- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Construction of Viral Vectors for Cell Type-specific CRISPR Gene Editing in the Adult Mouse Brain
Published: Vol 9, Iss 16, Aug 20, 2019 DOI: 10.21769/BioProtoc.3334 Views: 6915
Reviewed by: Arnau Busquets-GarciaValeria B Fernandez ValloneAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Dec 2018

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics








Abstract
Recently developed gene editing technologies based on engineered CRISPR/Cas9 systems enables researchers to disrupt genes in a cell type-specific manner in the adult mouse brain. Using these technologies, we recently showed that the dopamine beta-hydroxylase gene in Locus Coeruleus (LC) norepinephrine neurons plays a vital role in the maintenance of wakefulness. Our method consists of four steps, (1) crossing Cre-dependent spCas9 knockin mice with a Cre-driver mouse line to express spCas9 in the target neural populations, (2) cloning of sgRNA, (3) construction of an AAV (adeno associated virus) vector expressing dual sgRNA, and (4) virus packaging and stereotaxic injection of the virus into the target brain area. Here, we describe a detailed protocol of AAV vector construction for cell type-specific CRISPR gene editing in the adult mouse brain. The method adopts a dual-sgRNA strategy for efficient disruption of the target gene. At first, a few different sgRNAs targeting the same gene are cloned into a plasmid expressing spCas9. After evaluation of the sgRNAs by a T7 endonuclease assay, the two most efficient sgRNAs are cloned in tandem into an AAV vector using the Gibson Assembly method.
Keywords: CRISPR/Cas9Background
Interrogation of gene functions in specific cell subtypes in the brain remains a challenge. The conventional techniques such as RNAi-based methods lack cellular specificity and efficiency. On the other hand, recently developed gene editing techniques using the CRISPR/Cas9 system allows researchers efficient gene disruption in a cell type-specific manner. A research group led by Feng Zhang showed the disruption of multiple genes in the adult mouse brain by AAV-mediated delivery of sgRNA (Swiech et al., 2015). Also, the group established cre-dependent spCas9 knockin mice (Platt et al., 2014). In a recent study, we used an AAV vector carrying dual sgRNA to improve the efficiency of bi-allelic gene targeting (Yamaguchi et al., 2018). We here describe a protocol for the construction of dual sgRNA AAV vector (Figure 1).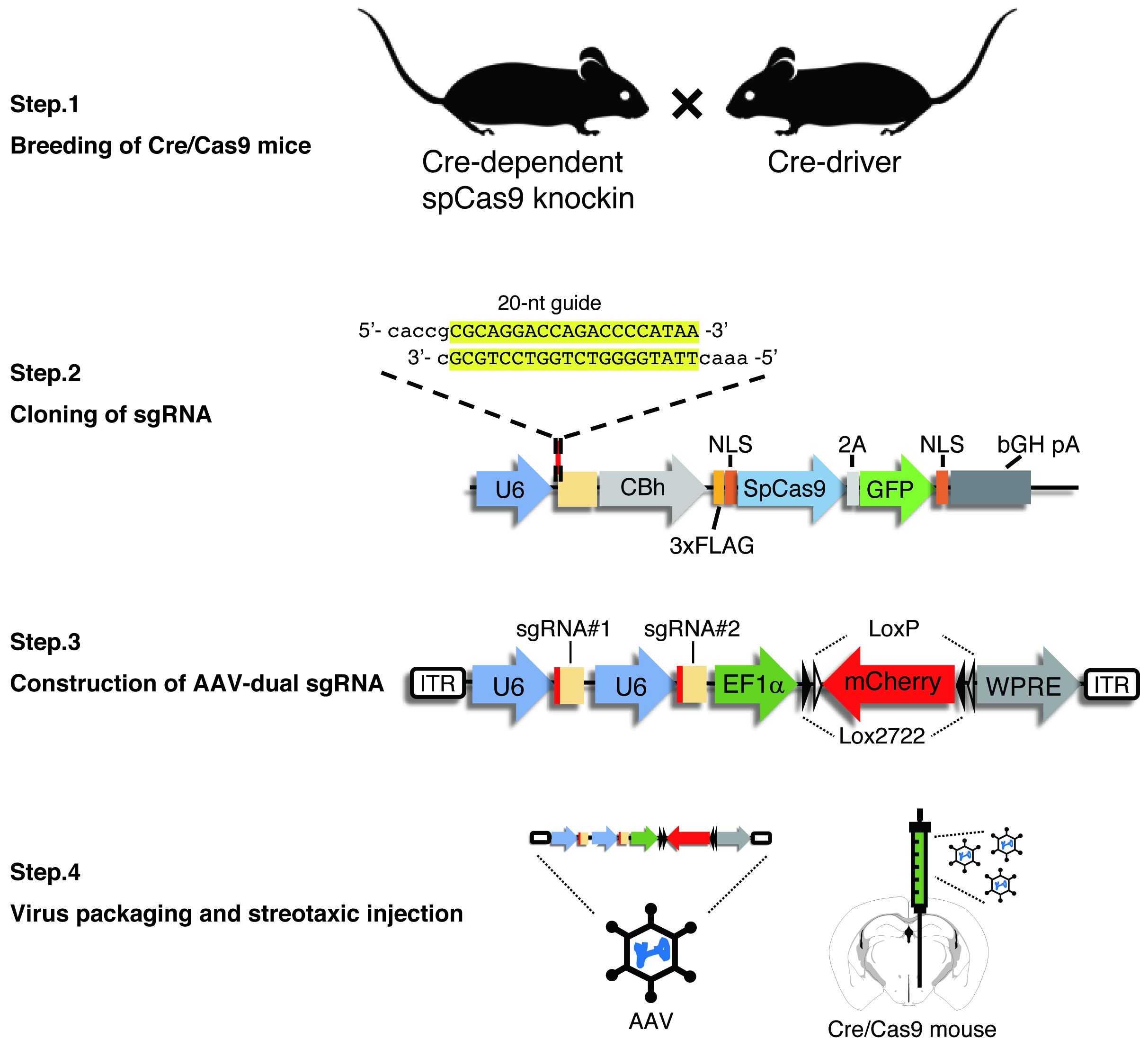
