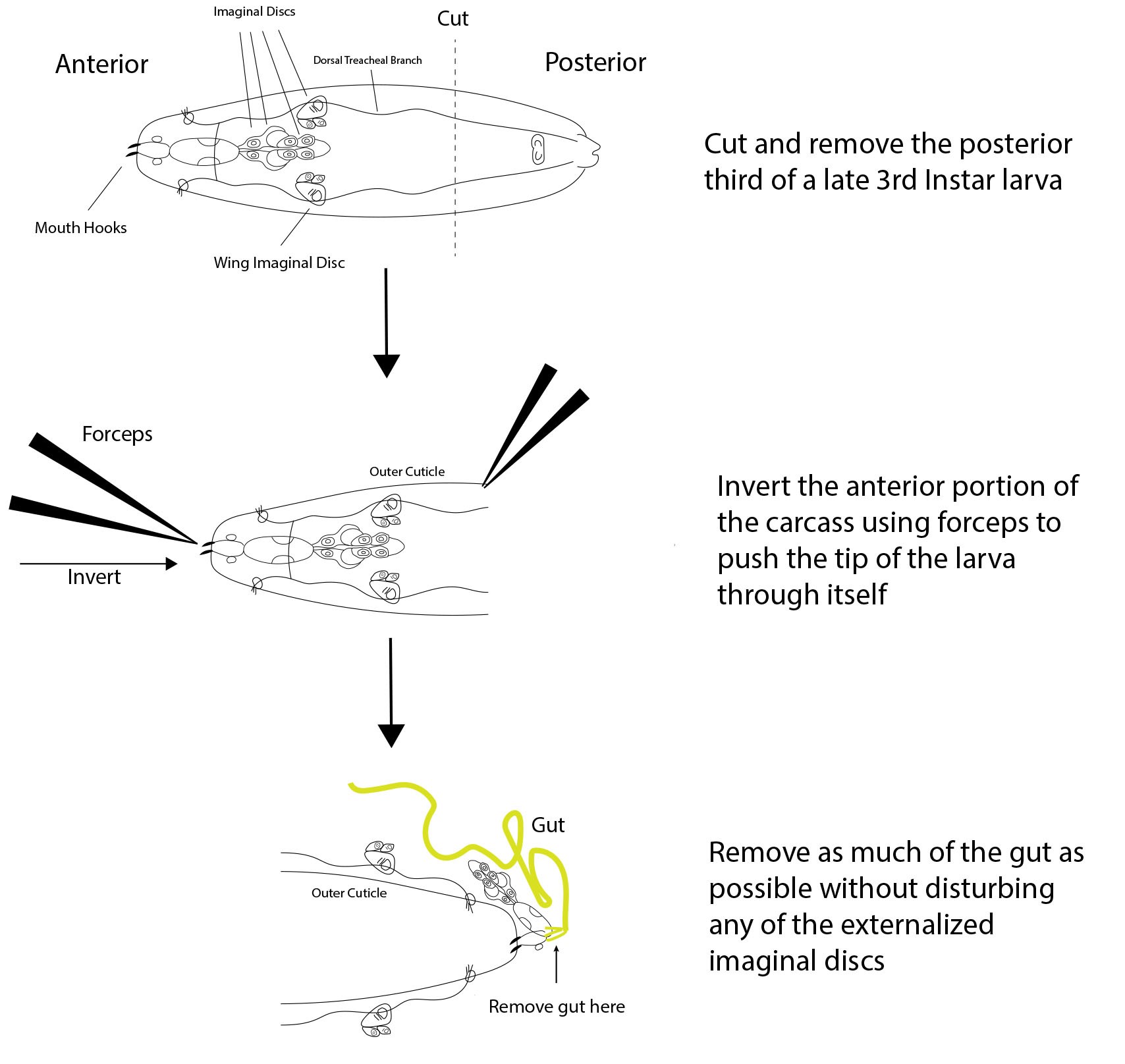
Figure 2. Dissection scheme of a late 3rd instar Drosophila Larva. A 3-step dissection is shown for a late 3rd instar larva. First, the posterior third of the larva is cut off and discarded, leaving just the desired anterior portion. Next, the anterior portion of the carcass is inverted using forceps to push the mouth hooks through the rest of the anterior portion, resulting in the externalization of all interior tissues including the wing imaginal discs. Finally, as much of the gut as possible is removed without disturbing the imaginal discs.