Figure 1. Schematic representation of all the procedure. Our method consists of four steps: (1) crossing Cre-dependent spCas9 knockin mice with a Cre-driver mouse line to express spCas9 in the target neural populations, (2) cloning of sgRNA into the spCas9 expressing plasmid, (3) construction of an AAV vector tandemly expressing dual sgRNA, and (4) AAV packaging and stereotaxic viral injection into the target brain area.
Materials and Reagents
- Pipette tips
- 24-well plate (coated) (Thermo Fisher Scientific, catalog number: 142475)
- 1 ml plastic tube
- NEB10-beta Competent E. coli (New England Biolabs, catalog number: C3019)
- pX458 (Addgene, catalog number: 48138)
- pAAV EF1α DIO mCherry (Addgene, catalog number: 20299)
- BbsI-HF (New England Biolabs, catalog number: R3539)
- MluI-HF (New England Biolabs, catalog number: R3198)
- DpnI (New England Biolabs, catalog number: R0176)
- Alkaline Phosphatase, Calf Intestinal (CIP) (New England Biolabs, catalog number: M0290)
- Guide oligonucleotides (see Procedure A)
- Quick ligation kit (New England Biolabs, catalog number: M2200)
- Gibson Assembly Master Mix (New England Biolabs, catalog number: E2611)
- PrimeSTAR HS DNA polymerase (Takara, catalog number: R010)
- dNTP Mix 10 mM each (the mix of 10 mM of dTTP, dCTP, dGTP and dATP) (GenScript, catalog number: C01689)
- QIAquick Gel Extraction kit (QIAGEN, catalog number: 28704)
- QIAprep Spin Miniprep kit (QIAGEN, catalog number: 27104)
- Tris-EDTA, (1x Solution) (Fisher BioReagents, catalog number: BP2473)
- LB Broth EZMix Powder (Sigma, catalog number: L7658)
- LB Agar (Fisher BioReagents, catalog number: BP1425)
- Ampicillin (Gemini Bio-Products, catalog number: 400-130P)
- Primer to verify the cloning of sgRNA into pX458: 5’-gactatcatatgcttaccgt-3’
- Primer to verify the tandem cloning of sgRNAs into pAAV: 5’-actgacgggcaccggagcca-3’
- Primers for Gibson Assembly:
Primer S1: 5’-taggggttcctgcggccgcacgcgtgagggcctatttc-3’
Primer AS1: 5’-ataggccctctctagaaaaaaagcaccgactc-3’
Primer S2: 5’-tttttctagagagggcctatttcccatg-3’
Primer AS2: 5’-atccatctttgcaaagcttacgcgtaaaaaagcaccgac-3’Optional (see Procedure C)
- NIH3T3 cell (ATCC, catalog number: CRL-1658)
- AAV vector (pX458-sgRNA or pX458 empty as negative control )
- Primers: You can design the primers to amplify the target exons by CHOPCHOP (see Procedure C)
- PrimeSTAR HS DNA polymerase
- FuGENE6 Transfection Reagent (Promega, catalog number: E2693)
- DMEM (1x) (Gibco, catalog number: 11995-065)
- Fetal Bovine Serum (Corning, catalog number: 35-015-CV)
- Penicillin/Streptomycin (Gibco, catalog number: 15070-063)
- PBS, pH 7.4 (Gibco, catalog number: 10010-023)
- TrypLE Select (1x) (Gibco, catalog number: 12563-011)
- QuickExtract DNA Extraction Solution (Epicentre, catalog number: QE0905T)
- T7 Endonuclease I (New England Biolabs, catalog number: M0302)
Equipment
- Pipettes (Gilson, Pipetman classic P10, P20, P200)
- Centrifuge (Eppendorf, Centrifuge 5424)
- Incubator (VWR, incubating 5000IR orbital shaker)
- Thermocycler (Applied Biosystems, GeneAmp PCR system 2700)
- Electrophoresis chamber (Labnet, ENDURO GEL XL)
- (Optional) CO2 Incubator (Binder, CB 170)
- (Optional) Clean bench (Labconco, Purifier classII biosafety cabinet)
- (Optional) Centrifugation (Thermo Scientific, Soryall legend XTR)
Procedure
- Design of sgRNA
Single guide RNAs (sgRNAs) with high efficiency and specificity can be designed by using gRNA design webtools. We recommend CHOPCHOP (http://chopchop.cbu.uib.no) because of its user-friendly interface (Labun et al., 2016). Also, you may find the guide design resources on the website of Feng Zhang’s lab (https://zlab.bio/guide-design-resources). The sgRNA should target early exons (near transcriptional start site) or essential exons of gene function. We usually choose 4 to 6 target sites on early exons which do not have the start codon because frameshift mutations near the start codon can lead to translation from other in-frame ATGs, resulting in the expression of a functional protein. If the target sequence is 5’-CGCAGGACCAGACCCCATAATGG-3’ [Protospacer adjacent motif (PAM) is labeled in blue], synthesize 5’-caccgCGCAGGACCAGACCCCATAA-3’ (sense) and 5’-aaacTTATGGGGTCTGGTCCTGCGc-3’ (anti-sense) to clone the guide sequence into pX458 (Ran et al., 2013) sgRNA scaffold (Figure 3A). cacc (lower case) in the first oligo and aaac (lower case) in the second oligo are overhangs for cloning into BbsI-site in pX458. We also add “g” between cacc and the target sequence in the first oligo because the human U6 promoter prefers “g” at the transcription start site for strong expression. Also, you may design primers using CHOPCHOP to amplify the target site for further evaluation of sgRNAs using the T7 endonuclease I assay (see Procedure C). - Cloning of sgRNAs into the spCas9 expressing plasmid (pX458)
- Digestion of pX458 with BbsI:
X μl pX458 (10 μg) 5 μl 10x NEB cut smart 1 μl BbsI-HF 24 - X μl MilliQ water 30 μl in total - Incubate at 37 °C overnight.
- Gel purify the plasmid using QIAquick Gel extraction kit. Elute the plasmid with 50 μl of 1x Tris-EDTA buffer.
- Annealing of oligonucleotides encoding sgRNAs:
1 μl oligonucleotide, sense (100 μM) 1 μl oligonucleotide, antisense (100 μM) 8 μl 1x Tris-EDTA buffer 10 μl in total - Incubate at 95 °C for 5 min. Then cool down to room temperature by switching off the thermocycler (30 min).