- Serial washing of dissected larvae
Note: Perform the following in a sterile Biosafety Cabinet.- Prepare 3 Eppendorf tubes with 1 ml of WM1 media in each.
- Wash scissor blades with 70% ethanol.
- Cut off ~1-2 mm of the end of a P-1000 pipette tip. The size of the cut tip opening should be large enough so that dissected larvae can fit into the pipette tip.
- Gently pipette and suck the dissected larvae from the bottom of the Eppendorf tube containing WM1 media into the pipette tip.
- Hold pressure on the pipette and let the dissected larvae settle to the bottom of the pipette tip, and then dispense them into one of the Eppendorf tubes with fresh WM1 media. Discard the remaining WM1 media in the pipette tip.
- Very gently invert this tube 3-5 times, and then let the dissected larvae settle to the bottom of the tube.
- Using a fresh pipette tip with the tip cut off in the same way as Step D2, repeat Steps D2-D6 twice with the remaining two Eppendorf tubes containing fresh WM1 media to wash the dissected larval carcasses (see Note 1).
- Ex vivo culture with furin inhibitors
- Prepare two plastic Petri dishes, one for the control group and one for the treatment group, as follows.
- To the treatment dish, add WM1 media and 10x Furin Inhibitor Cocktail (see Recipes) so that the cocktail will be at a 1x working concentration of 50 μM (i.e., add 200 μl of 10x Inhibitor Cocktail to 1.8 ml of WM1 media). Mix well.
Note: The total volume may have to be adjusted depending on the type of dish and number of carcasses, so the volume of 10x Inhibitor Cocktail used should be adjusted accordingly. We use a total volume of 2 ml in 35 mm Petri dishes. - To the control dish, use endotoxin-free water instead of Furin inhibitor cocktail. Mix well.
- Split the washed carcasses between the control and inhibitor-containing dishes.
- Cover each dish with the lid and place in a sterile 25 °C incubator.
- Incubate carcasses for 1, 2.5, and 5 h (see Note 2). For each time point, remove some (at least 5) of the carcasses using a pipette with a cut off tip while minimizing the amount of liquid removed. Place these carcasses into an Eppendorf tube containing 1 ml 1x PBS.
Note: The number of larvae initially dissected will depend on how many time points are being used for the ex vivo culture. We recommend using a minimum of 10 larvae per time point for each the control and treatment groups, but not to exceed 50 carcasses in a single petri dish.
- Antibody staining of ex vivo cultured larvae
- Gently invert the Eppendorf tube containing the larval carcasses 2-3 times.
- Let the carcasses settle to the bottom of the tube and remove the PBS from the tube by pipetting out as much liquid as possible without disturbing the carcasses with the pipette tip.
- Add 1 ml fresh 1x PBS to the carcasses.
- Repeat Steps F1-F3 twice to wash the carcasses to remove as much WM1 media and bacteria as possible.
- Add 1 ml 4% formaldehyde (see Recipes) to fix tissues.
- Rest the tube at a 45° angle so that the carcasses are settled in a single layer and fix for 25 min at room temperature without agitation.
- Remove the 4% formaldehyde as in Step F2.
- Wash 3 times with 1 ml 0.1% PBS-T (see Recipes). For each wash, leave on a nutating mixer for 5 min.
- Block for 1 h with rotation in 1 ml 5% Normal Goat Serum (see Recipes) at room temperature.
- Add α-HA (1:1,000) and α-Discs large (cell marker to visualize wing disc and ASP cells, 1:100) primary antibodies in 1 ml 5% Normal Goat Serum at their respective dilutions.