- Prepare the ligation reaction as following:
1 μl diluted (1:100 with 1x Tris-EDTA buffer) annealed oligonucleotides 1 μl BbsI-digested pX458 (50 ng/μl) 5 μl 2x ligation buffer 1 μl Quick ligase 2 μl MilliQ water 10 μl in total - Incubate at room temperature for 10 min.
- Add 5 μl of the ligation reaction to 50 μl of NEB10-beta.
- Next place on ice for 10 min.
- Heat shock at 42 °C for 30 s.
- Transfer onto ice for 3 min.
- Plate the reaction on an LB-Agar plate containing ampicillin (50 μg/ml).
- Incubate at 37 °C overnight.
- Inoculate 2 ml LB broth containing ampicillin (50 μg/ml) from a single colony and incubate at 37 °C overnight with agitation.
Note: 60-70% of colonies have a correct insertion in our experience. - Purify the plasmid from 1 ml of the overnight culture using QIAprep Spin Miniprep kit.
Elute the plasmid with 50 μl of buffer EB and determine the concentration - Verify the plasmid by sequencing with a primer (5’-gactatcatatgcttaccgt-3’).
- Digestion of pX458 with BbsI:
- (Optional) Evaluation of sgRNA by T7 endonuclease I assay
- Plate NIH3T3 cells at the density of 2.5 x 104 cells (500 μl/well, DMEM/10% FBS/1% Penicillin /1% Streptomycin) onto a well of 24-well plate.
- Incubate in a 37 °C CO2 incubator overnight.
- Transfection
- Add 1.5 μl of FuGENE6 reagent to 50 μl of serum-free DMEM in a 1 ml plastic tube and mix by vortexing.
- Incubate the mixture for 5 min at room temperature.
- Add 0.5 μg of pX458-sgRNA or control vector to the mixture and mix by vortexing.
- Incubate the mixture for 30 min at room temperature.
- Add the mixture to each well of a 24-well plate containing cells.
- Incubate in a 37 °C CO2 incubator for 24 h.
- Replace the cell culture media with fresh one (DMEM/10% FBS/1% Penicillin /1% Streptomycin).
- Repeat Steps C3d-C3e.
Note: Repeated transfection is essential for high expression of Cas9 and sgRNA to edit the target gene in NIH3T3 cells. - Incubate in a 37 °C CO2 incubator for 48 h.
- Remove the supernatant of cells by pipetting and wash cells with 500 μl of 1x PBS.
- Add 100 μl of QuickExtract to each well and collect the lysate containing genomic DNA to a 1 ml plastic tube.
- Incubate at 65 °C for 15 min and then at 98 °C for 10 min.
- Centrifuge at 20,000 x g for 10 min.
- Collect 60 μl of the supernatant (can be stored at -20 °C).
- PCR to amplify 200-300 bp fragments around the CRISPR binding site. The primers can be designed by CHOPCHOP.
6 μl lysate 1.5 μl dNTP (the mixture of dTTP, dCTP, dGTP and dATP, each at a final concentration of 10 mM) 12 μl 5x buffer 1.5 μl primer S (10 μM) 1.5 μl primer AS (10 μM) 1 μl PrimeSTAR HS DNA polymerase 36.5 μl MilliQ water 60 μl in total - Start PCR with the following settings:
- Denaturation at 95 °C for 2 min.
- 35 cycles of
95 °C for 10 s
55 °C for 5 s
72 °C for 45 s - Final elongation at 72 °C for 2 min.
- Store at 4 °C.
- Gel purify PCR product using QIAquick Gel extraction kit. Elute PCR product with 50 μl of buffer EB.
- Denaturarion and annealing of PCR product:
5 μl PCR product (200 ng)
2 μl 10x NEB buffer 2
12 μl MilliQ water
19 μl in total - 95 °C for 5 min.
- Cool down to room temperature by switching off the thermocycler (30 min).
- Denaturarion and annealing of PCR product:
- Add 1 μl of diluted (1:5) T7 endonuclease I (2.5 U/μl).