- Incubate overnight at 4 °C on a nutating mixer.
- Wash 3 times with 1 ml 0.1% PBS-T for 5 min each wash.
- Add the Alexa Fluor 555 Goat anti-Mouse and Alexa Fluor 647 Goat anti-Rat secondary antibodies (both 1:1,000) in 1 ml 0.1% PBS-T.
- Incubate on the nutating mixer for 2 h at room temperature.
- Wash 3 times with 1 ml 0.1% PBS-T for 15 min each wash.
- Mounting and imaging wing imaginal discs
- Place the stained carcasses from one group into a plastic Petri dish containing enough PBS-T such that the carcasses are submerged.
- Using the dissection microscope and forceps, remove the wing imaginal disc from the carcasses by cutting the connections to the cuticle and trachea surrounding the disc. Avoid touching the wing imaginal disc while removing it from the carcass (see [Purves and Brachmann, 2007] for a video showing an example of wing disc removal from a carcass).
- Pick up the wing disc by grabbing any associated tracheal branch with one of the forceps and transfer it to a slide with a drop of Vectashield mounting medium.
Note: If there is no available associated trachea to pick up the wing disc, grab the wing disc by the pouch of the disc (upper round part of the disc that does not contain Bnl-producing or receiving cells and will not be imaged). - After placing all wing discs from all carcasses on the slide, ensure that all wing discs are oriented with the associated trachea facing upwards (away from the slide).
- Place a coverslip on top of the wing discs by slowly lowering the coverslip down on one side to the other.
- Seal the edges of the coverslip to the slide using a thin layer of nail polish.
- Repeat Steps G1-G6 separately for each group and time point.
- Use the confocal microscope to image Bnl-receiving ASP cells and Bnl-producing wing disc source cells that are located just ahead of the tip of the ASP (Figure 1). Obtain z-stacks of the 488 (GFP), 647 (α-HA), and 561 (α-discs large) channels (see Note 3). An example is shown in Figure 3.