Note: Dilution of T7 endonuclease I is essential because an excess amount of the enzyme will result in non-specific cleavage of DNA. - Incubate at 37 °C for 30 min.
- Run on 2% agarose gel and verify bands in sgRNA transfected-wells compared with control wells (Figure 2).
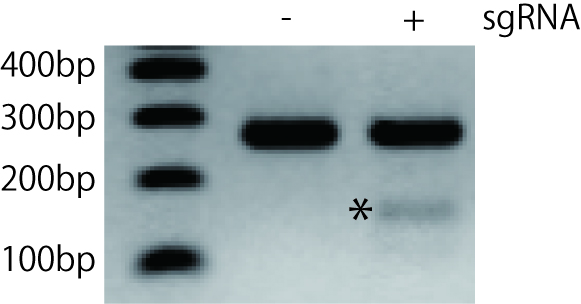
Figure 2. Agarose gel electrophoresis of T7 endonuclease I-digested PCR products derived from NIH3T3 cells transfected with sgRNA and Cas9. The target exon was PCR-amplified and gel purified. After the denaturation and annealing, the PCR product was digested by T7 endonuclease I and run on 2% agarose gel. The asterisk indicates cleaved DNA fragment. In this experiment, we found only one shorter fragment. However, you may find bands of two different sizes after the cleavage if you carefully design the primers. It can be useful to design PCR primers such that the indel resulting from genome editing is not in the middle of the amplicon, ultimately resulting in two cleaved bands of different sizes.
You can quantify the efficiency of gene modification based on the result of T7 endonuclease I assay. The percentage of cleavage on a gel could be converted to the percentage of gene modification by using an equation below.% gene modification = 100 x (1 - (1 - fraction cleaved)1/2 )(Guschin et al., 2010)
Also, you may evaluate the efficiency of gene editing by a webtool called Synthego’s ICE (https://ice.synthego.com/#/). Target exons from edited or unedited (control) cells are PCR-amplified and Sanger sequenced. You may submit the results of sequencing to the webtool and ICE compares these sequence traces to give a detailed analysis of gene editing.
- Plate NIH3T3 cells at the density of 2.5 x 104 cells (500 μl/well, DMEM/10% FBS/1% Penicillin /1% Streptomycin) onto a well of 24-well plate.
- Tandem cloning of two sgRNAs into pAAV EF1α DIO mCherry