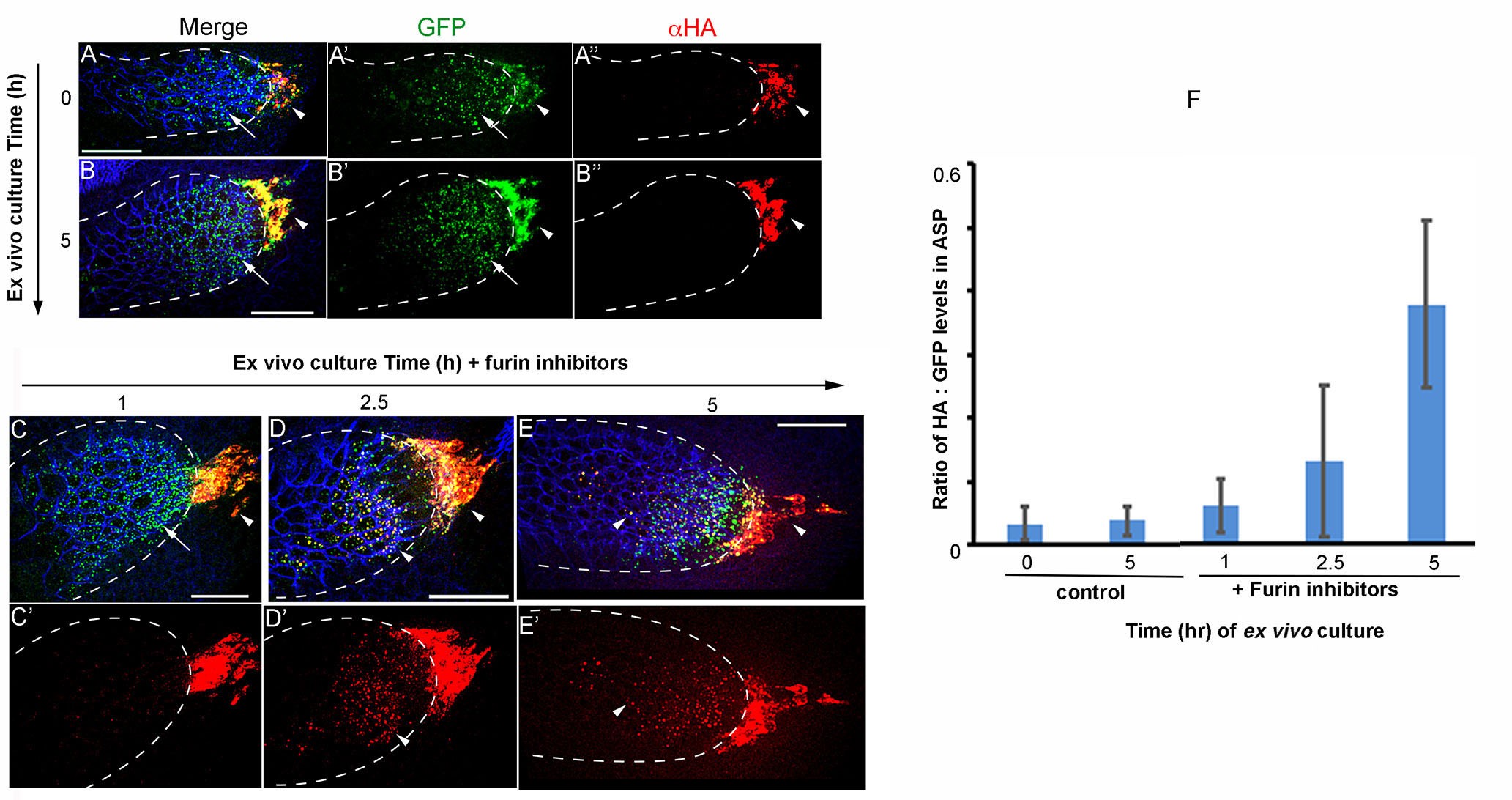
Figure 3. Ex vivo culture and Furin inhibitor assay on wing discs expressing Bnl:HA1GFP3. The α-HA stained (red) wing discs that expressed Bnl:HA1GFP3 under bnl-Gal4 were ex vivo-cultured for 0 (pre-treatment) and 5 h in the absence (A-B") and 1, 2.5 and 5 h in the presence of Furin inhibitors (C-E') as indicated; arrow, truncated Bnl:GFP3 derivative; arrowhead, uncleaved Bnl:HA1GFP3; blue, α-Discs large to mark cell outlines; merged (A, B, C-E) and either split green, red (A’-B”) or only red (C’-E’) channels were shown. (F) Graph comparing average levels of colocalized HA and GFP in ASPs cultured in the presence and absence of Furin inhibitors; samples were harvested at different time points from the continuous culture; N = 11 (0 h), 11 (1 h), 10 (2.5 h), 9 (5 h control), 12 (5 h test); P-values (ANOVA followed by Tukey HSD): P = 0.0001 for 5 h vs. either 0 h control, 5 h control, 1 h, or 2.5 h. Scale bars, 30 μm.
Data analysis
- Import and open one image file in ImageJ/Fiji.
- Create a flattened Z-stack that includes the entire depth of the tubular ASP recipient cells.
Note: Be sure not to include sections beneath the ASP in the flattened Z-stack, as the intensity quantitation can be affected by the presence of Bnl-expressing wing disc cells that are located under the ASP. - Use the polygon or freehand selection tool to draw a region of interest (ROI) around the ASP cells that contain Bnl:GFP signal.
Note: Be sure not to include Bnl-expressing wing disc cells that are located ahead of the tip of the ASP in the ROI. - Measure the intensity of both the α-HA channel and the GFP channel within this ROI and record these intensities in Excel.
- Move the polygon to an area of the wing disc where there is no GFP or HA signal.