In Procedure A, you clone several sgRNAs targeting the same gene. For the efficient gene disruption, you tandemly clone two different sgRNAs driven by independent human U6 promoters into an AAV vector (Figures 3B and 3C) using the Gibson Assembly method.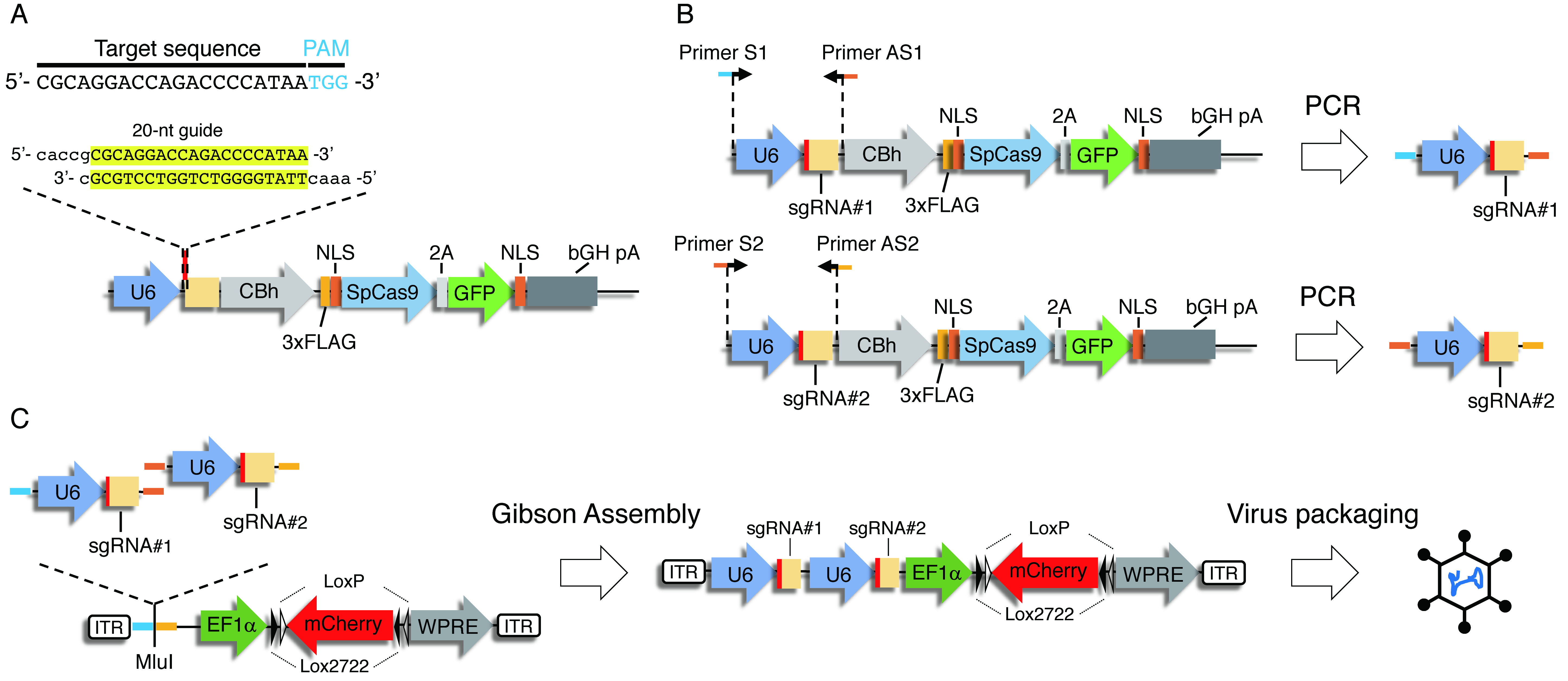
Figure 3. Construction strategy of a dual sgRNA viral vector. A. Cloning of guide oligonucleotides. Oligonucleotides with 20-nt guide sequence and overhangs for cloning into BbsI site in pX458 were annealed and cloned into BbsI-digested pX458. B. Cloning of U6 promoter-driven sgRNAs. U6 promoter followed by sgRNA1 or sgRNA2 were PCR amplified, respectively, by using primers with overhangs for Gibson Assembly. C. Gibson Assembly of the dual sgRNA virus vector. U6-gRNA1 and U6-gRNA2 were cloned into MluI-digested pAAV EF1 DIO mCherry using Gibson Assembly.- Digestion, dephosphorylation and purification of pAAV EF1α DIO mCherry with MluI.
- Digestion:
X μl pAAV EF1α DIO mCherry (10 μg) 5 μl 10x NEB cut smart 1 μl MluI-HF 24 - X μl MilliQ water 30 μl in total - Incubate at 37 °C overnight.
- Add 1 μl of CIP, and then incubate at 37 °C for 60 min.
- Gel purify the plasmid using QIAquick Gel extraction kit.
- Elute the plasmid with 50 μl of buffer EB.
- Digestion:
- Amplification of U6 promoter plus sgRNA#1 or sgRNA#2.
- PCR reaction#1 (Expected size is 393 bp):
3 μl pX458-sgRNA#1 (10 ng/μl) 1.5 μl dNTP, 10 mM each 12 μl 5x buffer 1.5 μl primer S1 (10 μM); 5’- taggggttcctgcggccgcacgcgtgagggcctatttc -3’ 1.5 μl primer AS1 (10 μM); 5’- ataggccctctctagaaaaaaagcaccgactc -3’ 1 μl PrimeSTAR HS DNA polymerase 39.5 μl MilliQ water 60 μl in total - PCR reaction#2 (Expected size is 388 bp):
3 μl pX458-sgRNA#2 (10 ng/μl) 1.5 μl dNTP, 10 mM 12 μl 5x buffer 1.5 μl primer S2 (10 μM); 5’- tttttctagagagggcctatttcccatg -3’ 1.5 μl primer AS2 (10 μM); 5’- atccatctttgcaaagcttacgcgtaaaaaagcaccgac -3’ 1 μl PrimeSTAR HS DNA polymerase 39.5 μl MilliQ water 60 μl in total
- PCR reaction#1 (Expected size is 393 bp):
- Start PCR with the following settings.