- Measure the intensity in both channels again to obtain a measurement of the background signal and record the background intensities in Excel.
- Subtract the background intensity values from the ASP intensity values for both channels.
- Obtain an HA:GFP ratio in the ASP by dividing the background-corrected HA channel intensity by the background-corrected GFP channel intensity.
- Repeat steps 1-8 until all images from all groups are analyzed.
- Perform the appropriate statistical test to determine if the HA:GFP ratios in the ASP are significantly different between control and treatment groups.
Notes
- We suggest trying only the dissection and culturing steps before doing the full experiment with inhibitors and staining to make sure that the culturing portion of the protocol is working. If the majority of the dissected larval carcasses turn completely black during the culturing period at any point, the tissues are dying and there is likely something wrong with one or multiple aspects of the protocol. This could be due to dissection technique, insufficient washing of the larvae before and/or after dissecting, or contaminated materials/equipment. Keeping as sterile an environment as possible during this process is critical for the success of the experiment, as bacterial infections can occur easily. Many of the bacteria likely originate from the dissected gut of the larva, hence the importance of removing the gut and washing the dissected larval carcass in fresh WM1 to eliminate as much bacteria as possible.
- The WM1 media volume for culturing should be just sufficient to submerge the larval carcasses. We evaluated wing discs expressing Bnl:HA1GFP3 cultured up to 16 h. Inter-organ transfer of the truncated Bnl:GFP3 portion from the wing disc to the growing ASP indicated that normal signaling activity persists even after 16 h of ex vivo conditions. However, due to the submerged culture conditions, there is an increase in hypoxia-induced changes in the tissue as clearly seen by the appearance of ectopic Bnl-expressing cells between 8 and 16 h of culture.
- It is critical that all imaging parameters (i.e., laser intensity, exposure time, gain, etc.) remain the same for each image and group. This ensures that the quantitation of fluorescent intensities is comparable between different groups.
Recipes
- WM1 media (Zartman et al., 2013)
Schneider’s medium
5% fly extract
40 μg/ml bovine insulin
1% penicillin/streptomycin- Combine ingredients to their appropriate concentration in Schneider’s medium (we normally make 250 ml total)
- Filter sterilize the media by passing it through a 250 ml filter unit
- Aliquot into 15 ml tubes
- 10x Furin Inhibitor Cocktail
- Dissolve individual Furin Inhibitors (I and II) separately in endotoxin-free water to a concentration of 1 mM
- Combine equal volumes of Furin Inhibitor I and Furin Inhibitor II (1 mM each) to create the 10x cocktail of inhibitors with a final concentration of 500 μM for each inhibitor. The working concentration of the cocktail is 50 μM
- 4% formaldehyde
Add 250 μl of 16% formaldehyde to 750 μl PBS in an Eppendorf tube - PBS with 0.1% Triton X-100 (PBS-T)
- Add 100 μl Triton X-100 to 100 ml PBS
- Shake vigorously to mix
- 5% Normal Goat Serum in PBS-T
- Add 10 ml of PBS to the Normal Goat Serum (NGS) vial that contains dehydrated NGS
- Resuspend the NGS powder
- Use a 10 ml syringe to remove the NGS solution
- While the NGS solution is in the syringe, attach a 0.2 μm sterile syringe filter onto the end of the syringe
- Aliquot the NGS solution through the filter into Eppendorf tubes
- Make 5% NGS by adding 50 μl of the filtered NGS to 950 μl PBS-T
Acknowledgments
This project was funded by the National Institutes of Health: grants K99/R00HL114867 and R35GM124878 to S. Roy.
Competing interests
Authors declare no competing interests.
References
- Anderson, E. N. and Wharton, K. A. (2017). Alternative cleavage of the bone morphogenetic protein (BMP), Gbb, produces ligands with distinct developmental functions and receptor preferences. J Biol Chem 292(47): 19160-19178.
- Constam, D. B. (2014). Regulation of TGFβ and related signals by precursor processing. Semin Cell Dev Biol 32: 85-97.
- Du, L., Sohr, A., Yan, G. and Roy, S. (2018). Feedback regulation of cytoneme-mediated transport shapes a tissue-specific FGF morphogen gradient. Elife 7. e38137.
- Khatib, A. M., and Geraldine, S. (2006). Growth Factors: To Cleave or not to Cleave. In: Regulation of Carcinogenesis, Angiogenesis and Metastasis by the Proprotein Convertases (PCs). Springer, Dordrecht. 121-135.
- Künnapuu, J., Bjorkgren, I. and Shimmi, O. (2009). The Drosophila DPP signal is produced by cleavage of its proprotein at evolutionary diversified furin-recognition sites. Proc Natl Acad Sci U S A 106(21): 8501-8506.
- Künnapuu, J., Tauscher, P. M., Tiusanen, N., Nguyen, M., Loytynoja, A., Arora, K. and Shimmi, O. (2014). Cleavage of the Drosophila screw prodomain is critical for a dynamic BMP morphogen gradient in embryogenesis. Dev Biol 389(2): 149-159.
- LeMosy, E. K. (2006). Proteolytic regulatory mechanisms in the formation of extracellular morphogen gradients. Birth Defects Res C Embryo Today 78(3): 243-255.
- Purves, D. C. and Brachmann, C. (2007). Dissection of imaginal discs from 3rd instar Drosophila larvae. J Vis Exp (2): 140.
- Rousselet, E., Benjannet, S., Marcinkiewicz, E., Asselin, M. C., Lazure, C. and Seidah, N. G. (2011). Proprotein convertase PC7 enhances the activation of the EGF receptor pathway through processing of the EGF precursor. J Biol Chem 286(11): 9185-9195.
- Sato, M., and Kornberg, T. B. (2002). FGF is an essential mitogen and chemoattractant for the air sacs of the Drosophila tracheal system. Developmental Cell 3:195-207.
- Sohr, A., Du, L., Wang, R., Lin, L. and Roy, S. (2019). Drosophila FGF cleavage is required for efficient intracellular sorting and intercellular dispersal. J Cell Biol 218(5): 1653.
- Sopory, S., Kwon, S., Wehrli, M. and Christian, J. L. (2010). Regulation of Dpp activity by tissue-specific cleavage of an upstream site within the prodomain. Dev Biol 346(1): 102-112.
- Tokhunts, R., Singh, S., Chu, T., D'Angelo, G., Baubet, V., Goetz, J. A., Huang, Z., Yuan, Z., Ascano, M., Zavros, Y., Therond, P. P., Kunes, S., Dahmane, N. and Robbins, D. J. (2010). The full-length unprocessed hedgehog protein is an active signaling molecule. J Biol Chem 285(4): 2562-2568.
- Tulin, S. and Stathopoulos, A. (2010). Analysis of Thisbe and Pyramus functional domains reveals evidence for cleavage of Drosophila FGFs. BMC Dev Biol 10: 83.
- Wharton, K. A. and Serpe, M. (2013). Fine-tuned shuttles for bone morphogenetic proteins. Curr Opin Genet Dev 23(4): 374-384.
- Zartman, J., S. Restrepo, and K. Basler. (2013). A high-throughput template for optimizing Drosophila organ culture with response-surface methods. Development. 140:667–674.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Sohr, A., Du, L. and Roy, S. (2019). Ex vivo Drosophila Wing Imaginal Disc Culture and Furin Inhibitor Assay. Bio-protocol 9(16): e3336. DOI: 10.21769/BioProtoc.3336.
- Sohr, A., Du, L., Wang, R., Lin, L. and Roy, S. (2019). Drosophila FGF cleavage is required for efficient intracellular sorting and intercellular dispersal. J Cell Biol 218(5): 1653.
Category
Developmental Biology > Cell signaling > Ligand
Cell Biology > Cell imaging > Confocal microscopy
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.