- Denaturation at 95 °C for 2 min.
- 35 cycles of
95 °C for 10 s
62 °C for 5 s
72 °C for 45 s - Final elongatation at 72 °C for 2 min.
- Store at 4 °C.
- Run on a 2% agarose gel.
- Gel purify the PCR product using QIAquick Gel extraction kit. Elute the PCR product with 50 μl of MilliQ water.
- Digestion of the residual template plasmid:
45 μl PCR amplicon#1 or #2 5 μl 10x NEB cut smart 1 μl DpnI 51 μl in total - Incubate at 37 °C for 30 min, 80 °C for 30 min.
- Gibson assembly:
1 μl CIP-treated MluI-digested pAAV EF1α DIO mCherry (100 ng/μl) 3 μl PCR amplicon#1 3 μl PCR amplicon#2 3 μl MilliQ water 10 μl Gibson Assembly mix 20 μl in total - Incubate at 50 °C for 15 min.
- Add 2 μl of ligation reaction to 50 μl of NEB10-beta.
- Follow Steps B9-B15.
- Verify the plasmid by sequencing with a primer (e.g., 5’-actgacgggcaccggagcca-3’).
- Proceed to virus packaging.
Note: We usually request virus packaging services from Stanford Neuroscience Gene Vector and Virus core. - Keep the virus stock at -80 °C.
Note: This strategy can disrupt genes in a cell type-specific manner. You can use the virus not only for adult mouse brain but also other organs.
- Digestion, dephosphorylation and purification of pAAV EF1α DIO mCherry with MluI.
Recipes
All materials and reagents are commercially available.
Acknowledgments
H.Y. was supported by Uehara memorial foundation research fellowship. L.d.L was supported by National Institute of Health Grants AG047671, MH087592, MH102638. This protocol was adapted from Yamaguchi et al. (2018) and includes a recent improvement of the method of our lab.
Competing interests
Competing interestsThe authors declare no competing financial interests.
References
- Labun, K., Montague, T. G., Gagnon, J. A., Thyme, S. B. and Valen, E. (2016). CHOPCHOP v2: a web tool for the next generation of CRISPR genome engineering. Nucleic Acids Res 44(W1): W272-276.
- Platt, R. J., Chen, S., Zhou, Y., Yim, M. J., Swiech, L., Kempton, H. R., Dahlman, J. E., Parnas, O., Eisenhaure, T. M., Jovanovic, M., Graham, D. B., Jhunjhunwala, S., Heidenreich, M., Xavier, R. J., Langer, R., Anderson, D. G., Hacohen, N., Regev, A., Feng, G., Sharp, P. A. and Zhang, F. (2014). CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell 159(2): 440-455.
- Ran, F. A., Hsu, P. D., Wright, J., Agarwala, V., Scott, D. A. and Zhang. F. (2013). Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8(11): 2281-2308.
- Swiech, L., Heidenreich, M., Banerjee, A., Habib, N., Li, Y., Trombetta, J., Sur, M. and Zhang, F. (2015). In vivo interrogation of gene function in the mammalian brain using CRISPR-Cas9. Nat Biotechnol 33(1): 102-106.
- Yamaguchi, H., Hopf, F. W., Li, S. B. and de Lecea, L. (2018). In vivo cell type-specific CRISPR knockdown of dopamine beta hydroxylase reduces locus coeruleus evoked wakefulness. Nat Commun 9(1): 5211.
- Guschin, D. Y., Waite, A. J., Katibah, G. E., Miller, J. C., Holmes, M. C. and Rebar, E. J. (2010). A rapid and general assay for monitoring endogenous gene modification. Methods Mol Biol 649: 247-256.
Article Information
Copyright
© 2019 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Yamaguchi, H. and de Lecea, L. (2019). Construction of Viral Vectors for Cell Type-specific CRISPR Gene Editing in the Adult Mouse Brain. Bio-protocol 9(16): e3334. DOI: 10.21769/BioProtoc.3334.
Category
Neuroscience > Neuroanatomy and circuitry > Animal model
Molecular Biology > DNA > Gene expression
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.